

Abstract
Dentin adhesives may undergo phase separation when bonding to wet demineralized dentin. We hypothesized that adhesives exhibiting phase separation will experience enhanced biodegradation of methacrylate ester groups. The objective of this project was to study the effect of enzyme-exposure on the release of methacrylic acid (MAA) and 2-hydroxyethyl methacrylate (HEMA) from adhesives formulated under conditions simulating wet bonding. HEMA/bisGMA(2,2-bis[4(2-hydroxy-3-methacryloyloxy-propyloxy)-phenyl] propane), 45/55 w/w ratio, was formulated with different water content: 0 Wt % (A00), 8 wt % (A08), and 16 wt % (A16). After a three day prewash, adhesive discs were incubated with/without porcine liver esterase (PLE) in phosphate buffer (PB, pH 7.4) at 37°C for 8 days. Supernatants were collected daily and analyzed for MAA and HEMA by HPLC. For all formulations, daily MAA release in the presence of PLE was increased compared to MAA release in PB. HEMA release in the presence of PLE was not detected while HEMA release was consistently measured in PB. A08 and A16 released significantly larger amounts of HEMA compared to A00. Analysis of the cumulative release of analytes showed that the leachables in PLE was significantly increased (p < 0.05) as compared with that released in PB indicating that MAA release was not only formed from unreacted monomers but from pendant groups in the polymer network. However, the levels of analytes HEMA in PB or MAA in PLE were increased in A08 and A16 as compared with A00, which suggests that there could be a greater loss of material in HEMA/bisGMA adhesives that experience phase separation under wet bonding conditions.
Keywords: dental/craniofacial material, stability, biodegradation, enzyme, phase separated polymer
INTRODUCTION
The main function of dentin adhesives is to provide bonding between materials, such as composite, and the tooth structure. As evidenced by clinical investigations, the durability of this bond is integral to the success of composites restorations, for example, the failure of the bond at the composite/tooth interface is a major factor in the premature breakdown of moderate to large composite restorations.1–4 The breakdown of the bond between the tooth and composite has been linked to the failure of our current adhesives to consistently seal and adhere to the dentin.4–9 Results from both in vitro and in vivo studies indicate that adhesive failure allows bacterial enzymes, oral fluids, and even bacteria to infiltrate the spaces between the tooth and composite.10 The penetration of these agents into the spaces between the tooth and composite undermines the restoration and leads to recurrent caries, hypersensitivity, and pulpal inflammation.11–13 Thus, the adhesive/dentin bond can be regarded as the first defense against substances that may penetrate and ultimately undermine the composite restoration in vivo.
Water is a major interfering factor when bonding adhesives and/or composites to the tooth.14 The water content of the dentin surfaces varies as a function of depth,15–18 nature of the substrate, that is, caries-affected or healthy dentin 19 and the presence of residual rinse water. Current commercial dentin adhesives combine the primer and adhesive in one bottle. In the presence of water, the adhesive may undergo phase separation. Within the wet demineralized dentin matrix, adhesive phase separation leads to very limited infiltration of bisGMA, the critical dimethacrylate component.20 Under these conditions, the adhesive/dentin bond has neither structural integrity nor durability. Adhesive phase separation inhibits the formation of an impervious, structurally integrated bond at the composite/tooth interface.8,9 Water is also known to facilitate the chemical degradation of adhesives. Water may be trapped within the matrix during photopolymerization in the mouth or it can enter the adhesive matrix by diffusion into the loosely cross-linked or hydrophilic HEMA-rich domains. The hydrophilic domain exhibits limited monomer/polymer conversion because of adhesive phase separation20 and lack of compatibility between the photoinitiator and hydrophilic phase.18 The poorly polymerized hydrophilic polymer domain degrades rapidly in the aqueous oral environment.9,21
The enzyme-catalyzed hydrolysis of ester linkages in methacrylate-based monomers and polymers has been largely reported from studies with dental composites.22–28 Esterases known to activate ester hydrolysis include salivary esterases, cholesterol esterase, pseudocholinesterase, porcine liver esterase, and acethylcholinesterase. In contrast to composites, there has been very limited investigation of the biodegradation of dentin adhesives in the presence of esterases.29,30 The objective of this project was to study the effect of enzyme exposure on the release of MAA and unpolymerized HEMA from HEMA/bisGMA adhesives formulated under conditions simulating wet bonding. The overall hypothesis was that the enzymatic biodegradation of HEMA/bisGMA formulations exhibiting heterogeneous phase structure would be enhanced as compared to homogeneous adhesive without phase separation. Adhesive phase separation has been modeled previously using HEMA/bisGMA resins formulated with water where a percent of water concentration at which phase separation occurs could be determined.9,20
MATERIALS AND METHODS
Materials
The model resin consisted of hydroxyethylmethacrylate (HEMA, Acros Organics, NJ) and 2,2-bis[4-(2-hydroxy-3-methacryloxypropoxy) phenyl]-propane (BisGMA, Poly-sciences, Washington, PA) at 45/55 wt/wt ratio. Distilled water at concentrations of 0, 8, 16 wt % was selectively added into the neat resins. The concentration of water was based on the total final weight of the model resin. Shaking and sonication were required to yield well-mixed resin solutions. A loss in clarity, as noted by visual examination, was interpreted as evidence of macro-phase separation. The following photoinitiators (from Aldrich, Milwaukee, WI) were used in this study: camphorquinone (CQ), ethyl-4-(dimethylamino) benzoate (EDMAB) at 0.5 and 0.5 mol %, respectively, with respect to the total amount of monomer. Porcine liver esterase (PLE, EC 3.1.1.1) was obtained from Sigma Chemical Co., St. Louis, MO. All other chemicals were reagent grade and used without further purification.
