

Abstract
The classic kidney disease of Human Immunodeficiency Virus (HIV) infection, HIV-associated nephropathy, is characterized by progressive acute renal failure, often accompanied by proteinuria and ultrasound findings of enlarged, echogenic kidneys. Definitive diagnosis requires kidney biopsy, which demonstrates collapsing focal segmental glomerulosclerosis with associated microcystic tubular dilatation and interstitial inflammation. Podocyte proliferation is a hallmark of HIV-associated nephropathy, although this classic pathology is observed less frequently in antiretroviral-treated patients. The pathogenesis of HIV-associated nephropathy involves direct HIV infection of renal epithelial cells, and the widespread introduction of combination antiretroviral therapy has had a significant impact on the natural history and epidemiology of this unique disease. These observations have established antiretroviral therapy as the cornerstone of treatment for HIV-associated nephropathy, in the absence of prospective clinical trials. Adjunctive therapy for HIV-associated nephropathy includes ACE inhibitors or angiotensin receptor blockers, as well as corticosteroids in selected patients with significant interstitial inflammation or rapid progression.
Keywords: HIV-associated nephropathy, focal segmental glomerulosclerosis, HIV, kidney
Introduction
Within a few years of the first published descriptions of the acquired immunodeficiency syndrome (AIDS), kidney disease was recognized as a complication of infection with Human Immunodeficiency Virus (HIV).1–3 Early reports from New York and Miami described an aggressive form of collapsing focal segmental glomerulosclerosis (FSGS) in African-Americans and Haitian immigrants with advanced AIDS. Now known as HIV-associated nephropathy (HIVAN), this unique kidney disease quickly became a leading cause of end-stage renal disease (ESRD) among young African-Americans, paralleling the growth of the HIV epidemic in this population.4 5 This review will focus on the clinical presentation, histopathology, and changing epidemiology of HIVAN in the era of combination antiretroviral therapy (ART).
Clinical presentation of HIV-associated nephropathy
In 1984, physicians in New York City reported a series of patients with advanced AIDS and rapidly progressive glomerular disease.1 All of the affected patients in this initial report were African-Americans or Haitian immigrants, although the significance of race was not fully appreciated until several years later.6 The classic clinical presentation of HIVAN was characterized by rapidly progressive renal failure, accompanied by moderate to nephrotic range proteinuria, bland urinary sediment, and ultrasound findings of large, highly echogenic kidneys. Progression to ESRD or death was nearly universal, and by 1999 “AIDS nephropathy” had become the third leading cause of ESRD among African-Americans between the ages of 20 and 64 years.4
Although cases have been reported in the setting of asymptomatic HIV infection or acute HIV seroconversion,7 HIVAN was originally described in patients with AIDS1 2 3 and is still generally recognized as a complication of advanced HIV disease.8 In a series of 107 HIV-infected patients who underwent kidney biopsy between 1995 and 2002, patients with classic HIVAN were more likely to have a CD4 cell count < 200 cells/mm3 compared to patients with an alternative diagnosis (70% versus 31%), although there were no significant differences in HIV RNA level or ART use.8 The combination of nephrotic range proteinuria and a CD4 cell count < 200 cells/mm3 was observed in half of the patients with HIVAN, compared to only 15% of those with another diagnosis. While the combination of nephrotic range proteinuria and a CD4 cell count < 200 cells/mm3 had limited sensitivity and specificity for the diagnosis of HIVAN, the absence of both findings may be more useful to exclude the diagnosis, with a negative predictive value reaching 90%.8 In an earlier study, these investigators found that the degree of echogenicity on renal ultrasound may also have some predictive value for the diagnosis or exclusion of HIVAN, but only at the extremes of echogenicity. Kidney size did not distinguish HIVAN from other renal diagnoses in this study, possibly because patients with HIVAN presented with more advanced kidney disease.9 While clinical characteristics may help to refine the differential diagnosis in patients with contraindications to kidney biopsy, more than 30% of patients with suspected HIVAN based on these and other clinical criteria will have an alternative diagnosis on kidney biopsy.8,10
Pathology of HIV-associated nephropathy
In the 1980's, the classic pathologic features of HIVAN began to emerge from autopsy and renal biopsy studies performed in institutions with a high prevalence of HIV/ AIDS.1 11 12 13 On gross examination at autopsy, the kidneys were often enlarged, pale and swollen, with combined kidney weights as high as 500 g.14 Renal enlargement may persist even in the setting of ESRD, owing to the presence of numerous tubular microcysts distending the parenchyma.
Light microscopy
In the acute phase, untreated HIVAN typically manifests a dramatic pattern of collapsing FSGS.11 12 Glomerular capillary lumina are occluded by an implosive wrinkling and collapse of the glomerular basement membrane that is more often global than segmental, without predilection for the perihilar segments (Figure 1 and Figure 2). The acute nature of the glomerular injury is evidenced by the lack of appreciable increase in intracapillary or mesangial matrix. The glomerular collapse is accompanied by prominent hypertrophy and hyperplasia of the overlying podocytes, which have enlarged, open vesicular nuclei with frequent nucleoli, occasional binucleate forms, and rare mitotic figures. Visceral epithelial cells may be so crowded as to obliterate the urinary space, forming pseudocrescents (Figure 3). The podocyte cytoplasm is typically vacuolated, containing prominent intracytoplasmic protein resorption (hyaline) droplets (Figure 4). As the lesions evolve, the glomerular tuft retracts into a tight solidified ball crowned by overlying enlarged, vacuolated visceral epithelial cells. At this stage, the urinary space often appears dilated, containing a proteinaceous filtrate. Unlike FSGS, not otherwise specified (NOS), the collapsing variant of FSGS usually lacks hyalinosis, endocapillary foam cells, and adhesions to Bowman's capsule in the acute phase.15 However, repeat biopsies and postmortem studies have shown that collapsing lesions may evolve into a more typical pattern of FSGS (NOS).
Figure 1.
