

Abstract
Huntington’s disease (HD) is a fatal neurodegenerative disorder caused by the presence of an abnormally expanded polyglutamine domain in the N-terminus of huntingtin. We developed a recombinant adeno-associated viral serotype 5 (rAAV5) gene transfer strategy to posttranscriptionally suppress the levels of striatal mutant huntingtin (mHtt) in the R6/1 HD transgenic mouse via RNA interference. Transient cotransfection of HEK293 cells with plasmids expressing a portion of human mHtt derived from R6/1 transgenic HD mice and a short-hairpin RNA directed against the 5′ UTR of the mHtt mRNA (siHUNT-1) resulted in reduction in the levels of mHtt mRNA (−75%) and protein (−60%). Long-term in vivo rAAV5-mediated expression of siHUNT-1 in the striatum of R6/1 mice reduced the levels of mHtt mRNA (−78%) and protein (−28%) as determined by quantitative RT-PCR and Western blot analysis, respectively. The reduction in mHtt was concomitant with a reduction in the size and number of neuronal intranuclear inclusions and a small but significant normalization of the steady-state levels of preproenkephalin and dopamine- and cAMP-responsive phosphoprotein 32 kDa mRNA. Finally, bilateral expression of rAAV5-siHUNT-1 resulted in delayed onset of the rear paw clasping phenotype exhibited by the R6/1 mice. These results suggest that a reduction in the levels of striatal mHtt can ameliorate the HD phenotype of R6/1 mice.
Keywords: brain, adeno-associated virus, gene therapy, viral gene transfer, RNA interference, short hairpin RNA, small interfering RNA, transcriptional dysregulation, polyglutamine, neuronal intranuclear inclusions
INTRODUCTION
Huntington’s disease (HD) is an inherited, autosomal dominant neurological disorder characterized by progressive development of motor abnormalities and cognitive impairments starting in midlife. HD is caused by the expression of an abnormally expanded polyglutamine domain (pQ) in the N-terminus of huntingtin (Htt), which is the product of the Htt gene [1]. The presence of a pQ domain in mutant Htt (mHtt) is thought to be responsible for the initiation of a cascade of pathological molecular changes that result in dysfunction [2,3] and progressive loss of the γ-amino butyric acid-producing medium spiny neurons of the caudate and putamen [4]. Although HD was first described over a century ago, and the gene for Htt was discovered in 1993, there is currently no effective therapy and the disease inevitably leads to death within 10 to 15 years of symptom onset [4].
Huntingtin is a large cytoplasmic protein that is localized to many subcellular compartments and is present at high concentrations in the brain and testis [5,6]. Recent evidence suggests that Httmay function as a scaffolding protein that is involved in several cellular processes, including vesicle transport, protein trafficking, and transcriptional regulation [2,7–10]. The specific role of Htt in each of these processes has not been fully elucidated. Ablation of the Htt gene in the mouse results in abnormal brain development, increased apoptotic cell death in the brain, and death by embryonic day 8 [11–13]. Conditional deletion of the Htt gene during the perinatal period causes abnormal brain development and neurodegeneration in a pattern reminiscent of that observed in late-stage HD [14]. These observations suggest that functional Htt is necessary for neurogenesis and development. Homozygous knock-in HD mice with mHtt in both copies of the mouse Hdh locus, however, are viable [15,16] indicating that mHtt complements normal Htt function early in development. It is unclear whether Htt is required for proper neuronal function in the adult brain, although there is recent evidence that mHtt loses some aspects of Htt function [2,10,17]. While it is clear that HD is a late-onset disorder, indicating that expanded pQ confers a toxic gain of function to mHtt, it is an open question whether impairing mHtt expression postnatally will be beneficial in modifying HD.
Along these lines, mice that carry a Htt transgene with an expanded CAG repeat under the control of a doxycycline-responsive promoter develop a HD-like phenotype that can be reversed following conditional suppression of the mHtt transgene [18]. This study showed that the expression of mHtt was necessary to maintain the progression of HD and that blockade of mHtt expression led to a reversal of HD-like symptoms including a clearance of neuronal intranuclear inclusions (NII) and behavioral improvements. This observation led to the hypothesis that suppression of mHtt activity could potentially ameliorate the HD phenotype in affected individuals. Several strategies for inhibiting mHtt expression or aggregation, including small molecules, are under development [19]. For example, intracellular single-chain antibodies that interfere with aggregation of mHtt have been designed [20,21]. These intrabodies have been engineered into lentiviral vectors and have been shown to affect striatal cell culture models of HD positively [22].
As an alternative strategy, the goal of this study was to achieve posttranscriptional gene silencing of mHtt in the striatum of the R6/1 HD transgenic mouse. This mouse model recapitulates many of the aspects of human HD, including the formation of NIIs, progressive loss of the steady-state mRNA levels of a subset of neuronal genes, and development of an HD-like neurological behavioral phenotype [23–28]. Our strategy was based on the use of recombinant adeno-associated virus serotype-5 (rAAV5) vectors to deliver anti-mHtt short-hairpin RNA molecules into the striatum of the R6/1 HD transgenic mouse.
It has been demonstrated that rAAV5 vectors can efficiently mediate the widespread and long-term expression of transgenes in striatal neurons in the rodent and nonhuman primate. It has also been demonstrated that in vitro and in vivo expression of short, hairpin-like double-stranded RNA (shRNA) molecules in neuronal cells results in sequence-specific silencing of targeted genes via the RNA interference (RNAi) pathway [29–33]. Indeed, a recent study showed that in vivo rAAV1-mediated cerebellar expression of a shRNA directed against mutant ataxin-1 in a mouse model of spinocerebellar ataxia type-1 (SCA-1) resulted in greater than 50% reduction of the target protein, rescue of Purkinje cells, and improvement in the abnormal motor behavior of SCA-1 mice [34]. In a landmark study, rAAV1 was used to deliver shRNAs in 4-week-old HD-N171-82Q mice and led to a reduction of NII formation and behavioral improvements 3 months after vector injection [30].