Adhesive Disc Preparation and Characterization
The preparation of the cylindrical specimens, which were used in this investigation, has been described previously.31 In brief, the model resins were injected into circular aluminum molds (ID 4.0 mm) and sealed with a cleaned cover glass. Each specimen was light-cured for 20 s using a dental curing light (Spectrum® 800, Densply, Milford, DE) operated at 550 mW/cm2. After 24 h, the cover slips were carefully peeled off and the cylindrical specimens (4.0 mm diameter × 1.0 mm thickness) were obtained for the degradation study.
The degree of conversion (DC) was determined from the surface of randomly selected discs using micro-Raman spectroscopy. Spectra were collected using a Horiba Jobin Yvon LabRam HR Raman spectrometer. The DC was based on the ratio of the Raman spectral features: 1640 cm−1 (C=C)/1608 cm−1 (deformation of phenyl) calculated from three separate spectra. Adhesive sample discs were weighed before and after biodegradation to determine mass loss. For SEM analysis, the biodegraded specimens were mounted on aluminum stubs, sputter coated with 20 nm of gold-palladium and imaged at a variety of magnifications in a Philips XL30 ESEM-FEG (Philips, Eindhoven, The Netherlands) at 5–15 kV.
Enzymatic Biodegradation and Analysis of MAA and HEMA
Five adhesive discs with a surface area of about 2 mm2 (2 mm2/mL) were placed in sterile bottles and pre-washed in 0.01M phosphate buffered saline (PBS), pH 7.4, for 3 days to remove most of the unpolymerized monomers. Following the prewash, adhesive discs were incubated in 1 mL 0.2M phosphate buffer solution containing porcine liver esterase (PLE, EC 3.1.1.1., Sigma E 3019), 30 U/mL, at 37°C for 8 days with shaking; concurrent analysis without enzyme consisted of incubations of test specimens in 0.2M phosphate buffer (PB). Daily changes with PLE enzyme were necessary to maintain its optimum activity. PLE was selected for its nonspecific effect on ester bonds and its optimum activity was routinely checked at zero and 24 h using ethyl butyrate. One Unit PLE hydrolyzed 1.0 µM of ethyl butyrate to butyric acid and ethanol per minute at pH 8.0/25°C, after 24 h the activity was 96–98%.
Daily changes of fresh enzyme allowed daily collection of the aqueous phase supernatants, which on collection was immediately centrifuged to remove the enzyme (15 min at 10,000g). The supernatants were then stored at −20°C until ready for HPLC analysis. The HPLC (Waters 500 system, C18 column) conditions included CH3CN:10 mM potassium phosphate buffer (80:20, v/v), 1 mL/min flow rate, 20 µL injection volume and UV detection at 208 nm.24 Samples were thawed and centrifuged again (15 min at 10,000g) before injection into the HPLC system for analysis. The retention times for MAA and HEMA were 2.2 and 2.9 min, respectively. The supernatants of test specimens were evaluated against a HEMA or MAA standard curve to determine the release of each chemical. At the end of 8 days, the adhesive discs were oven-dried in a vacuum (45°C) for 4 weeks and weighed again to determine mass loss.
Statistical Analysis
Data were obtained in µg/mL and were reported in µg/mg, relative to the weighed adhesive for data normalization. One way ANOVA and Scheffe multiple comparison test (p < 0.05) were used to detect significant differences in (a) the concentration of each analyte in the same medium at different times and (b) the concentration of each analyte release in the presence and the absence of enzyme for the three different concentrations of water in the adhesive.
RESULTS
Adhesive Characterization
The appearance of adhesives cured in the absence of water (A00) was transparent while those cured in the presence of water (A08 and A16) were opaque (Table I). Under the conditions of this study, the degree of conversion for all adhesives was in the range of 73.6 to 81.3% (Table I). Macro/micro-level phase separation was observed by SEM in a previous study.32,33 Adhesive A16 discs with 16% water concentration in their formula caused the polymerized resin to separate from water droplets. Void size ranged from submicron to several microns as shown on the resin surface (Figure 1). Even for adhesive A08, our previous work using tapping mode atomic force microscopy (TMAFM)/Imaging technique, showed that nano-scale phase separation is a general feature of model crosslinked polymethacrylate, even for visibly void-free polymerized resin.
TABLE I.
Adhesive Disc Appearance and Degree of Conversion (DC) of disc surfaces (Mean ± SD)
| Adhesive ID | A00 | A08 | A16 |
|---|---|---|---|
| Adhesive appearance | 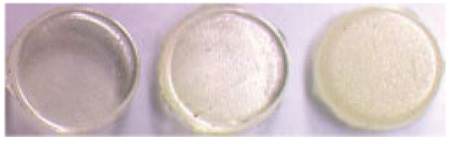 |
||
| Bottom surface DC (%) | 76.5 ± 1.1 | 76.0 ± 1.2 | 77.8 ± 1.4 |
| Top surface DC (%) | 81.3 ± 0.1a | 73.6 ± 0.4 | 76.8 ± 1.2 |
Significantly different from all other DC values, one way ANOVA, Scheffe test, n = 3.