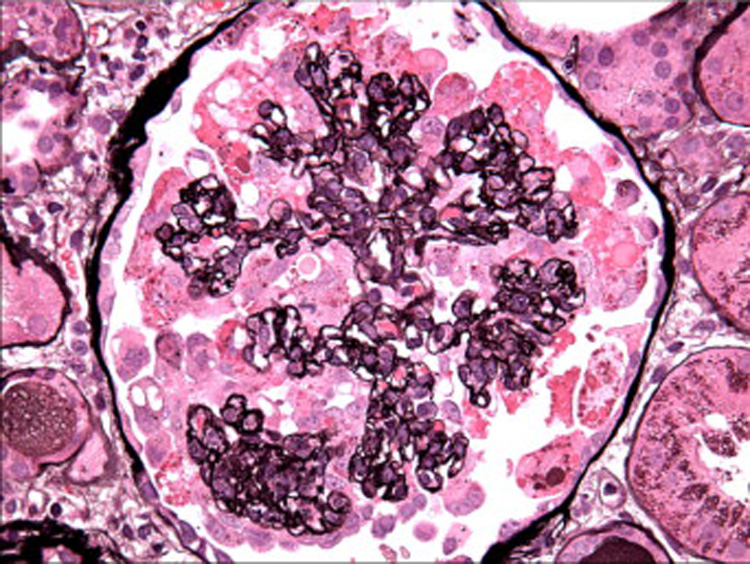
The glomerular capillary lumina are globally obliterated by collapse of glomerular basement membranes with hypertrophy and hyperplasia of overlying podocytes (Jones methenamine silver, x400). [COLOR PRINT/ ONLINE]
Figure 2.
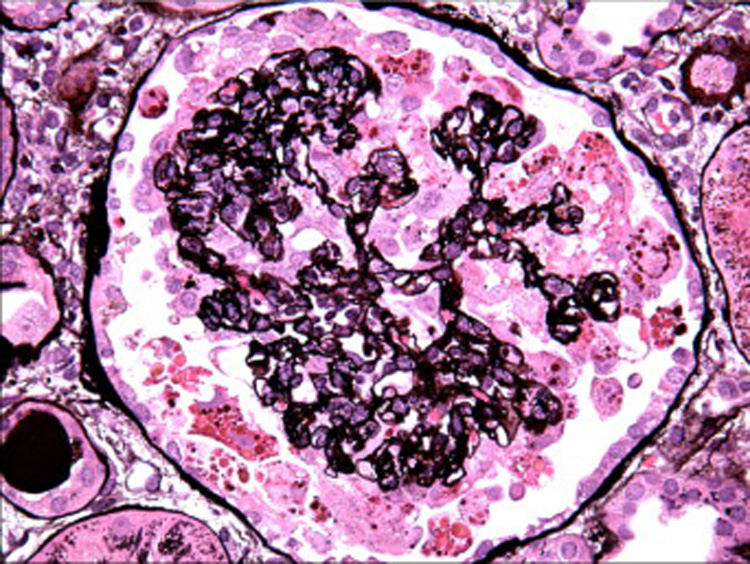
The podocytes surrounding the collapsed tuft form a corona of hypertrophied cells with numerous protein resorption droplets. Some of the podocytes appear detached from the tuft and suspended in the urinary space (Jones methenamine silver, x400). [COLOR PRINT/ ONLINE]
Figure 3.
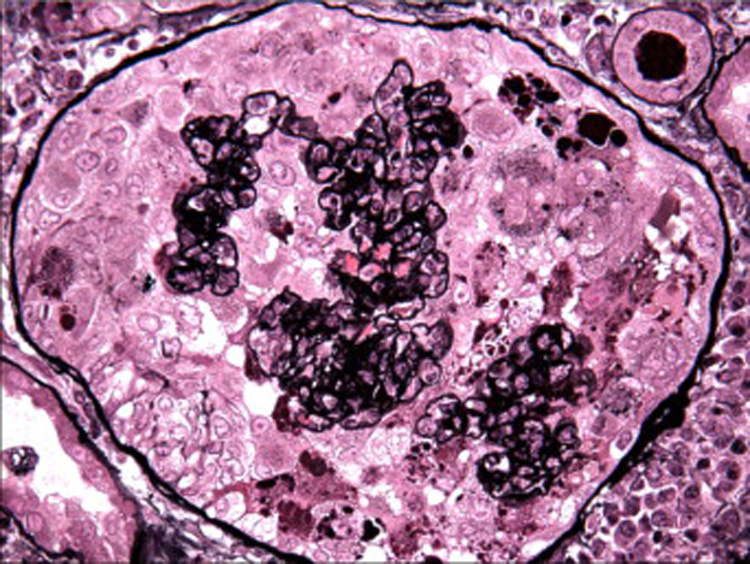
The podocyte hyperplasia forms pseudocrescents that obliterate the urinary space, (Jones methenamine silver, x400). [COLOR PRINT/ ONLINE]
Figure 4.
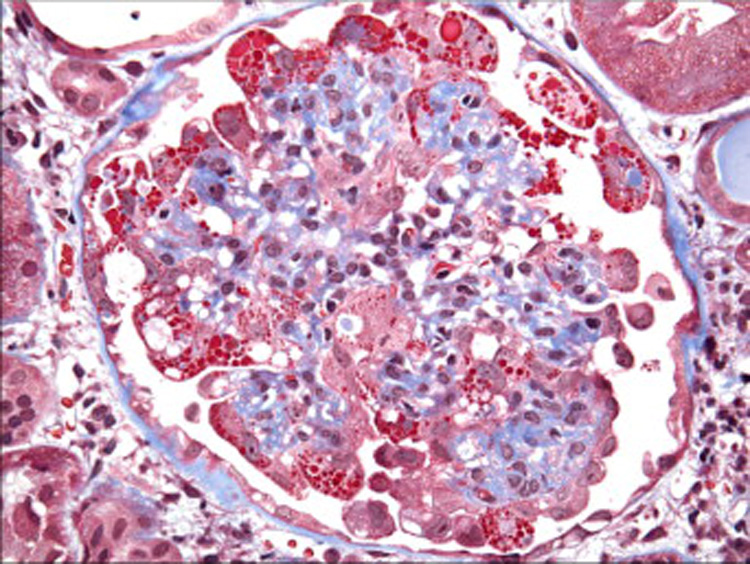
Overlying the collapsed tuft, the enlarged podocytes contain abundant trichromred protein resorption droplets (Masson trichrome, x400). [COLOR PRINT/ ONLINE]
Tubulo-interstitial disease is an invariable component of HIVAN and often appears out of proportion to the degree of glomerular injury. In addition to tubular atrophy, interstitial fibrosis, edema, and inflammation, there are widespread tubular degenerative and regenerative changes.11 12 These include tubular epithelial simplification and hypertrophy with enlarged hyperchromatic nuclei, prominent nucleoli, mitotic figures, and focal apoptosis. The HIV-1 gene Vpr has been implicated in these changes, based on recent work demonstrating that Vpr expression impairs cytokinesis in tubular epithelial cells in vitro.16 Distended tubules containing loose proteinaceous casts form “tubular microcysts”, which may be numerous10 (Figure 5). Interstitial leukocytes consist predominantly of T lymphocytes, with renal CD4/CD8 ratios ranging from 0.35 to approximately 1.12,17 Monocytes/ macrophages, plasma cells, and B-cells comprise a relatively small component of the infiltrate.