Using a similar strategy, we examined whether rAAV5-mediated delivery of anti-mutant Htt shRNAs could reduce levels of striatal mHtt and slow the progression of the HD-like behavioral phenotype in the aggressive R6/1 mouse model of HD. We found that long-term in vivo expression of two different rAAV5-shRNA vectors led to significant reduction in striatal mHtt mRNA and protein levels. This reduction was concomitant with a decrease in both the size and the number of NIIs. Additionally, the suppression of striatal mHtt expression by one shRNA (siHUNT-1) was associated with a trend toward normalization of the steady-state mRNA levels of preproenkephalin and dopamine-and cAMP-responsive phosphoprotein 32 kDa, which progressively decline in both mouse HD models and human HD. Finally, long-term bilateral expression of siHUNT-1 in the striatum resulted in a mild behavioral improvement of R6/1 mice.
RESULTS
In Vitro Activity of Anti-huntingtin Short-Hairpin RNAs
R6/1 transgenic HD mice express exon 1 of human HD with ≈ 115 CAG repeats [24]. We designed shRNAs (Fig. 1B) that could target specific sequences of exon 1 of human HD, but did not have significant sequence similarity to the endogenous mouse Htt mRNA. We generated rAAV5-based DNA constructs expressing two anti-mHtt shRNA molecules (Figs. 1A and 1B). We tested the mHtt silencing activity of these constructs in vitro by transiently cotransfecting each construct with a eukaryotic expression vector containing the R6/1 Hdh transgene (mHtt) into HEK293 cells. We performed transfections using a 1:4 and a 1:8 target vector-to-shRNA vector ratio. Forty-eight hours posttransfection, we analyzed mHtt mRNA and protein expression levels. Northern blot analysis of total RNA obtained from HEK293 cells cotransfected with a shRNA vector targeting nucleotides in the 5′ UTR of the mHtt mRNA (siHUNT-1) or a shRNA vector targeting a region immediately upstream of the CAG repeat domain (siHUNT-2) resulted in greater than 75% (P < 0.001) reduction of mHtt mRNA compared to samples from cells that expressed mHtt and a GFP-only vector (Fig. 1C). Increasing the shRNA-to-CMV-R6/1 vector ratio did not further lower steady-state levels of mHtt, suggesting that the shRNA-dependent mHtt reduction observed in HEK293 cells was maximal for these vectors. Cotransfection with a control shRNA vector targeted to the dog rhodopsin mRNA (siRho-1) did not affect the levels of mHtt mRNA (Fig. 1C), demonstrating that siHUNT-1 and siHUNT-2 exerted a sequence-specific effect on mHtt mRNA.
FIG. 1. rAAV-shRNA constructs mediate the silencing of mHtt in vitro.
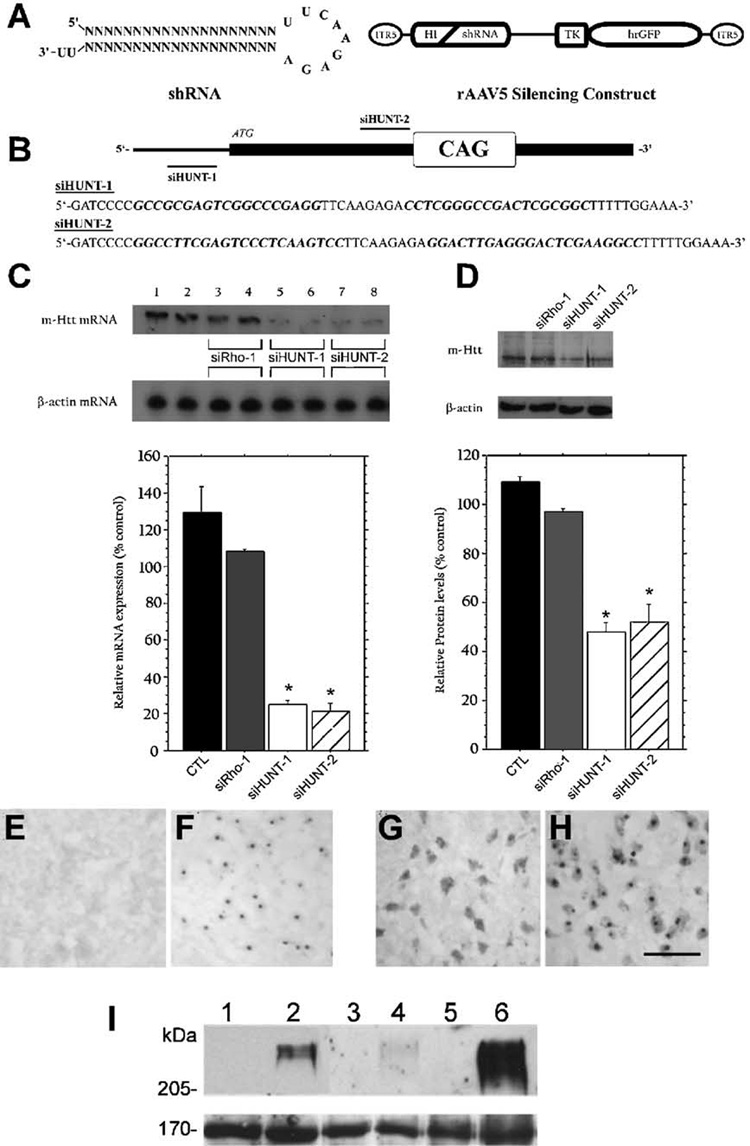
(A) The human H1 RNA promoter unit was used to drive the intracellular expression of shRNAs directed against the R6/1 mHtt transgene mRNA from within the context of rAAV serotype-5 vectors. Expression of hrGFP was used to positively identify transduced cells. (B) Schematic representation of the 5′ end of the mRNA encoded by mHtt and the regions targeted by siHUNT-1 and siHUNT-2 shRNAs. (C) Northern blot analysis of mHtt mRNA obtained from CMV-R6/1-transfected (lanes 1 and 2) and CMV-R6/1 and shRNA-cotransfected HEK293 cells (lanes 3–8). Coexpression of CMV-R6/1 with either siHUNT-1 or siHUNT-2 shRNA, at either a 1:4 (lanes 5 and 7) or a 1:8 (lanes 6 and 8) target-to-shRNA ratio, resulted in significantly reduced levels of mHtt mRNA (1.1-kb band) compared to cotransfection with the control shRNA, siRho-1 (lanes 3 and 4). Comparisons of the average optical density signal from three independent experiments performed with a 1:8 target-to-shRNA ratio showed that expression of siHUNT-1 and siHUNT-2 reduced mHtt mRNA (normalized to β-actin mRNA) by ~75% compared to siRho-1 (*P < 0.001). (D) Western blot analysis of mHtt protein (~70 kDa) in total protein isolated from transfected HEK293 cells using