Figure 1.

Representative SEM micrographs of A00, A08, and A16. Adhesive discs A16 with 16% water concentration in their formula caused the polymerized resin to separate from water droplets. Void size ranged from submicron to several microns as shown on the resin surface.
The final mass of adhesive type per treatment with and without PLE enzyme was not significantly (p > 0.05) different from its initial mass. However, except for A16, there were significant differences (ANOVA/Scheffe post hoc testing, p < 0.05, n = 3) in mass loss for most adhesives formulated with water compared to the control adhesive, A00 (Table II).
TABLE II.
Adhesive Mass Loss after Biodegradation (Mean ± SD)
| Adhesive ID | E | Initial Mass (mg) | Final Mass (mg) | Mass Loss (mg) |
|---|---|---|---|---|
| A00 | (−) | 100.7 ± 1.1a | 99.8 ± 0.5a | 0.22 ± 0.27a |
| (+) | 98.3 ± 1.7a | 97.2 ± 2.1a | 1.03 ± 0.41a | |
| A08 | (−) | 84.5 ± 1.9b | 81.4 ± 1.8b | 3.09 ± 0.13b |
| (+) | 81.1 ± 1.3b | 77.6 ± 1.3b | 3.56 ± 0.06b | |
| A16 | (−) | 77.3 ± 3.0b,c | 75.6 ± 2.3b,c | 1.73 ± 0.29a |
| (+) | 74.1 ± 0.6c | 71.4 ± 0.6c | 2.65 ± 0.21b |
Values are expressed as mean ± SD of n = 3. Within values of initial mass, final mass and mass loss, similar superscripted letters (a,b,c) indicate no significant differences among values (ANOVA/Scheffe post hoc testing, p > 0.05) n = 3, vertical comparisons). Final mass values per treatment with (+) or without (−) enzyme esterase (E) was not significantly different from its initial mass (Paired t-test, p > 0.05, horizontal comparison).
Release of MAA and HEMA in Prewash Eluates
The concentration of HEMA release in the prewash step was greater than MAA levels for all adhesive formulations. HEMA levels were six-, nine-, and sevenfold larger than MAA levels for A00, A08, and A16, respectively. Adhesives formulated with water facilitated the release of HEMA in the prewash step. The release of HEMA was significantly increased (p < 0.05) for adhesives A08 (3.69 µM/mL) and A16 (2.99 µM/mL) compared with HEMA release from the control A00 (0.64 µM/mL).
Release of Residual HEMA in the Biodegradation Studies
The daily HEMA release measured in buffer [HEMA (in PB)] is presented in Table III. Release of residual monomer HEMA was consistently measured from all adhesive types incubated in buffer without enzyme. For adhesives formulated with water, the daily content of HEMA in PB was higher for A08 than A16 in the first days and then decreased leveling off by the fourth day (Figure 2, bottom). Because of high release on the first days, the average 8-day cumulative release of HEMA for the adhesive A08 (11.88 ± 0.45 µM/mL) was significantly higher (p < 0.05) than A16 (8.25 ± 0.57 µM/mL), and both significantly (p < 0.05) increased relative to A00 (1.01 ± 0.07 µM/mL). However, there was no HEMA release detected from adhesives incubated in the presence of enzyme.
TABLE III.
AA and HEMA Release in Daily Eluates from A00, A08, or A16 Adhesives Incubated with (+) and Without (−) esterase (E)
| Analytes from A00 (µmoles/mL) | Analytes from A08 (µmoles/mL) | Analytes from A16 (µmoles/mL) | Alle | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Days | MAA (−)E | MAA (+)E | HEMA (−)E | HEMA (−)E | MAA (−)E | MAA (+)E | HEMA (−)E | MAA (−)E | MAA (+)E | HEMA (+)E |
| 1 | 0.48 ± 0.01a | 1.01 ± 0.05a | 0.21 ± 0.00a | 2.17 ± 0.16a | 0.79 ± 0.03a | 5.37 ± 0.88a | 2.64 ± 0.48a | 0.78 ± 0.03a | 3.62 ± 0.15a | ND |
| 2 | 0.47 ± 0.01a | 0.87 ± 0.01b | 0.20 ± 0.00a | 1.25 ± 0.06b | 0.73 ± 0.01a | 3.25 ± 0.08b | 2.71 ± 0.01a | 0.80 ± 0.07a | 2.34 ± 0.20b | ND |
| 3 | 0.45 ± 0.02a | 0.87 ± 0.02b | 0.13 ± 0.01b | 0.90 ± 0.23c | 0.88 ± 0.03a | 2.72 ± 0.04b | 1.97 ± 0.13b | 0.89 ± 0.17a | 2.46 ± 0.18b | ND |
| 4 | 0.60 ± 0.02b | 0.97 ± 0.03a | 0.12 ± 0.03b | 0.89 ± 0.07c | 0.84 ± 0.14a | 2.46 ± 0.11b | 1.06 ± 0.12c | 0.92 ± 0.07a | 2.34 ± 0.01b | ND |
| 5 | 0.80 ± 0.09b | 1.33 ± 0.05c | 0.09 ± 0.04b | 0.88 ± 0.05c | 0.97 ± 0.29a | 2.37 ± 0.08b | 1.08 ± 0.04c | 0.73 ± 0.01a | 2.18 ± 0.08b | ND |
| 6 | 0.54 ± 0.01a | 1.37 ± 0.05c | 0.08 ± 0.01b | 1.01 ± 0.03c | 0.70 ± 0.01a | 2.11 ± 0.09c | 0.71 ± 0.01c | 0.79 ± 0.01a | 1.26 ± 0.05c | ND |
| 7 | 0.63 ± 0.02b | 0.80 ± 0.03a | 0.08 ± 0.04b | 0.67 ± 0.03c | 0.63 ± 0.02a | 1.81 ± 0.05c | 0.76 ± 0.06c | 0.70 ± 0.01a | 1.72 ± 0.07c | ND |
| 8 | 0.53 ± 0.01a | 0.92 ± 0.07a | 0.11 ± 0.01b | 0.48 ± 0.04d | 0.68 ± 0.01a | 1.79 ± 0.32c | 0.94 ± 0.05c | 0.66 ± 0.04a | 1.56 ± 0.10c | ND |
Values are expressed as mean ± SD of n = 3. For each column, values with similar superscripted letter (a,b,c,d) indicate these values are not tatistically significantly different from one another (ANOVA/Scheffe post hoc testing, p > 0.05).