Figure 5.
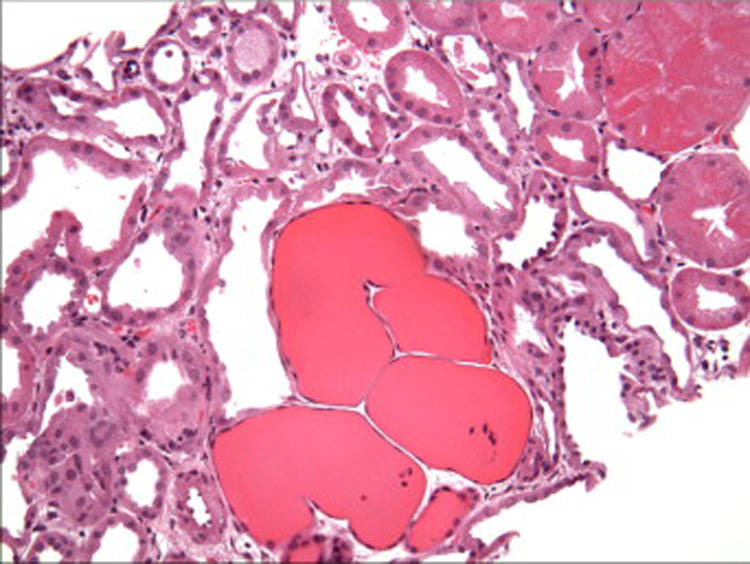
There are focally distended tubules forming microcysts that contain loose proteinaceous casts. Adjacent cortical tubules display degenerative changes (H&E, x200). [COLOR PRINT/ ONLINE]
Immunofluorescence
By immunofluorescence, there are segmental to global deposits of IgM, C3, and less commonly C1, in the collapsing segments. The non-collapsed glomeruli may display weaker mesangial staining for IgM and C3. Glomerular deposits of IgG and IgA are not identified. Visceral epithelial protein reabsorption droplets often stain for IgG, IgA, and albumin, with similar staining in the tubular epithelial protein droplets, consistent with increased intracellular protein trafficking.
Electron microscopy
At the ultrastructural level, the collapsed lobules display wrinkling and little or no thickening of glomerular basement membranes. The overlying podocytes are markedly hypertrophied with severe foot process effacement, focal detachment, and increased numbers of organelles including electron dense protein resorption droplets, electron lucent transport vesicles, and abundant rough endoplasmic reticulum (Figure 6). The actin cytoskeleton usually appears disrupted, giving the cells a relatively open-appearing cytoplasm (Figure 7). Non-collapsed capillaries also display severe foot process effacement, typically >50%, and often exceeding 90% of the capillary surface area (Figure 7). No electron dense deposits are observed, with the exception of rare small paramesangial electron densities corresponding to the mesangial deposits of IgM.
Figure 6.

The collapsed capillaries have wrinkled glomerular basement membranes and are surrounded by hyperplastic visceral epithelial cells that obliterate the urinary space, in continuity with adjacent parietal epithelial cells (electron micrograph, x3000).
Figure 7.

A high power view shows podocyte hypertrophy with open-appearing cytoplasm and complete effacement of foot processes. One of the podocytes contains prominent intracytoplasmic protein resorption droplets (electron micrograph, x5000).
Endothelial tubulo-reticular inclusions, also known as “interferon footprints”, are commonly identified as 24 nm tubular structures located in dilated cisternae of smooth endoplasmic reticulum (Figure 8). These structures are a marker of HIV infection that can be found in endothelial cells and lymphocytes throughout the body. It is important to recognize that they are not a specific feature of the nephropathy and may be found in HIV-infected patients without HIVAN, as well as in patients with systemic lupus erythematosus, hepatitis C, and other viral infections. Although it has not been studied systematically, endothelial tubulo-reticular inclusions appear to be less common in renal biopsies from patients with HIVAN who are receiving ART. This observation is consistent with a reduction in viral burden and associated cytokine dyregulation that would be predicted to occur in the setting of ART. Endothelial tubulo-reticular inclusions are also readily identified in collapsing glomerulopathy secondary to interferon therapy, but they are typically lacking in idiopathic collapsing FSGS, as well as those cases associated with pamidronate toxicity or parvovirus B19 infection.
Figure 8.

A large tubulo-reticular inclusion is seen within a glomerular endothelial cell (electron micrograph, x40,000).
Tubular epithelial cells often display enlarged regenerative nuclei with prominent nucleoli (Figure 9), whereas the cells lining tubular microcysts are typically flattened (Figure 10). Other ultrastructural findings in HIVAN include increased numbers of nuclear bodies within tubular and interstitial cells.12,18 These intranuclear structures, which measure from 0.5 to 1.5 microns in diameter, are of uncertain significance. They are not specific for HIVAN, and may be upregulated in diverse cell types in response to viral infections, hormonal activation, and neoplastic transformation. Granular degeneration of the nuclear chromatin of tubular and interstitial cells and the presence of cylindrical confronting cisternae may also be observed.12,18
Figure 9.

A tubule is lined by hyperplastic epithelial cells with enlarged regenerative nuclei containing prominent nucleoli (electron micrograph, x4000).
Figure 10.

In an area of back-to-back tubular microcyst formation, the lining epithelial cells of 2 individual microcysts appear flattened and compressed by the voluminous proteinaceous electron dense cast material, (electron micrograph, x3000).