a human-specific anti-Htt antibody (Hum-1). Analysis of the average optical density signal of mHtt normalized to h-actin revealed that mHtt was reduced by intracellular expression of both siHUNT-1 (N60% reduction, *P < 0.0001) and siHUNT-2 (>55% reduction) compared to cells cotransfected with siRho-1. Error bars represent + standard error of the mean (+SEM). (E–I) Hum-1 antibody (G, H), like antiubiquitin antibody (E, F), detects NIIs in R6/1 (F, H) but not wild-type (E, G) mice. Striatal sections from 15-week-old mice are shown. Hum-1 antibody crossreacts with cytoplasmic proteins in striatal neurons of both wild-type (G) and R6/1 (H) mice. Scale bar, 50 µm. (I) Western blot analysis of total (lanes 1 and 2), cytoplasmic (lanes 3 and 4), and nuclear (lanes 5 and 6) protein derived from 15-week-old wild-type (lanes 1, 3, and 5) and R6/1 mice (lanes 2, 4, and 6) using Hum-1 demonstrated that the antibody detects a high-molecular-weight aggregate, which is enriched in the nucleus, in R6/1 transgenic mice. The bottom blot shows a 170-kDa cross-reacting band that is present in all samples and was used as loading control.
To determine whether siHUNT-1- and siHUNT-2-dependent reduction in mHtt mRNA was associated with decreased levels of mHtt protein in transfected HEK293 cells, we generated a rabbit polyclonal antibody against a specific peptide in the amino-terminus of human Htt (Hum-1) and used this antibody in Western blot analysis. Hum-1 detected NIIs and a high-molecular-weight band of greater than 250 kDa in R6/1 but not wild-type mice (Figs. 1E–1I). The ~250-kDa Hum-1-immunoreactive band was enriched in nuclear fractions of striatal tissue isolated from R6/1 mice (Figs. 1E–1H). Immunoblot analysis of total protein isolated from HEK293 cells transfected with CMVR6/1 or CMV-R6/1 and anti-mHtt shRNA constructs using Hum-1 showed that the reduction in mRNA levels was associated with a significant decline in the levels of mHtt protein (Fig. 1D). Coexpression of the mHtt vector with siHUNT-1 shRNA reduced the levels of mHtt protein to ≈ 40% (P < 0.001) of that observed in cells cotransfected with a GFP-only vector. We also observed ≈ 55%(P < 0.001) reduction in the levels of mHtt protein in samples from cells transfected with siHUNT-2 shRNA. mHtt protein levels were unaffected by the expression of siRho-1 shRNA molecules (Fig. 1D). These in vitro results demonstrate that intracellular expression of both siHUNT-1 and siHUNT-2 results in the specific and efficient knockdown of mHtt mRNA and protein levels in cultured HEK293 cells.
Long-Term Striatal Expression of rAAV5-shRNAs in the R6/1 Mouse
We next investigated whether rAAV5-mediated long-term striatal expression of siHUNT-1 or siHUNT-2 shRNA could induce RNA interference of the mHtt transgene in the R6/1 transgenic HD mouse. To test the in vivo activity of both shRNAs, we divided 6- to 8-week-old R6/1 mice and wild-type littermates into four groups and injected them unilaterally at two different sites in the right striatum (Fig. 2A). Each intrastriatal injection delivered 2 µl of high-titer rAAV5-siHUNT-1 (R6/1 n = 12, wild type n = 6), rAAV5-siHUNT-2 (R6/1 n = 13, wild type n = 5), rAAV5-siRho-1 (R6/1 n = 4), or rAAV5-GFP (R6/1 n = 5, wild type n = 5) viral vectors. Ten weeks postsurgery, we obtained fresh-frozen coronal brain sections and determined viral transduction, mHtt mRNA levels, and protein expression levels relative to the left untreated hemisphere.
FIG. 2. Experimental design.

(A) R6/1 and littermate control animals were injected unilaterally with rAAV5-siHUNT-1, -siHUNT-2, -siRho-1, or -GFP viral vectors (annotated below the line). (B) Experimental design for bilateral injections was identical to that shown above. Behavioral tests were performed weekly between the time points indicated by the arrows above and below the timeline.