HEMA was not detected (ND) in all the supernatants of resins incubated with esterase.
Figure 2.

Adhesives formulated with water (A08 and A16) exhibited significantly increased levels of net MAA in PLE (top) and HEMA in PB (bottom) relative to the adhesives formulated without water (A00). Furthermore, the daily release of net MAA in PLE exceeds that of HEMA release in PB for all adhesives.
Release of MAA
MAA release in daily eluates from A00, A08, and A16 adhesives is presented in Table III. In systems with buffer (−E), the daily release of MAA remained fairly similar, however, levels of MAA from A08 and A16 were increased compared to the control A00. As expected, MAA release from all the formulations in the presence of enzyme (+E) was significantly increased (p < 0.05) compared with MAA levels in phosphate buffer (−E). In systems with esterase, the daily levels of net MAA for A08 and A16 were significantly increased in the first days and then decreased sharply, leveling off to similar levels by the third day. The plot of the net cumulative release of MAA in the presence of the enzyme PLE is depicted in Figure 2 (top) and was obtained by subtracting the MAA levels measured in buffer [MAA(in PLE) − MAA(in PB)]. Because of the large amount of MAA release in the first day (Table III), the total cumulative release of MAA for A08 (21.87 µM/mL) was significantly greater than A16 (17.48 µM/mL) and both showed significantly greater (p < 0.05) cumulative MAA release than A00 (8.14 µm/mL). Moreover, the cumulative HEMA and MAA release measured in the presence of esterase [MAAin PLE] was also significantly different (p < 0.05) from the sum of analytes [MAA in PB + HEMA in PB] in buffer over the 8 day period of incubation as seen in Table III.
DISCUSSION
The structure of methacrylate adhesives suggests a general mechanism for their chemical and enzymatic degradation in oral fluids. On prolonged exposure of the restoration to oral fluids, water begins to penetrate the resin. Water initially enters the matrix by diffusion into loosely cross-linked or hydrophilic domains or may be trapped within the matrix during photopolymerization in the moist environment of the mouth. Portions of the matrix may also be directly exposed to oral fluids, particularly at the gingival margin of Class II and V composite restorations. The presence of water promotes the chemical hydrolysis of ester bonds in methacrylate materials. This reaction is expected to be relatively slow at the neutral pH typical of saliva, but excursions in pH caused by foods or cariogenic bacteria may lead to transient acid or base catalysis. The carboxylate and alcohol degradation products of ester hydrolysis are more hydrophilic than the parent ester, further enhancing the local ingress of water. Over years of exposure to salivary fluids, local domains of the methacrylate network may become sufficiently degraded and/or hydrophilic to permit access by esterases which greatly accelerate ester bond hydrolysis.
In this study, the resins HEMA/bisGMA polymerized in the presence of water (A08 and A16) were considered models of polymers with phase separation as has been observed under wet bonding protocols and also models of enhanced local ingress of water that may permit accelerated ester bond hydrolysis by esterases with subsequent elution of the breakdown product MAA.1,34 The effect of water contained in dentin on the quality of the bonding interface has been investigated for one-bottle adhesives.35,36 The water concentration values (0, 8, and 16 wt %) were selected on the basis of our previous work.31,32 It is noted that the formulations containing 0 or 8% water present one solution phase prior to photopolymerization. Based on the finding of nano-phase separation in dentin adhesive, the 8 wt % concentration of water is still below that required for visible macro-phase separation in HEMA/bisGMA formulations with a mass ratio of 45/55. This concentration was controlled to maintain visually homogeneous specimens prior to photopolymerization and simulated the situation in which the homogeneous adhesives confront the threshold of water/monomers (liquid/liquid) phase separation. Nanolevel phase separation was found for A08 formulation.32
Wet bonding means that the dentin is kept fully hydrated throughout the bonding procedure; the surface morphology of the demineralized layer does not change because the water supporting the collagen matrix is not removed.37 During acid etching, the mineral phase is extracted from a zone that measures between 1 and ~10 µm of the dentin surface.38–40 The composition of the exposed substrate differs radically from mineralized dentin. For example, mineralized dentin is 50% mineral, 30% collagen, and 20% water by volume,41 whereas demineralized dentin is 30% collagen and 70% water.38,42 With removal of the mineral phase, the collagen fibers are suspended in water. The only mechanism available for adhesive resin infiltration is diffusion of the resin into the water that is in the spaces of the substrate and along the collagen fibers.43,44 As we know, the adhesive/primer combination in one-bottle dentin adhesives must infiltrate the water-laden collagen fibril matrix before polymerization. Water is actually a major interfering factor when bonding adhesive and/or composites to the tooth.8,20,45–47 This relationship was the basis for our investigations of the behavior of the monomer in the presence of water.