Podocyte dysregulation in HIV-associated nephropathy
In the course of normal glomerular development, podocytes undergo a choreographed program of proliferation and progressive maturation, culminating in a fully differentiated, quiescent phenotype. In the mature podocyte, the expression of WT-1, a zinc finger transcription factor that downregulates proliferation, coincides with podocyte exit from the cell cycle, the expression of cyclin kinase inhibitors, and the acquisition of maturity markers.19 20 In HIVAN and collapsing forms of primary FSGS, injured podocytes revert to a developmental program that includes downregulation of cyclin kinase inhibitors, entry into the cell cycle, and loss of mature phenotypic markers.21 22 23 24 Because it involves a loss of podocyte expression of the developmental regulatory protein WT-1, this altered podocyte phenotype has been termed “dysregulated.”21
The injured podocytes express the proliferation marker Ki-67 and lose maturity markers, such as CD10/CALLA, C3b receptor, GLEPP-1, podocalyxin, synaptopodin, and WT-1.21 Of note, a reduction in synaptopodin expression, in contrast to other markers, was also observed in histologically unaffected glomeruli, suggesting that it precedes collapse.21 By contrast, these podocyte phenotypic changes were not identified in minimal change disease or membranous glomerulopathy, despite similar levels of proteinuria and similar degrees of foot process effacement. In collapsed glomeruli, the endothelial expression of podocalyxin was preserved despite severe structural alterations of the capillary tuft and complete loss of podocyte expression.21 These findings point to derangement of the podocyte phenotype occurring as a primary event, rather than as a consequence of collapse.
Each phase of the cell cycle is governed by positive regulatory proteins (cyclins and cyclin-dependent kinases) and negative regulatory proteins (cyclin kinase inhibitors). Progression through the cell cycle requires the activation of cyclin-dependent kinases by complexing with specific partner cyclins. The Cip/Kip family of cyclin kinase inhibitors (p21, p27, p57) function in both G1 and S phase, and p21 also inhibits G2/M phase complexes. The cyclin kinase inhibitors p27 and p57 are constitutively expressed in mature podocytes, whereas p21 is not.22 In HIVAN, as well as in idiopathic collapsing FSGS, there is decreased podocyte expression of p27 and p57, accompanied by de novo expression of p21 and Ki-67.22 23 Interestingly, a generalized reduction in p27 and p57 can be identified in podocytes of some histologically unaffected glomeruli.22 These findings support a paradigm wherein the reduction in p27 and p57 precedes the development of collapsing lesions and is permissive for the proliferative podocyte phenotype. Similar alterations in podocyte phenotype, as well as a dedifferentiated tubular epithelial phenotype, have been found in the HIV-1 transgenic murine model of HIVAN, supporting a pathogenic role for HIV viral gene expression.24 The pathologic alterations can be attenuated or reversed by administration of the cyclin kinase inhibitor R-roscovitine, indicating a critical role for cell cycle dysregulation in this model.25 How specific viral proteins may mediate podocyte dysregulation is discussed in a companion article on HIVAN pathogenesis.
The origin of the proliferating glomerular epithelial cells has been debated. Studies of human HIVAN by Dijkman et al. have suggested that parietal epithelial cells proliferate to repopulate the injured visceral epithelial cells.26 In that study, many of the proliferating epithelial cells in areas of pseudocrescent formation expressed the parietal epithelial cell marker CK8 and lacked podocyte specific markers. In addition, cell bridges to the CK8-positive parietal lining could be observed in serial sections, and no glomerular epithelial cells co-expressed podocyte markers and CK8.26 These findings are consistent with emerging evidence that the parietal epithelium at the glomerular hilus may provide a niche for podocyte progenitor cells.27 On the other hand, studies have clearly established the ability of podocytes to enter the cell cycle. Transgenic mice expressing HIV-1 Nef under the podocin promoter demonstrate expression of the cell cycle markers Ki-67 and phospho-Stat3 in podocytes.28 The relative contribution of podocytes and parietal cells to glomerular epithelial cell proliferation in HIVAN and other forms of collapsing glomerulopathy remains to be fully defined.
Differential diagnosis of renal biopsy findings
A biopsy picture of collapsing glomerulopathy is not specific for HIVAN. Differential diagnosis of the collapsing variant of FSGS includes primary (idiopathic) FSGS,29 parvovirus B19 infection,30 SV40 infection,31 acute CMV infection,32 erythrophagocytosis syndrome,33 interferon therapy,34 pamidronate toxicity,35 acute vaso-occlusive injury,36 rare familial forms,37 and glomerular injury in the renal allograft associated with microvascular disease.38
Renal biopsy in the HIV-infected patient is required to establish a diagnosis of HIVAN and exclude other causes of renal dysfunction and proteinuria, including a variety of HIV-related glomerular diseases, non-HIV-related renal diseases, and medication nephrotoxicity, many of which are reviewed in detail in companion articles. Other glomerular lesions encountered in the HIV-infected patient are listed in Table 1. Immune complex-mediated glomerular disease is more common in the Caucasian population, whereas HIVAN predominantly affects African Americans. It is particularly challenging for the renal pathologist to distinguish HIVAN from other forms of FSGS, including secondary FSGS from hypertensive arterionephrosclerosis or pre-existing primary FSGS, which are also more common in black patients. Recent biopsy series in HIV-infected patients indicate an increasing prevalence of FSGS (NOS) in parallel with a reduction in HIVAN, suggesting modification of the collapsing pattern of HIVAN by ART.39,40 To illustrate the changing epidemiology of HIVAN, the renal biopsy incidence of HIVAN was 65% in a series of 112 patients reported in 1997,41 but only 35% in a recent series of 152 biopsies.39 Because endothelial tubulo-reticular inclusions also appear to be reduced by ART, the biopsy picture of HIVAN may be attenuated in ART-treated patients, approximating the morphologic appearance of FSGS (NOS). In addition, as patients live longer with HIV infection, they may develop other non-HIV-related kidney diseases common in the aging population, such as diabetic nephropathy and arterionephrosclerosis of aging or hypertension, which may also exhibit focal sclerosing features.
TABLE 1.