Injection of rAAV5-GFP, rAAV5-siRho-1, and anti-mHtt rAAV5 shRNA vectors resulted in widespread and intense neuronal transduction as was previously observed in the rat striatum using pseudotyped rAAV5 vectors [35]. In situ hybridization (ISH) analysis performed on coronal sections against the rAAV5-encoded hrGFP mRNA revealed a nonuniform but widespread rAAV5 transduction (Fig. 3A). hrGFP mRNA was efficiently expressed along the dorsal–ventral and rostral–caudal axis of the striatum. A strong hybridization signal was detected on either side of the corpus callosum but not in the white matter tracts (Fig. 3A), suggesting that the majority of hrGFP signal was concentrated in neuronal cell bodies [35–37]. hrGFP protein expression was intense and paralleled the widespread pattern of rAAV5 transduction detected by ISH analysis (Fig. 3B). Native hrGFP epifluorescence was observed in all sections that contained rAAV5-transduced striatal tissue, such as the one shown in Fig. 3B. Long-term striatal expression of control rAAV5 vectors (rAAV5-siRho-1, rAAV5-GFP) in the R6/1 mouse was not associated with any abnormal changes in cellular morphology or astrocyte activation as determined by cresyl violet and GFAP staining (data not shown). These observations demonstrated that the striatum of the R6/1 mouse can tolerate strong, widespread, and long-term rAAV5-mediated expression of hrGFP and shRNA molecules.
FIG. 3. Long-term in vivo striatal expression of rAAV5-siHUNT1 and rAAV-siHUNT2 decreases mHtt trans-gene mRNA expression.
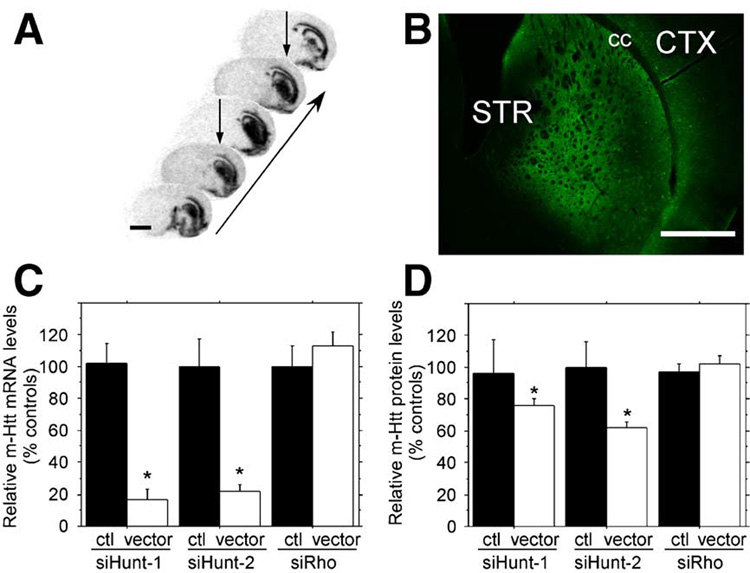
(A) In situ hybridization with a probe against the virally encoded hrGFP mRNA. Coronal sections were obtained 10 weeks postsurgery. The images are arranged on a rostral–caudal axis to illustrate the extent of the transduced area as indicated by the arrow. Two separate injections were placed into the same hemisphere (arrows). These sections were obtained from one animal and are representative of all other study animals. Scale bar, 2 mm. (B) Photomicrograph of native hrGFP protein expression from the striatum of a representative injected animal demonstrates that the vector-derived mRNA shown in (A) is transcribed into protein. STR, striatum; cc, corpus callosum; and CTX, cortex. Scale bar, 1 mm. (C) Real-time quantitative PCR analysis of mHtt mRNA knockdown. Total RNA was extracted from the striata of R6/1 mice. The levels of striatal mHtt mRNA on the uninjected (black bars) striata and striata injected with rAAV5 vectors (white bars) are shown. rAAV5-siHUNT-1, rAAV5-siHUNT-2, and rAAV5 siRho were measured, normalized to the levels of HPRT mRNA, and reported as a percentage of control. Expression of rAAV5-siHUNT-1 resulted in a significant 75% reduction of mHtt mRNA expression (F[1,11] = 40.0, *P < 0.001). Striatal rAAV5-siHUNT-2 treatment resulted in significant 78% reduction of striatal mHtt mRNA expression (F[1.8] = 19.1, *P = 0.002). In contrast, striatal injection of the control shRNA rAAV5-siRho did not reduce striatal mHtt mRNA levels (F[1,8] = 0.71, P > 0.40). (D) Similar to striatal mRNA analysis in (C), total protein was obtained from the injected and untreated control striata of R6/1 mice. Similar to the mRNA levels, there was a significant rAAV5-sitHUNT-1-mediated 24.5% reduction in the levels of a Hum-1-immunoreactive high-molecular-weight band (simple main effects, F[1,18] = 5.2, *P < 0.04). The rAAV5-siHUNT-2-treated group displayed a significant 38% reduction of striatal mHtt (simple main effects, F[1,18] = 8.4, *P = 0.01), while the rAAV-siRho control groups did not have significant reductions of striatal mHtt (simple main effects, F[1,18] = 0.50, P > 0.40) compared to R6/1 control striata. Error bars represent +SEM.
To evaluate the silencing activity of rAAV5-siHUNT-1 and rAAV5-siHUNT-2 in vivo, we extracted total cellular RNA from rAAV5-shRNA-transduced striatal regions and subjected the RNA to reverse-transcription reactions optimized to convert mHtt transgene mRNA to cDNA. We determined the levels of mHtt mRNA using real-time quantitative PCR. Baseline levels of striatal mHtt mRNA were established by analyzing total RNA from striatal tissue contralateral to the rAAV5-shRNA injection. mHtt mRNA levels from control and rAAV5-shRNA-injected striatum were normalized to hypoxanthine phosphoribosyl transferase (HPRT) mRNA levels and subjected to one-way analysis of variance (ANOVA). Long-term expression of rAAV5-siHUNT-1 or rAAV5-siHUNT-2 in the striatum of the R6/1 mouse resulted in highly significant reductions in the levels of mHtt mRNA (Fig. 3C), demonstrating that intrastriatal delivery of rAAV5 anti-mHtt shRNAs in the R6/1 mouse can induce efficient knockdown of mHtt mRNA levels. Moreover, this reduction was specific to the expression of rAAV5-siHUNT-1 or rAAV5-siHUNT-2 since mHtt mRNA levels were unchanged following the expression of rAAV5-siRho-1 (Fig. 3C).