In this project, we analyzed the formation of MAA as a hydrolytic biodegradation product from HEMA/bisGMA adhesive discs (A00, A08, and A16), which were prewashed to eliminate most of the un-reacted monomer. Prewash was done based on the observation that components that are leached during laboratory tests would not necessarily be found clinically because of chemical changes occurring with time during intraoral surface interactions with saliva, food and enzymes. The prewash step indicated that adhesives simulating wet bonding protocols, A08 and A16, allowed the release of large amounts of HEMA compared with the control adhesive A00. Furthermore, the amounts of HEMA released were seven- to eightfold higher than MAA release, which suggests that HEMA hydrolysis in water in the prewash step is relatively slow. The measurement of MAA release in the prewash indicated a general hydrolytic instability of the adhesives.
In the enzymatic biodegradation studies, the release of MAA and HEMA (Table III) were measured to evaluate the overall biodegradation of the polymer in the presence or absence of porcine liver esterase. In systems without esterase, MAA release [MAA(in PB)] was similar in daily eluates up to 8 days for all adhesives (Table III). However, HEMA release in the system without esterase [HEMA(in PB)] was higher for adhesives A08 and A16 compared with A00 (Figure 2, bottom). In comparison, in systems with esterase, the levels of [MAAin PLE] were significantly increased relative to systems without esterase, and the levels of MAA release were high in the first days and decreased in time. This was particularly evident with adhesives formulated with water, A08 and A16 (Figure 2, top). On the other hand, no HEMA release was observed with any of the adhesives incubated in the presence of esterase; these results indicate accelerated hydrolysis of residual unpolymerized HEMA to MAA.
To indicate that esterase significantly accelerated the overall polymer biodegradation process, we analyzed our results in regards to the formation of MAA and the release of HEMA (Table III). If we assume all the MAA measured came from the hydrolysis of unpolymerized HEMA then the concentration of MAA in the presence of esterase [MAAin PLE] would be similar to the sum of analytes in buffer, [MAA(in PB) + HEMA(in PB)]. However, our results as presented in Figure 3, illustrate that the cumulative MAA release measured in the presence of esterase [MAAin PLE] was significantly different (p < 0.05) from the sum of analytes [MAA in PB + HEMA in PB] in buffer over the 8 day period of incubation. While this was seen for all adhesives, the adhesives formulated with water, A08 and A16, exhibited increased release of the analytes compared to A00, which is in line with the increase release of HEMA from these adhesives. Another way to appreciate accelerated ester bond hydrolysis in the polymer is that the net MAA concentration in PLE [MAA(in PLE) − MAA(in PB)] (Figure 2, top) is relatively larger than the HEMA release in PB. This is seen in Figure 2, which top and bottom figures were plotted on similar Y-axis scale.
Figure 3.

The cumulative release of MAA in the presence of porcine liver esterase (PLE) was significantly different (p < 0.05) from the sum of analytes [MAA + HEMA] released in the buffer (PB). These differences indicate that MAA formation in the 8-day enzymatic biodegradation study is attributable to hydrolysis of unpolymerized HEMA, pendant groups of bisGMA in the polymer and/or unpolymerized bisGMA. In addition, adhesives simulating wet bonding, A08 and A16 exhibited significantly larger release of analytes compared with the control A00.
The effect of PLE on the overall biodegradation of the polymer could be further evaluated with other enzymes that have greater affinity for bisGMA. Previous studies indicated that in contrast to HEMA, bisGMA has greater susceptibility to hydrolysis by cholesterol esterase and acetyl cholinesterase,24 Biodegradation of HEMA/bisGMA adhesives in the presence of either enzyme would be more clinically relevant since these were found to simulate salivary enzyme activity.24,48
Theoretically, the hydrolysis from unreacted pendant groups in a HEMA/bisGMA polymer network would release ethylene glycol if derived from poly-HEMA repeated units; and MAA, bisGMA diol, or monomethacrylate alcohol of bisGMA if derived from poly-bisGMA pendant groups. From these hydrolytic reactions, acidic groups would remain in the polymer. This process of polymer biodegradation indicates that MAA is released from pendant methacrylate groups in polymerized bisGMA and not from poly(HEMA) units. Since the increase of MAA in the presence of esterase [MAA (in PLE)] was 1.2- to 1.5-fold higher relative to the sum of analytes in buffer, [MAA(in PB) + HEMA(in PB)], the results suggest partial biodegradation of the HEMA/bisGMA polymer.