GLOMERULAR LESIONS OCCURRING IN HIV-INFECTED PATIENTS
| HIVAN (collapsing glomerulopathy) |
| Focal segmental glomerulosclerosis (FSGS), not otherwise specified (NOS) |
| Minimal change disease |
| Immune complex-mediated glomerulonephritis |
| Lupus-like nephritis |
| IgA nephropathy |
| Membranoproliferative glomerulonephritis (associated with hepatitis C or B) |
| Membranous glomerulopathy (associated with hepatitis B or C, or neoplasia) |
| Acute post-infectious glomerulonephritis |
| Fibrillary and immunotactoid glomerulonephritis (often associated with hepatitis C) |
| Diabetic nephropathy |
| Amyloidosis, AA type (associated with IVDU) |
| Thrombotic microangiopathy |
Treatment of HIV-associated nephropathy
In the absence of randomized clinical trials, the treatment of HIVAN is based on small, uncontrolled studies, epidemiologic data, and pathogenic insights. The pathogenesis of HIVAN is reviewed in a companion article in this issue, and is known to involve direct HIV infection and gene expression in renal epithelial cells, as well as host factors that affect susceptibility. Consistent with the direct pathogenic role of HIV infection, the introduction of combination ART in 1996 was followed by a decline in the incidence of HIVAN42 43 and in the number of new cases of ESRD attributed to “AIDS nephropathy” in the United States (US).5 These suggestive epidemiologic data are supported by small, uncontrolled studies demonstrating improved renal survival with ART,44 45 and by case reports documenting renal recovery and histological improvement following the initiation of ART7 46. In a retrospective study of 42 patients with biopsy-proven HIVAN from 6 US academic medical centers, ART use was associated with delayed progression to ESRD (HR 0.24, 95% CI 0.07–0.84).44 A similar improvement in renal survival with ART was demonstrated in a single-center retrospective study of 36 patients with biopsy-proven HIVAN (HR 0.30, 95% CI 0.09–0.98).45 Although these estimates were adjusted for demographic and clinical characteristics, it is likely that there are also important unmeasured differences between patients who received ART and those who did not receive ART in these non-randomized studies.
Recognizing that randomized controlled trials comparing ART to placebo are no longer ethically tenable, recently updated expert guidelines consider HIVAN an indication for the initiation of ART, regardless of CD4 cell count.47 48 The guidelines also recommend adjunctive therapy with ACE inhibitors or angiotensin receptor blockers as tolerated,47 based on evidence of benefit from cohort studies in patients with HIVAN and from randomized clinical trials in other glomerular diseases.49 The addition of corticosteroids may be considered in patients with aggressive disease or a prominent interstitial inflammatory component, based on uncontrolled clinical studies and in vitro evidence that HIV infection induces a local inflammatory reaction in tubular epithelial cells.50–52
The management of ESRD in patients with HIV infection is discussed in detail elsewhere in the issue. With improvements in the survival of HIV-positive dialysis patients,53 patients with HIVAN who are approaching ESRD should be offered a choice between hemodialysis and peritoneal dialysis, which offer similar survival in adults with HIV infection.54 Selected patients with remote HIVAN and well-controlled HIV infection may also be candidates for kidney transplantation.55
Epidemiology of HIV-associated nephropathy in the antiretroviral era
Although the incidence of ESRD attributed to HIVAN reached a plateau in the US following the introduction of combination ART, 800–900 new cases are reported to the US Renal Database System (USRDS) each year, and the prevalence of HIV-related ESRD continues to increase.5,56 At the end of 2005, more than 2,700 individuals were living with ESRD attributed to HIVAN in the US, compared to only 150 cases at the end of 1990.5,56 This trend is projected to continue, in large part because of improvements in the survival of HIV-positive dialysis patients, but also because of the disproportionate burden of HIV infection and AIDS among African-Americans. Assuming a stable annual mortality rate of 24% and linear growth of the HIV epidemic among African-Americans, it is projected that nearly 10,000 individuals will be living with ESRD attributed to HIVAN by the year 2020.56 These projections were based on diagnoses of “AIDS nephropathy” reported to the USRDS at the discretion of the treating physician. While these cases likely reflect a heterogeneous population of HIV-positive patients with HIVAN and other glomerular diseases, these projections underestimate the true prevalence of HIV infection in the ESRD population. Unfortunately, the USRDS no longer collects data on HIV infection as a comorbid condition in incident ESRD patients, and future estimates from the USRDS will be limited to ESRD that is attributed to HIVAN.
In addition to the continued growth of the HIV-positive ESRD population in the US, there is also alarming potential for an epidemic of HIVAN in sub-Saharan Africa. Nearly 90% of the ESRD attributed to HIVAN in the US occurs in African-Americans,5 and a similar racial predisposition has been observed in other countries.57 58 Emerging data suggest a high prevalence of kidney disease among HIV-infected individuals in sub-Saharan Africa, ranging from 6% among Kenyan patients without other risk factors for kidney disease, to as high as 38% in a Nigerian cohort.59–61 Although kidney biopsies are performed less frequently in resource-limited settings, HIVAN was the most common diagnosis identified in published biopsy series from Nigeria and South Africa.60 61,62 With expanding access to ART and prolonged survival of HIV-infected patients, HIVAN will likely be an important contributor to the growing public health burden of chronic kidney disease in sub-Saharan Africa.62,63
Acknowledgments
Grant Support: Supported in part by NIH grants P01DK56492-05 and K23DK077568.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Christina M. Wyatt, Assistant Professor, Department of Medicine, Division of Nephrology Mount Sinai School of Medicine, New York, NY.
Paul E. Klotman, Professor and Chair, Samuel Bronfman Department of Medicine Mount Sinai School of Medicine, New York, NY.
Vivette D. D’Agati, Professor, Department of Pathology Columbia University Medical Center, New York, NY.