We measured the relative intensity of the high-molecular-weight Hum-1-immunoreactive band in Western blots to compare the levels of mHtt protein in transduced and control R6/1 striata. We normalized the levels of mHtt protein to a cross-reactive band that was recognized by the Hum-1 antibody and was present at constant levels in protein extracts isolated from R6/1 and wild-type mice. rAAV5-siHUNT-1 and rAAV5-siHUNT-2 reduced striatal mHtt protein levels but not to the same degree observed with striatal mHtt mRNA levels. Thus, striatal rAAV5-siHUNT-1 expression reduced aggregated striatal mHtt protein approximately 25% compared to aggregated mHtt from control striata or from rAAV5-siRho-treated mice (Fig. 3D). In addition, rAAV5-siHUNT-2 reduced aggregated mHtt protein 38% relative to untreated control striata or rAAV5-siRho injected mice (Fig. 3D). Expression of the control shRNA, siRho, had no effect on striatal mHtt expression.
There is a discrepancy between the magnitude of siHUNT-1- and si-HUNT-2-mediated mHtt mRNA reduction and the magnitude of reduction of mHtt protein from the same striata. One potential reason for this discrepancy between mRNA and protein is the differential kinetics of turnover of mRNA versus protein. Furthermore, for protein measurement we assayed a high-molecular-weight species that was resolved on our gels and not total mHtt protein. In summary, long-term intrastriatal expression of rAAV5-siHUNT-1 and -siHUNT-2 shRNAs significantly reduced striatal mHtt mRNA levels and also resulted in diminished expression of striatal mHtt aggregates.
Reduction in the Size and Amount of NIIs is Observed in R6/1 Mice Treated with rAAV5-siHUNT-1 or rAAV5-siHUNT-2
One of the pathological markers of disease progression in mouse models of pQ disease such as HD is the presence of NIIs. Studies have shown that suppressing the expression of the mutant pQ-containing protein in neurons results in the clearance of NIIs [18,34]. We examined whether shRNA treatment altered the number or size of NIIs in R6/1 mice. We reasoned that using an anti-Htt antibody (Hum-1) instead of an anti-ubiquitin antibody would allow us to correlate the levels of mHtt directly with the presence or absence of NIIs. Immuno-histochemical analysis of coronal brain sections from untreated R6/1 mice subjected to Hum-1 and anti-ubiquitin immunohistochemistry revealed a similar pattern of immunoreactivity between both antibodies with respect to the size and distribution of NIIs in the R6/1 mouse brain (Figs. 1E–1H). Histological analysis of sections obtained from R6/1 mice of various ages showed that Hum-1-positive NIIs become visible in the light microscope at about 8 weeks after birth (data not shown), in agreement with the time that ubiquitinpositive NIIs become visible in the R6/1 mouse [28]. Hum-1- (data not shown) and anti-ubiquitin- [28] immunoreactive NIIs increase in size and staining intensity during the progression of HD, suggesting that protein deposition in NIIs is cumulative.
We analyzed Hum-1-immunostained coronal sections from unilaterally injected R6/1 mice and compared the relative area and intensity of NIIs present in four brain regions including the left primary motor cortex, right primary motor cortex, left medial striatum, and right medial striatum (Fig. 4A). Three of these regions, left primary motor cortex, right primary motor cortex, and left medial striatum, corresponded to nontransduced control tissue, while the right medial striatum exhibited strong rAAV5-mediated transduction (Fig. 3A, 3B, and Fig. 4A). Comparison of Hum-1 staining in the left and right primary motor cortex revealed no differences in the area or intensity of NIIs in any of the R6/1 mice that were untreated or treated with rAAV5-siHUNT-1 (Figs. 4D and E), siHUNT-2, or siRho-1 (data not shown). Areas shown in Figs. 4D and 4E were taken from the same R6/1 mouse section and are representative of all other rAAV5-siHUNT-1, -siHUNT-2, -siRho-1, and untreated R6/1 mice. Hum-1-immunoreactive NIIs in the left (Figs. 4F, 4J, and 4N) and right (Figs. 4G, 4K, and 4O) medial striatum were smaller and had less intense staining than those present in the cortical regions of the same R6/1 mouse (Figs. 4D vs 4F). A comparison of the nontransduced left and transduced right medial striatum regions showed a marked reduction in the intensity of Hum-1 NII staining in the rAAV5-siHUNT-1- (Fig. 4G) and rAAV5-siHUNT-2- (Fig. 4K) transduced right medial striatum compared to the left medial striatum (Figs. 4F and 4J, respectively) of the same animal. The reduction in Hum-1 NII staining intensity was more pronounced in sections obtained from rAAV5-siHUNT-2-treated compared to siHUNT-1-treated R6/1 mice. Sections from R6/1 mice treated with either rAAV5-GFP (data not shown) or rAAV5-siRho-1 (Figs. 4N and 4O) showed no significant difference in the pattern of Hum-1 NII staining between the left and the right medial striatum regions (Figs. 4N vs 4O). The observation of rAAV5-siHUNT1- and rAAV5-siHUNT-2-mediated reduction of striatal Hum-1 NII staining further corroborates the immunoblot data indicating reduced striatal mHtt protein in vector-treated striata.
FIG. 4. Analysis of mHtt protein aggregates after in vivo expression of rAAV5-shRNAs.