From the above analyses, all the adhesives investigated A00, A08, and A16, underwent hydrolytic instability in the presence of PLE. Since adhesives formulated with water A08 and A16 release greater amounts of analytes compared to A00 (Figure 3), they reflect their higher release of HEMA in the elution process in the aqueous incubation system. The implications of this effect suggest that the overall loss of material could be greater in adhesives that experience phase separation under wet bonding conditions.
Differences in MAA release between the control A00 and adhesives A08 and A16 can be attributed to phase separation that occurred in the adhesives formulated with water (Figure 1). Because of phase separation, poorly polymerized hydrophilic polymer domains degrade rapidly in the aqueous oral environment.9,21 Under these conditions, adhesives formulated with water provide models to study the effect of water on structural integrity. However, increasing water content in the resins from 8 to 16% did not increase MAA release (A08 > A16 > A00, Figure 2, top). Comparisons of mass loss with and without esterase for each adhesive type indicated that A16 was the only adhesive with a significant mass loss after biodegradation with esterase (Table II). Overall, our mass loss analysis reflected the trend that adhesives A08 and A16 exhibited greater MAA and HEMA release than A00 (Figure 2 and Figure 3). However, these mass losses did not change the final weight of the sample discs (Table II) suggesting that weight loss analysis may not be a suitable method for evaluating biodegradation. The differences between the A08 and A16 require further investigation to develop a comprehensive argument, however this difference was not the focus of this study. The hypothesis is accepted, that is, the enzymatic biodegradation of HEMA/bisGMA formulations exhibiting heterogeneous phase structure (nano-level A08 or macro-level A16) would be enhanced as compared with homogeneous adhesive without phase separation (A00).
Results with our model adhesives for phase separation lay the groundwork for future investigations focused on determining the esterase resistance of new water-compatible adhesives with different hydrophilic/hydrophobic properties. This type of comparison could provide more information on the release of HEMA and MAA from esterase resistant HEMA/bisGMA-based adhesives where the concentration of MAA release in systems with esterase would be less than or equal to the sum of MAA and HEMA release in systems without esterase. This in turn could reflect more biocompatible and/or durable adhesives.
CONCLUSIONS
Adhesives formulated with water to simulate phase separation, A08 and A16, exhibited significantly larger release of HEMA in PB compared to adhesives without phase separation, A00. This trend was reflected for MAA release in the presence of esterase; MAA release from adhesives A08 and A16 was significantly increased compared with A00. Mass loss analysis within groups of adhesive types strengthened this trend. These results indicate that adhesives formulated in the presence of water to simulate wet bonding exhibited greater release of HEMA, which underwent enhanced hydrolysis of its ester group to MAA in the presence of PLE. Within differences among adhesive types, the cumulative release of MAA in the presence of PLE was significantly increased (p < 0.05) as compared with the sum of MAA and HEMA released in PB indicating that MAA release was not only formed from HEMA but perhaps from pendant groups in the polymer network. Thus, PLE significantly accelerated the overall biodegradation process of A00, A08, and A16 adhesives during the 8 days of enzymatic biodegradation. The increased levels of analytes obtained with A08 and A16 relative to A00 suggest that phase-separated HEMA/bisGMA adhesives may experience accelerated degradation under clinical conditions.
Acknowledgments
Contract grant sponsor: National Institute of Dental and Craniofacial Research/National Institutes of Health; contract grant numbers: R01 DE14392, K25DE015281, R13 DK069504
REFERENCES
- 1.De Munck J, Van Landuyt K, Peumans M, Poitevin A, Lambrechts P, Braem M, Van Meerbeek B. A critical review of the durability of adhesion to tooth tissue: Methods and results. J Dent Res. 2005;84:118–132. doi: 10.1177/154405910508400204. [DOI] [PubMed] [Google Scholar]
- 2.Van Nieuwenhuysen JP, D’Hoore W, Carvalho J, Qvist V. Long-term evaluation of extensive restorations in permanent teeth. J Dent. 2003;31:395–405. doi: 10.1016/s0300-5712(03)00084-8. [DOI] [PubMed] [Google Scholar]
- 3.Owens BM, Johnson WW. Effect of insertion technique and adhesive system on microleakage of Class V resin composite restorations. J Adhes Dent. 2005;7:303–308. [PubMed] [Google Scholar]
- 4.Van Meerbeek B, Van Landuyt K, De Munck J, Hashimoto M, Peumans M, Lambrechts P, Yoshida Y, Inoue S, Suzuki K. Technique-sensitivity of contemporary adhesives. Dent Mater J. 2005;24:1–13. doi: 10.4012/dmj.24.1. [DOI] [PubMed] [Google Scholar]