References
- 1.Rao TK, Filippone EJ, Nicastri AD, Landesman SH, Frank E, Chen CK, Friedman EA. Associated focal and segmental glomerulosclerosis in the acquired immunodeficiency syndrome. N Engl J Med. 1984;310:669–673. doi: 10.1056/NEJM198403153101101. [DOI] [PubMed] [Google Scholar]
- 2.Pardo V, Aldana M, Colton RM, Fischl MA, Jaffe D, Moskowitz L, Hensley GT, Bourgoignie JJ. Glomerular lesions in the acquired immunodeficiency syndrome. Ann Intern Med. 1984;101:429–434. doi: 10.7326/0003-4819-101-4-429. [DOI] [PubMed] [Google Scholar]
- 3.Gardenswartz MH, Lerner CW, Seligson GR, Zabetakis PM, Rotterdam H, Tapper ML, Michelis MF, Bruno MS. Renal disease in patients with AIDS: a clinicopathologic study. Clin Nephrol. 1984;21:197–204. [PubMed] [Google Scholar]
- 4.Ross MJ, Klotman PE. Recent progress in HIV-associated nephropathy. J Am Soc Nephrol. 2002;13:2997–3004. doi: 10.1097/01.asn.0000040750.40907.99. [DOI] [PubMed] [Google Scholar]
- 5.U.S. Renal Data System. USRDS 2007 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. 2007
- 6.Cantor ES, Kimmel PL, Bosch JP. Effect of race on expression of acquired immunodeficiency syndrome-associated nephropathy. Arch Intern Med. 1991;151:125–128. [PubMed] [Google Scholar]
- 7.Winston JA, Bruggeman LA, Ross MD, Jacobson J, Ross L, D'Agati VD, Klotman PE, Klotman ME. Nephropathy and establishment of a renal reservoir of HIV type 1 during primary infection. N Engl J Med. 2001;344:1979–1984. doi: 10.1056/NEJM200106283442604. [DOI] [PubMed] [Google Scholar]
- 8.Atta MG, Choi MJ, Longenecker JC, Haymart M, Wu J, Nagajothi N, Racusen LC, Scheel PJJ, Brancati FL, Fine DM. Nephrotic range proteinuria and CD4 count as noninvasive indicators of HIV-associated nephropathy. Am J Med. 2005:118–1288. doi: 10.1016/j.amjmed.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 9.Atta MG, Longenecker JC, Fine DM, Nagajothi N, Grover DS, Wu J, Racusen LC, Scheel PJJ, Hamper UM. Sonography as a predictor of human immunodeficiency virus-associated nephropathy. J Ultrasound Med. 2004;23:603–610. doi: 10.7863/jum.2004.23.5.603. quiz 612-3. [DOI] [PubMed] [Google Scholar]
- 10.D'Agati V, Appel GB. Renal pathology of human immunodeficiency virus infection. Semin Nephrol. 1998;18:406–421. [PubMed] [Google Scholar]
- 11.Cohen AH, Nast CC. HIV-associated nephropathy. A unique combined glomerular, tubular, and interstitial lesion. Mod Pathol. 1988;1:87–97. [PubMed] [Google Scholar]
- 12.D'Agati V, Suh JI, Carbone L, Cheng JT, Appel G. Pathology of HIV-associated nephropathy: a detailed morphologic and comparative study. Kidney Int. 1989;35:1358–1370. doi: 10.1038/ki.1989.135. [DOI] [PubMed] [Google Scholar]
- 13.Strauss J, Abitbol C, Zilleruelo G, Scott G, Paredes A, Malaga S, Montane B, Mitchell C, Parks W, Pardo V. Renal disease in children with the acquired immunodeficiency syndrome. N Engl J Med. 1989;321:625–630. doi: 10.1056/NEJM198909073211001. [DOI] [PubMed] [Google Scholar]
- 14.AH C, CC N. Renal injury associated with human immunodeficiency vius infection. In: Charles JJ, L OJ, M SM, G SF, editors. Heptinstall's Pathology of the Kidney. Philadelphia: Lippincott Williams & Wilkins; 2007. [Google Scholar]
- 15.D'Agati VD, Fogo AB, Bruijn JA, Jennette JC. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am J Kidney Dis. 2004;43:368–382. doi: 10.1053/j.ajkd.2003.10.024. [DOI] [PubMed] [Google Scholar]
- 16.Rosenstiel P, Letourneau TGA, Chan JC, Husain M, D'Agati V, Klotman ME, Klotman PE. HIV-1 vpr impairs cytokinesis in human proximal tubule cells leading to multinucleation and hypertrophy: implications for HIVAN pathogenesis. Kidney Int. 2008 doi: 10.1038/ki.2008.303. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rey L, Viciana A, Ruiz P. Immunopathological characteristics of in situ T-cell subpopulations in human immunodeficiency virus-associated nephropathy. Hum Pathol. 1995;26:408–415. doi: 10.1016/0046-8177(95)90142-6. [DOI] [PubMed] [Google Scholar]
- 18.Chander P, Soni A, Suri A, Bhagwat R, Yoo J, Treser G. Renal ultrastructural markers in AIDS-associated nephropathy. Am J Pathol. 1987;126:513–526. [PMC free article] [PubMed] [Google Scholar]
- 19.Nagata M, Nakayama K, Terada Y, Hoshi S, Watanabe T. Cell cycle regulation and differentiation in the human podocyte lineage. Am J Pathol. 1998;153:1511–1520. doi: 10.1016/s0002-9440(10)65739-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83:253–307. doi: 10.1152/physrev.00020.2002. [DOI] [PubMed] [Google Scholar]
- 21.Barisoni L, Kriz W, Mundel P, D'Agati V. The dysregulated podocyte phenotype: a novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. 1999;10:51–61. doi: 10.1681/ASN.V10151. [DOI] [PubMed] [Google Scholar]
- 22.Shankland SJ, Eitner F, Hudkins KL, Goodpaster T, D'Agati V, Alpers CE. Differential expression of cyclin-dependent kinase inhibitors in human glomerular disease: role in podocyte proliferation and maturation. Kidney Int. 2000;58:674–683. doi: 10.1046/j.1523-1755.2000.00213.x. [DOI] [PubMed] [Google Scholar]
- 23.Barisoni L, Mokrzycki M, Sablay L, Nagata M, Yamase H, Mundel P. Podocyte cell cycle regulation and proliferation in collapsing glomerulopathies. Kidney Int. 2000;58:137–143. doi: 10.1046/j.1523-1755.2000.00149.x. [DOI] [PubMed] [Google Scholar]
- 24.Barisoni L, Bruggeman LA, Mundel P, D'Agati VD, Klotman PE. HIV-1 induces renal epithelial dedifferentiation in a transgenic model of HIV-associated nephropathy. Kidney Int. 2000;58:173–181. doi: 10.1046/j.1523-1755.2000.00152.x. [DOI] [PubMed] [Google Scholar]
- 25.Gherardi D, D'Agati V, Chu TH, Barnett A, Gianella-Borradori A, Gelman IH, Nelson PJ. Reversal of collapsing glomerulopathy in mice with the cyclin-dependent kinase inhibitor CYC202. J Am Soc Nephrol. 2004;15:1212–1222. doi: 10.1097/01.asn.0000124672.41036.f4. [DOI] [PubMed] [Google Scholar]