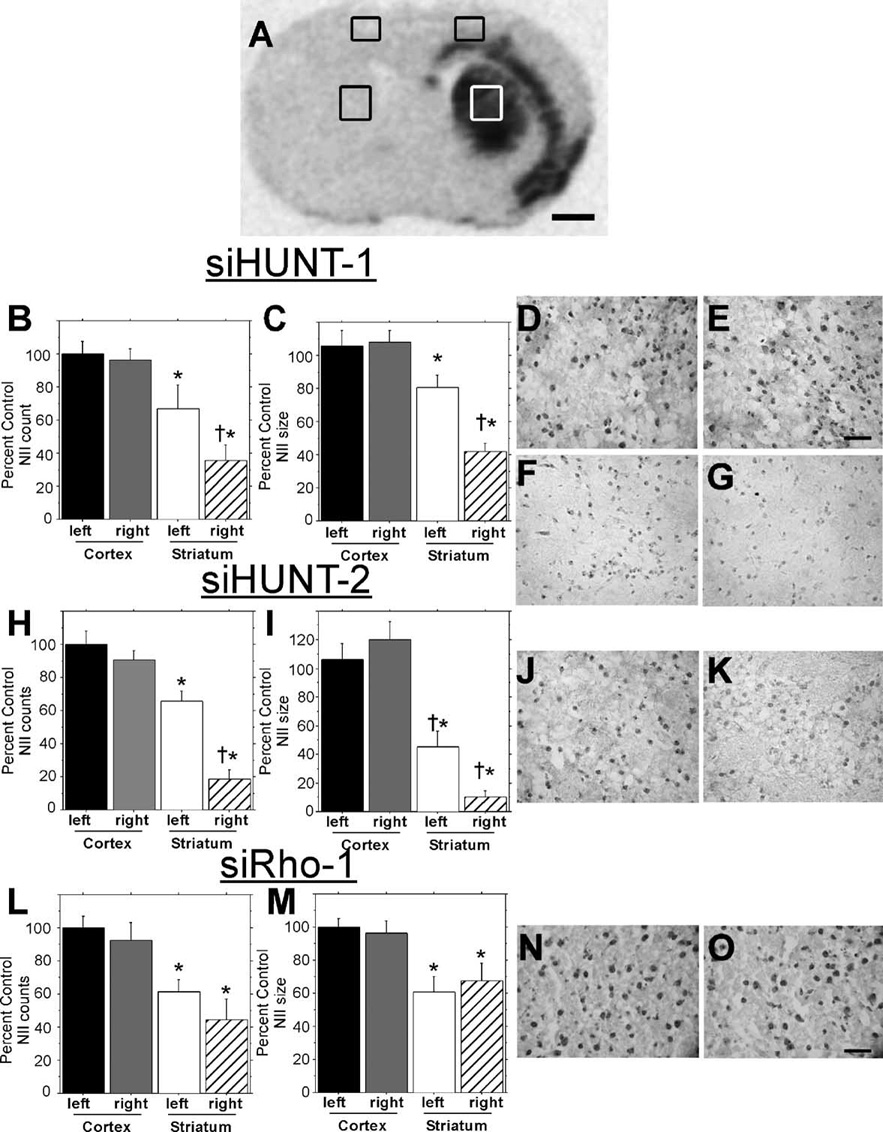
(A) Coronal section from rAAV5-siHunt-1-injected R6/1 mouse processed for in situ hybridization with a probe against vector-specific sequences (as shown in Fig. 3A) showing the areas of Hum-1 staining used for analysis of NII number (B, H, L) and size (C, I, M). Analysis of NIIs in (B–G) siHUNT-1-, (H–K) siHUNT-2-, and (L–O) siRho-1-treated R6/1 mice. Number and size of NIIs (+SEM) are expressed as percentage of left primary motor cortex; the cortex in either hemisphere was not transduced by rAAV and served as a region control (*P < 0.05 compared to left motor cortex; †P < 0.05 compared to left striatum). Photomicrographs of (D) untreated left and (E) siHUNT-1-treated right cortical Hum-1-stained coronal sections from the area shown in the box in (A). Cortical NII size and distribution were similar in siHUNT-2- and siRho-1-treated animals (data not shown). Scale bar, 50 µm, applies to D and E. Representative photomicrographs from the control left striatal area depicted in (A) from (F) rAAV5-siHUNT-1-, (J) siHUNT-2-, and (N) siRho-1-treated mice. Representative photomicrographs from the right striatum of (G) rAAV5-siHUNT-1-, (K) siHUNT-2-, and (O) siRho-1- treated mice. It should be noted that adjacent untransduced striatal areas had Hum-1-positive NII frequency and size that were indistinguishable from those seen in the opposite control striatum. Scale bar, 50 µm, applies to (F, G, J, K, N, and O).
To quantify the relative area and amount of NIIs present in shRNA-treated striata, we analyzed the same relative four regions shown in Fig. 4A in siHUNT-1-treated (n = 11), siHUNT-2-treated (n = 6), and siRho-1-treated (n = 4) R6/1 mice. The mean relative area (µm2) and number of particle (NII) counts within a specified area range were calculated for each of the four regions in all of the unilaterally injected R6/1 mice using Image J software and reported as percentage of the nontransduced left primary motor cortex (Figs. 4B, 4C, 4H, 4I, 4L, and 4M). Cortical NII numbers were unaffected by striatal injection of rAAV5-siHUNT1 (Fisher’s paired least significant difference (PLSD) post hoc test, P = 0.8) and the frequency of NIIs in cortex was greater than in the striatum of the R6/1 mouse (F[1,20] = 22.0, *P < 0.001). Treatment with rAAV5-siHUNT-1 significantly reduced the frequency of striatal NIIs by 31% compared to the control uninjected striatum (Fisher’s PLSD post hoc test, †P = 0.04). Similar to NII numbers, NIIs were smaller on average in the striatum, regardless of treatment, compared to the NII size in cortex (F[1,20] = 38.4,*P < 0.001). rAAV5-mediated siHUNT-1 expression reduced the size of the remaining NIIs by 39% compared to the untreated control striatum (post-hoc, †P = 0.0013). The frequency of NIIs in the cortex of siHUNT-2-treated R6/1 mice was greater than the frequency measured in the striatum (F[1,20] = 68.4, *P < 0.0001). rAAV5-mediated siHUNT-2 expression further reduced the number of striatal NIIs by 47% compared to the left striatum (Fisher’s PLSD post hoc test, †P < 0.0001). Hum-1-positive cortical NIIs were significantly larger than striatal NIIs in all siHUNT-2-treated mice (F[1,20] = 71.1, *P < 0.0001). Striatal siHUNT-2 expression further reduced striatal NII size by 35% compared to the untreated striatum (Fisher’s PLSD post hoc test, †P < 0.03). As with the other vector-treated groups, cortical NIIs were more frequent (F[1,12] = 20.5, *P = 0.0007) and larger (F[1,12] = 16.8, *P < 0.002) than striatal NIIs in siRho-1-treated mice. However, there was no difference in NII number (Fisher’s post hoc test, P > 0.2) or size (Fisher’s PLSD post hoc test, P > 0.5) between siRho-1-treated and untreated striata (Figs. 4L and 4M). Our results show that long-term expression of rAAV5-siHUNT-1 or rAAV5-siHUNT-2, but not control siRho-1, in the R6/1 striatum led to a significant reduction in the area and frequency of striatal NIIs present within the region of viral transduction.