- 5.Meiers JC, Kresin J. Cavity disinfectants and dentin bonding. Oper Dent. 1996;21:153–159. [PubMed] [Google Scholar]
- 6.Hashimoto M, Ohno H, Kaga M, Endo K, Sano H, Oguchi H. Resin-tooth adhesive interfaces after long-term function. Am J Dent. 2001;12:211–215. [PubMed] [Google Scholar]
- 7.Murray PE, Windsor LJ, Smyth TW, Hafez AA, Cox CF. Analysis of pulpal reactions to restorative procedures, materials, pulp capping, and future therapies. Crit Rev Oral Biol Med. 2002;13:509–520. doi: 10.1177/154411130201300607. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Spencer P. Interfacial chemistry of Class II composite restoration: Structure analysis. J Biomed Mat Res. 2005;75:580–587. doi: 10.1002/jbm.a.30451. [DOI] [PubMed] [Google Scholar]
- 9.Spencer P, Wang Y, Bohaty B. Interfacial chemistry of moisture- aged class II composite restorations. J Biomed Mater Res B Appl Biomater. 2006;77:234–240. doi: 10.1002/jbm.b.30434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gallo LM, Nickel JC, Iwasaki LR, Palla S. Stress-field translation in the healthy human temporomandibular joint. J Dent Res. 2000;79:1740–1746. doi: 10.1177/00220345000790100201. [DOI] [PubMed] [Google Scholar]
- 11.Van Meerbeek B, De Munck J, Yoshida Y, Inoue S, Vargas M, Vijay P, Van Landuyt K, Lambrechts P, Vanherle G. Buonocore memorial lecture. Adhesion to enamel and dentin: Current status and future challenges. Oper Dent. 2003;28:215–235. [PubMed] [Google Scholar]
- 12.Bergenholtz G, Cox CF, Loesche WJ, Syed SA. Bacterial leakage around dental restorations: Its effect on the dental pulp. J Oral Pathol. 1982;11:439–450. doi: 10.1111/j.1600-0714.1982.tb00188.x. [DOI] [PubMed] [Google Scholar]
- 13.Hashimoto M, Ohno H, Kaga M, Endo K, Sano H, Oguchi H. In vivo degradation of resin-dentin bonds in humans over 1 to 3 years. J Dent Res. 2000;79:1385–1391. doi: 10.1177/00220345000790060601. [DOI] [PubMed] [Google Scholar]
- 14.Ferracane JL. Hygroscopic and hydrolytic effects in dental polymer networks. Dent Mater. 2006;22:211–222. doi: 10.1016/j.dental.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 15.Roulet JF, Degrange M, editors. Adhesion: The Silent Revolution in Dentistry. 1st ed. Chicago: Quintessence Publishing Co; 1999. p. 263. [Google Scholar]
- 16.Marshall GW, Marshall SJ, Kinney JH, Balooch M. The dentin substrate: Structure and properties related to bonding. J Dent. 1997;25:441–458. doi: 10.1016/s0300-5712(96)00065-6. [DOI] [PubMed] [Google Scholar]
- 17.Pereira PNR, Okuda M, Sano H, Yoshikawa T, Burrow MF, Tagami J. Effect of intrinsic wetness and regional difference on dentin bond strength. Dent Mater. 1999;15:46–53. doi: 10.1016/s0109-5641(99)00013-5. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Spencer P, Hager C, Bohaty B. Comparison of interfacial characteristics of adhesive bonding to superficial versus deep dentin using SEM and staining techniques. J Dent. 2006;34:26–34. doi: 10.1016/j.jdent.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 19.Ito S, Saito T, Tay FR, Carvalho RM, Yoshiyama M, Pashley DH. Water content and apparent stiffness of non-caries versus caries-affected human dentin. J Biomed Mater Res B Appl Biomater. 2005;72:109–116. doi: 10.1002/jbm.b.30130. [DOI] [PubMed] [Google Scholar]
- 20.Spencer P, Wang Y. Adhesive phase separation at the dentin interface under wet bonding conditions. J Biomed Mater Res. 2002;62:447–456. doi: 10.1002/jbm.10364. [DOI] [PubMed] [Google Scholar]
- 21.Breschi L, Mazzoni A, Ruggeri A, Cadenaro M, Di Lenarda R, De Stefano Dorigo E. Dental adhesion review: Aging and stability of the bonded interface. Dent Mater. 2008;24:90–101. doi: 10.1016/j.dental.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 22.Santerre JP, Shajii L, Tsang H. Biodegradation of commercial dental composites by cholesterol esterase. J Dent Res. 1999;78:1459–1468. doi: 10.1177/00220345990780081201. [DOI] [PubMed] [Google Scholar]
- 23.Santerre JP, Cai K, Banh M. Isolation of human salivary esterase activity involved in composite biodegradation. Brisbane. 2006 [Google Scholar]
- 24.Yourtee DM, Smith RE, Russo KA, Burmaster S, Cannon JM, Eick JD, Kostoryz EL. The stability of methacrylate biomaterials when enzyme challenged: Kinetic and systematic evaluations. J Biomed Mater Res. 2001;57:522–531. doi: 10.1002/1097-4636(20011215)57:4<522::aid-jbm1198>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 25.Finer Y, Jaffer F, Santerre JP. Mutual influence of cholesterol esterase and pseudocholinesterase on the biodegradation of dental composites. Biomaterials. 2004;25:1787–1793. doi: 10.1016/j.biomaterials.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 26.Santerre JP, Cai K, Banh M. Isolation of human saliva esterase activity involved in composite biodegradation. J Dent Res. 2006;85 (Spec Iss B): Abstract 1971. [Google Scholar]