- 26.Dijkman HB, Weening JJ, Smeets B, Verrijp KC, van Kuppevelt TH, Assmann KK, Steenbergen EJ, Wetzels JF. Proliferating cells in HIV and pamidronate-associated collapsing focal segmental glomerulosclerosis are parietal epithelial cells. Kidney Int. 2006;70:338–344. doi: 10.1038/sj.ki.5001574. [DOI] [PubMed] [Google Scholar]
- 27.Bariety J, Mandet C, Hill GS, Bruneval P. Parietal podocytes in normal human glomeruli. J Am Soc Nephrol. 2006;17:2770–2780. doi: 10.1681/ASN.2006040325. [DOI] [PubMed] [Google Scholar]
- 28.Husain M, D'Agati VD, He JC, Klotman ME, Klotman PE. HIV-1 Nef induces dedifferentiation of podocytes in vivo: a characteristic feature of HIVAN. AIDS. 2005;19:1975–1980. doi: 10.1097/01.aids.0000191918.42110.27. [DOI] [PubMed] [Google Scholar]
- 29.Valeri A, Barisoni L, Appel GB, Seigle R, D'Agati V. Idiopathic collapsing focal segmental glomerulosclerosis: a clinicopathologic study. Kidney Int. 1996;50:1734–1746. doi: 10.1038/ki.1996.493. [DOI] [PubMed] [Google Scholar]
- 30.Moudgil A, Nast CC, Bagga A, Wei L, Nurmamet A, Cohen AH, Jordan SC, Toyoda M. Association of parvovirus B19 infection with idiopathic collapsing glomerulopathy. Kidney Int. 2001;59:2126–2133. doi: 10.1046/j.1523-1755.2001.00727.x. [DOI] [PubMed] [Google Scholar]
- 31.Li RM, Branton MH, Tanawattanacharoen S, Falk RA, Jennette JC, Kopp JB. Molecular identification of SV40 infection in human subjects and possible association with kidney disease. J Am Soc Nephrol. 2002;13:2320–2330. doi: 10.1097/01.asn.0000028249.06596.cf. [DOI] [PubMed] [Google Scholar]
- 32.Tomlinson L, Boriskin Y, McPhee I, Holwill S, Rice P. Acute cytomegalovirus infection complicated by collapsing glomerulopathy. Nephrol Dial Transplant. 2003;18:187–189. doi: 10.1093/ndt/18.1.187. [DOI] [PubMed] [Google Scholar]
- 33.Thaunat O, Delahousse M, Fakhouri F, Martinez F, Stephan JL, Noel LH, Karras A. Nephrotic syndrome associated with hemophagocytic syndrome. Kidney Int. 2006;69:1892–1898. doi: 10.1038/sj.ki.5000352. [DOI] [PubMed] [Google Scholar]
- 34.Shah M, Jenis EH, Mookerjee BK, Schriber JR, Baer MR, Herzig GP, Wetzler M. Interferon-alpha-associated focal segmental glomerulosclerosis with massive proteinuria in patients with chronic myeloid leukemia following high dose chemotherapy. Cancer. 1998;83:1938–1946. [PubMed] [Google Scholar]
- 35.Markowitz GS, Appel GB, Fine PL, Fenves AZ, Loon NR, Jagannath S, Kuhn JA, Dratch AD, D'Agati VD. Collapsing focal segmental glomerulosclerosis following treatment with high-dose pamidronate. J Am Soc Nephrol. 2001;12:1164–1172. doi: 10.1681/ASN.V1261164. [DOI] [PubMed] [Google Scholar]
- 36.Greenberg A, Bastacky SI, Iqbal A, Borochovitz D, Johnson JP. Focal segmental glomerulosclerosis associated with nephrotic syndrome in cholesterol atheroembolism: clinicopathological correlations. Am J Kidney Dis. 1997;29:334–344. doi: 10.1016/s0272-6386(97)90193-1. [DOI] [PubMed] [Google Scholar]
- 37.Avila-Casado MC, Vargas-Alarcon G, Soto ME, Hernandez G, Reyes PA, Herrera-Acosta J. Familial collapsing glomerulopathy: clinical, pathological and immunogenetic features. Kidney Int. 2003;63:233–239. doi: 10.1046/j.1523-1755.2003.00713.x. [DOI] [PubMed] [Google Scholar]
- 38.Nadasdy T, Allen C, Zand MS. Zonal distribution of glomerular collapse in renal allografts: possible role of vascular changes. Hum Pathol. 2002;33:437–441. doi: 10.1053/hupa.2002.124333. [DOI] [PubMed] [Google Scholar]
- 39.Berliner AR, Fine DM, Lucas GM, Rahman MH, Racusen LC, Scheel PJ, Atta MG. Observations on a Cohort of HIV-Infected Patients Undergoing Native Renal Biopsy. Am J Nephrol. 2008;28:478–486. doi: 10.1159/000112851. [DOI] [PubMed] [Google Scholar]
- 40.Fine DM, Perazella MA, Lucas GM, Atta MG. Kidney biopsy in HIV: beyond HIV-associated nephropathy. Am J Kidney Dis. 2008;51:504–514. doi: 10.1053/j.ajkd.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 41.D'Agati V, Appel GB. HIV infection and the kidney. J Am Soc Nephrol. 1997;8:138–152. doi: 10.1681/ASN.V81138. [DOI] [PubMed] [Google Scholar]
- 42.Ahuja TS, Borucki M, Funtanilla M, Shahinian V, Hollander M, Rajaraman S. Is the prevalence of HIV-associated nephropathy decreasing? Am J Nephrol. 1999;19:655–659. doi: 10.1159/000013537. [DOI] [PubMed] [Google Scholar]
- 43.Lucas GM, Eustace JA, Sozio S, Mentari EK, Appiah KA, Moore RD. Highly active antiretroviral therapy and the incidence of HIV-1-associated nephropathy: a 12-year cohort study. AIDS. 2004;18:541–546. doi: 10.1097/00002030-200402200-00022. [DOI] [PubMed] [Google Scholar]
- 44.Szczech LA, Gupta SK, Habash R, Guasch A, Kalayjian R, Appel R, Fields TA, Svetkey LP, Flanagan KH, Klotman PE, Winston JA. The clinical epidemiology and course of the spectrum of renal diseases associated with HIV infection. Kidney Int. 2004;66:1145–1152. doi: 10.1111/j.1523-1755.2004.00865.x. [DOI] [PubMed] [Google Scholar]
- 45.Atta MG, Gallant JE, Rahman MH, Nagajothi N, Racusen LC, Scheel PJ, Fine DM. Antiretroviral therapy in the treatment of HIV-associated nephropathy. Nephrol Dial Transplant. 2006;21:2809–2813. doi: 10.1093/ndt/gfl337. [DOI] [PubMed] [Google Scholar]
- 46.Wali RK, Drachenberg CI, Papadimitriou JC, Keay S, Ramos E. HIV-1-associated nephropathy and response to highly-active antiretroviral therapy [letter] Lancet. 1998;352:783–784. doi: 10.1016/S0140-6736(98)24037-2. [DOI] [PubMed] [Google Scholar]
- 47.Gupta SK, Eustace JA, Winston JA, Boydstun II, Ahuja TS, Rodriguez RA, Tashima KT, Roland M, Franceschini N, Palella FJ, Lennox JL, Klotman PE, Nachman SA, Hall SD, Szczech LA. Guidelines for the management of chronic kidney disease in HIV-infected patients: recommendations of the HIV Medicine Association of the Infectious Diseases Society of America. Clin Infect Dis. 2005;40:1559–1585. doi: 10.1086/430257. [DOI] [PubMed] [Google Scholar]
- 48.Panel on Antiretroviral Guidelines for Adult and Adolescents. [Accessed Dec 30, 2007];Department of Health and Human Services; Guidelines for the use of antiretroviral agents in HIV-infected adults and adolescents. 2007 December 1;:1–136. Available at http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf.