Reduced Levels of Striatal mHtt Affect Levels of Striatal-Specific Transcripts
Expression of exon 1 of mHtt leads to a decrease in the level of a subset of striatal-specific mRNAs of the R6/1 mouse [38–42]. It has been demonstrated that some of these transcripts are also reduced in HD patients. At the very least, evaluation of these striatal-specific transcripts can serve as an important metric of disease progression in R6/1 mice. To examine the effects that silencing striatal mHtt expression might have on this molecular phenotype, we performed in situ hybridization analysis of transcripts on coronal sections obtained from unilaterally injected wild-type and R6/1 mice. Sections were hybridized with oligonucleotide probes against preproenkephalin (ppEnk), dopamine- and cAMP-responsive phosphoprotein, 32 kDa (DARPP-32), phosphodiesterase 10A (PDE10A), phosphodiesterase 1B (PDE1B), nerve growth factor-inducible A (NGFi-A), and the dopamine type 2 (D2) receptor (Table 1).
TABLE 1.
In situ hybridization probes
| Oligonucleotide probe used to detect | Sequence |
|---|---|
| siHUNT-1 and-2 | 5′-CTTACACCCACTCGTGCAGGCTGCCCAGGG-3′ |
| pTRUF11 | 5′-GGCGGAGCGGGGCACGGGGCGAAGGCAGCG-3′ |
| DARPP-32 | 5′-TCCACTTGGTCCTCAGAGTTTCCATCTCTC-3′ |
| ppEnk | 5′-ATCTGCATCCTTCTTCATGAAACCGCCATACCTCTTGGCAAGGATCTC-3′ |
| NGFi-A | 5′-CCGTTGCTCAGCAGCATCATCTCCTCCAGTTTGGGGTAGTTGTCC-3′ |
| D2 receptor | 5′-GGCAGGGTTGGCAATGATACACTCATTCTGGTCTGTATT-3′ |
| PDE10A | 5′-GACCAATGTCAAAGTGGAATAGCTCGATGTCCCGGC-3′ |
| PDE1B | 5′-CATGTAGCGCAGCAGAGACCGTAGCTTAATCCACA-3′ |
| β-Actin | 5′-GCCGATCCACACGGAGTACTTGCGCTCAGGAGGAGCAATGATCT-3′ |
Densitometric analysis of rAAV5-siHUNT-1-injected R6/1 mice showed an increase in the mean levels of striatal ppEnk (24%) and DARPP-32 (16%) mRNA in the transduced striatum (Figs. 5A and 5B) compared to the contralateral uninjected side. There was no significant side-to-side difference in the levels of PDE10A, PDE1B, NGFi-A, or D2 receptor mRNAs in R6/1 mice treated with rAAV5-siHUNT-1 (data not shown).
FIG. 5. ISH analysis of ppEnk and DARPP-32 transcripts.
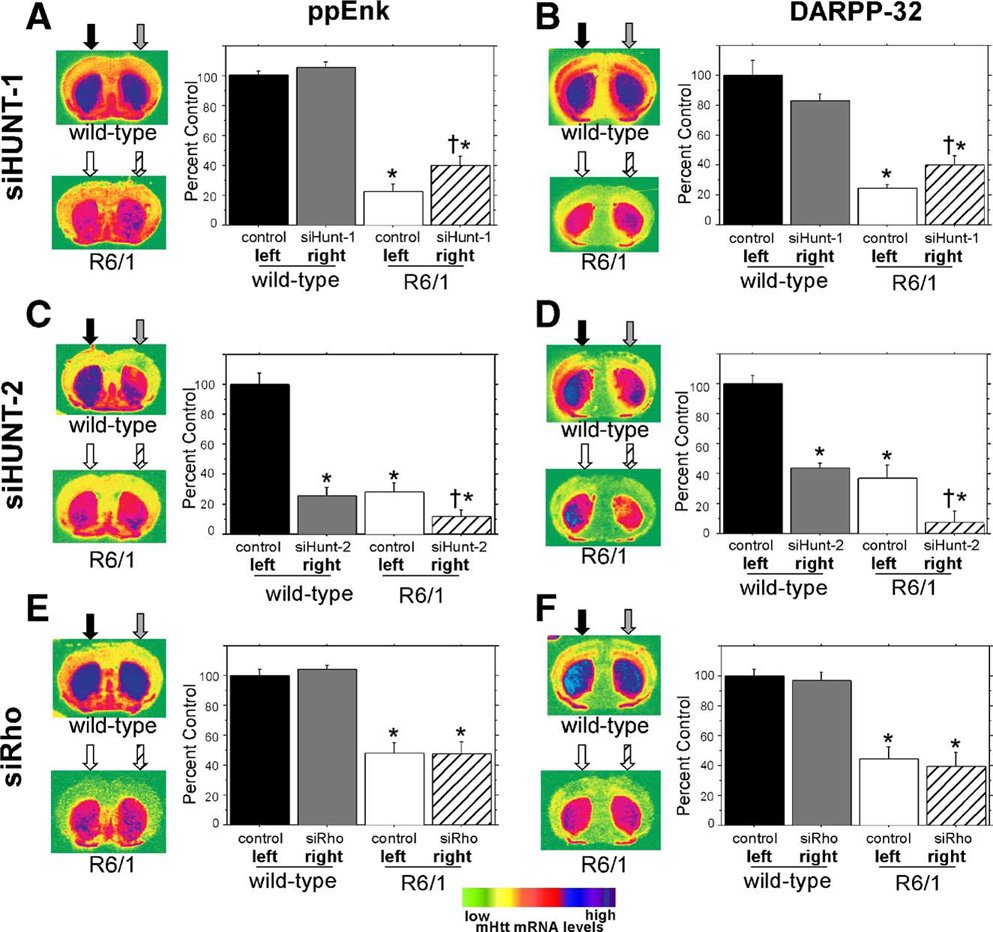
Coronal sections shown on the left of (A), (C), and (E) were hybridized to a ppEnk-specific oligonucleotide probe (Table 1), while sections in (B), (D) and (F) were hybridized to a DARPP-32-specific oligonucleotide probe (Table 1). The arrows above the coronal sections show the approximate vector injection site and the fill patterns of the arrows correspond to the fill patterns of the histograms to show which treatment was received in each hemisphere. To better demonstrate the anatomical pattern of striatal transcripts the coronal sections were false colored. The scale is shown at the bottom. (A) Evaluation of long-term intrastriatal rAAV-siHUNT-1 expression on striatal ppEnk levels in R6/1 mice and wild-type littermate controls. Quantitative analysis of normalized ppENK levels confirm that ppENK levels are reduced in 22-week-old R6/1 mice (F[1,66] = 99.5, *P < 0.0001). rAAV-siHUNT-1 injection into wild-type control striata does not lead to any alteration in striatal ppENK levels compared to untreated control left striata (Fisher’s PLSD post hoc test, P > 0.7). In contrast, in