- 27.Finer Y, Santerre JP. Salivary esterase activity and its association with the biodegradation of dental composites. J Dent Res. 2004;83:22–26. doi: 10.1177/154405910408300105. [DOI] [PubMed] [Google Scholar]
- 28.Finer Y, Santerre JP. Influence of silanated filler content on the biodegradation of bisGMA/TEGDMA dental composite resins. J Biomed Mater Res A. 2007;81:75–84. doi: 10.1002/jbm.a.31004. [DOI] [PubMed] [Google Scholar]
- 29.Bean TA, Zhuang WC, Tong PY, Eick JD, Yourtee DM. Effect of esterase on methacrylates and methacrylate polymers in an enzyme simulator for biodurability and biocompatibility testing. J Biomed Mater Res. 1994;28:59–63. doi: 10.1002/jbm.820280108. [DOI] [PubMed] [Google Scholar]
- 30.Armstrong SR, Jessop JL, Vargas MA, Zou Y, Qian F, Campbell JA, Pashley DH. Effects of exogenous collagenase and cholesterol esterase on the durability of the resin-dentin bond. J Adhes Dent. 2006;8:151–160. [PubMed] [Google Scholar]
- 31.Ye Q, Spencer P, Wang Y. Nanoscale patterning in cross-linked methacrylate copolymer networks: An atomic force microscopy study. J Appl Polym Sci. 2007;106:3843–3851. doi: 10.1002/app.27044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ye Q, Wang Y, Spencer P. Nanophase separation in polymers exposed to simulated oral environment. J Biomed Mater Res Appl Biomater. doi: 10.1002/jbm.b.31047. Forthcoming. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ye Q, Park JG, Topp E, Wang Y, Misra A, Spencer P. Effect of Nanophase Separation on the In Vitro Performance of Model Dentin Adhesive. J Dent Res. doi: 10.1177/154405910808700911. Forthcoming. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eick JD, Miller RG, Robinson SJ, Bowles CQ, Gutshall PL, Chappelow CC. Quantitative analysis of the dentin adhesive interface by Auger spectroscopy. J Dent Res. 1996;75:1027–1033. doi: 10.1177/00220345960750040501. [DOI] [PubMed] [Google Scholar]
- 35.Yoshida E, Uno S, Nodasaka Y, Kaga M, Hirano S. Relationship between water status in dentin and interfacial morphology in all-in-one adhesives. Dent Mater. 2007;23:556–560. doi: 10.1016/j.dental.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 36.Zhang ZX, Huang C, Zheng TL, Wang S, Cheng XR. Effects of residual water on microtensile bond strength of one-bottle dentin adhesive systems with different solvent bases. Chin Med J (Engl) 2005;118:1623–1628. [PubMed] [Google Scholar]
- 37.Kinney JH, Balooch M, Marshall SJ, Marshall GW. Atomic force microscope study of dimensional changes in dentine during drying. Arch Oral Biol. 1993;38:1003–1007. doi: 10.1016/0003-9969(93)90114-2. [DOI] [PubMed] [Google Scholar]
- 38.Eick JD, Gwinnet AJ, Pashley DH, Robinson SJ. Current concepts on adhesion to dentin. Crit Rev Oral Biol Med. 1997;8:306–335. doi: 10.1177/10454411970080030501. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y, Spencer P. Quantifying adhesive penetration in adhesive/dentin interface using confocal Raman microspectroscopy. J Biomed Mater Res. 2002;59:46–55. doi: 10.1002/jbm.1215. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y, Spencer P. Overestimating hybrid layer quality in polished adhesive/dentin interfaces. J Biomed Mater Res. 2004;68:735–746. doi: 10.1002/jbm.a.20105. [DOI] [PubMed] [Google Scholar]
- 41.Marshall J, Dentin GW. Microstructure and characterization. Quint Int. 1993;24:606–617. [PubMed] [Google Scholar]
- 42.Pashley DH, Ciucchi B, Sano H, Horner JA. Permeability of dentin to adhesive agents. Quint Int. 1993;24:618–631. [PubMed] [Google Scholar]
- 43.Radovic I, Vulicevic ZR, Garcia-Godoy F. Morphological evaluation of 2- and 1-step self-etching system interfaces with dentin. Oper Dent. 2006;31:710–718. doi: 10.2341/05-145. [DOI] [PubMed] [Google Scholar]
- 44.Salim DA, Andia-Merlin RY, Arana-Chavez VE. Micromorphological analysis of the interaction between a one-bottle adhesive and mineralized primary dentine after superficial deproteination. Biomaterials. 2004;25:4521–4527. doi: 10.1016/j.biomaterials.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 45.Wang Y, Spencer P. Hybridization efficiency of the adhesive dentin interface with wet bonding. J Dent Res. 2003;82:141–145. doi: 10.1177/154405910308200213. [DOI] [PubMed] [Google Scholar]
- 46.Spencer P, Wang Y, Bohaty B. Interfacial chemistry of moisture- aged class II composite restorations. J Biomed Mater Res B Appl Biomater. 2006;77:234–240. doi: 10.1002/jbm.b.30434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y, Spencer P, Yao X, Ye Q. Effect of co-initiator and water on the photoreactivity and photopolymerization of HEMA/Camphoroquinone-based reactant mixtures. J Biomed Mater Res. 2006;78:580–587. doi: 10.1002/jbm.a.30733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finer Y, Santerre JP. The influence of resin chemistry on a dental composite’s biodegradation. J Biomed Mater Res. 2004;69:233–246. doi: 10.1002/jbm.a.30000. [DOI] [PubMed] [Google Scholar]