- 49.Wei A, Burns GC, Williams BA, Mohammed NB, Visintainer P, Sivak SL. Long-term renal survival in HIV-associated nephropathy with angiotensin-converting enzyme inhibition. Kidney Int. 2003;64:1462–1471. doi: 10.1046/j.1523-1755.2003.00230.x. [DOI] [PubMed] [Google Scholar]
- 50.Smith MC, Austen JL, Carey JT, Emancipator SN, Herbener T, Gripshover B, Mbanefo C, Phinney M, Rahman M, Salata RA, Weigel K, Kalayjian RC. Prednisone improves renal function and proteinuria in human immunodeficiency virus-associated nephropathy. Am J Med. 1996;101:41–48. doi: 10.1016/s0002-9343(96)00065-4. [DOI] [PubMed] [Google Scholar]
- 51.Eustace JA, Nuermberger E, Choi M, Scheel PJ, Jr., Moore R, Briggs WA. Cohort study of the treatment of severe HIV-associated nephropathy with corticosteroids. Kidney Int. 2000;58:1253–1260. doi: 10.1046/j.1523-1755.2000.00280.x. [DOI] [PubMed] [Google Scholar]
- 52.Ross MJ, Fan C, Ross MD, Chu TH, Shi Y, Kaufman L, Zhang W, Klotman ME, Klotman PE. HIV-1 infection initiates an inflammatory cascade in human renal tubular epithelial cells. J Acquir Immune Defic Syndr. 2006;42:1–11. doi: 10.1097/01.qai.0000218353.60099.4f. [DOI] [PubMed] [Google Scholar]
- 53.Ahuja TS, Collinge N, Grady J, Khan S. Changing trends in the survival of dialysis patients with human immunodeficiency virus in the United States. J Am Soc Nephrol. 2002;13:1889–1893. doi: 10.1097/01.asn.0000019773.43765.bf. [DOI] [PubMed] [Google Scholar]
- 54.Ahuja TS, Collinge N, Grady J, Khan S. Is dialysis modality a factor in survival of patients with ESRD and HIV-associated nephropathy? Am J Kidney Dis. 2003;41:1060–1064. doi: 10.1016/s0272-6386(03)00204-x. [DOI] [PubMed] [Google Scholar]
- 55.Wyatt CM, Murphy B. Kidney transplantation in HIV-infected patients. Semin Dial. 2005;18:495–498. doi: 10.1111/j.1525-139X.2005.00095.x. [DOI] [PubMed] [Google Scholar]
- 56.Schwartz EJ, Szczech LA, Ross MJ, Klotman ME, Winston JA, Klotman PE. Highly active antiretroviral therapy and the epidemic of HIV+ end-stage renal disease. J Am Soc Nephrol. 2005;16:2412–2420. doi: 10.1681/ASN.2005040340. [DOI] [PubMed] [Google Scholar]
- 57.Lopes GS, Marques LP, Rioja LS, Basilio-de-Oliveira CA, Oliveira AV, Nery AC, Santos Oda R. Glomerular disease and human immunodeficiency virus infection in Brazil. Am J Nephrol. 1992;12:281–287. doi: 10.1159/000168461. [DOI] [PubMed] [Google Scholar]
- 58.Laradi A, Mallet A, Beaufils H, Allouache M, Martinez F. HIV-associated nephropathy: outcome and prognosis factors. Groupe d' Etudes Nephrologiques d'Ile de France. J Am Soc Nephrol. 1998;9:2327–2335. doi: 10.1681/ASN.V9122327. [DOI] [PubMed] [Google Scholar]
- 59.Wools-Kaloustian K, Gupta SK, Muloma E, Owino-Ong'or W, Sidle J, Aubrey RW, Shen J, Kipruto K, Zwickl BE, Goldman M. Renal disease in an antiretroviral-naive HIV-infected outpatient population in Western Kenya. Nephrol Dial Transplant. 2007;22:2208–2212. doi: 10.1093/ndt/gfm223. [DOI] [PubMed] [Google Scholar]
- 60.Emem CP, Arogundade F, Sanusi A, Adelusola K, Wokoma F, Akinsola A. Renal disease in HIV-seropositive patients in Nigeria: an assessment of prevalence, clinical features and risk factors. Nephrol Dial Transplant. 2008;23:741–746. doi: 10.1093/ndt/gfm836. [DOI] [PubMed] [Google Scholar]
- 61.Han TM, Naicker S, Ramdial PK, Assounga AG. A cross-sectional study of HIV-seropositive patients with varying degrees of proteinuria in South Africa. Kidney Int. 2006;69:2243–2250. doi: 10.1038/sj.ki.5000339. [DOI] [PubMed] [Google Scholar]
- 62.Gerntholtz TE, Goetsch SJ, Katz I. HIV-related nephropathy: a South African perspective. Kidney Int. 2006;69:1885–1891. doi: 10.1038/sj.ki.5000351. [DOI] [PubMed] [Google Scholar]
- 63.Arogundade FA, Barsoum RS. CKD prevention in Sub-Saharan Africa: a call for governmental, nongovernmental, and community support. Am J Kidney Dis. 2008;51:515–523. doi: 10.1053/j.ajkd.2007.12.006. [DOI] [PubMed] [Google Scholar]