R6/1 mice, striatal rAAV5-siHUNT-1 treatment led to a mild 24% increase in striatal ppEnk levels compared to the ppEnk levels observed in the control striatum (Fisher’s PLSD post hoc test, †P = 0.01). (B) Evaluation of long-term intrastriatal rAAV-siHUNT-1 injection on steady-state striatal DARPP-32 mRNA levels. Similar to ppENK mRNA levels, DARPP-32 mRNA was reduced in R6/1 animals compared to littermate controls (F[1,66] = 114.1, *P > 0.0001). rAAV5-siHUNT-1 injection did not affect striatal DARPP-32 levels in wild-type controls (Fisher’s PLSD post hoc test, *P > 0.1). Striatal rAAV5-siHUNT-1 treatment resulted in a significant 16% increase in DARPP-32 mRNA levels (Fisher’s PLSD post hoc, †P < 0.03) in R6/1 mice compared to DARPP-32 mRNA levels in the untreated striatum. (C) Analysis of the effect of intrastriatal injection of rAAV-siHUNT-2 on striatal ppEnk transcript levels. In stark contrast to the pattern seen with rAAV5-siHUNT-1 injection (A), rAAV5-siHUNT-2 long-term expression led to a 75% reduction of striatal ppENK levels in the injected striatum of wild-type littermate controls (Fisher’s PLSD post hoc test, *P < 0.0001). Likewise, rAAV5-siHUNT-2 treatment also resulted in a further 16% reduction in striatal ppENK levels in R6/1 mice (Fisher’s PLSD post hoc test, †P < 0.05). (D) Analysis of the effect of intrastriatal injection of rAAV-siHUNT-2 on striatal DARPP-32 mRNA levels. Similar to the pattern seen in response to rAAV5-siHUNT-2 expression striatal ppEnk levels (C), rAAV5-siHUNT-2 long-term expression led to a 44% reduction of striatal DARPP-32 mRNA levels in the injected striatum of wild-type littermate controls (Fisher’s PLSD post hoc test, *P = 0.0005). In addition, rAAV5-siHUNT-2 treatment resulted in a further 70% reduction in striatal DARPP-32 mRNA levels in R6/1 mice (Fisher’s PLSD post hoc test, †P < 0.01). This further reduction in the treated striatum of the R6/1 mice is particularly apparent in the right side of the coronal section shown to the left of the histogram. (E) Evaluation of the effect of the long-term expression of the control shRNA, siRho, in the striatum of wild-type littermate controls and R6/1 mice on striatal ppEnk levels. rAAV5-siRho injections had no effect on striatal ppEnk transcript levels in either wild-type controls (Fisher’s PLSD post hoc test, P > 0.9) or R6/1 mice (Fisher’s PLSD post hoc test, P > 0.6). (F) Evaluation of the effect of the long-term expression of the control shRNA, siRho, in the striatum of wild-type littermate controls and R6/1 mice on striatal DARPP-32 mRNA levels. rAAV5-siRho injections had no effect on striatal DARPP-32 mRNA levels in either wild-type controls (Fisher’s PLSD post hoc test, P > 0.7) or R6/1 mice (Fisher’s PLSD post hoc test, P > 0.6). Error bars are +SEM.
In contrast, expression of rAAV5-siHUNT-2 in R6/1 mice resulted in a marked decrease in the mean levels of both ppEnk (16%) and DARPP-32 (30%) mRNAs compared to the uninjected left side (Figs. 5C and 5D). More importantly, this decrease was also evident in unilaterally injected wild-type mice for both ppEnk (75%) and DARPP-32 (56%) mRNAs (Figs. 5C and 5D). This pronounced reduction in mRNA levels was also observed in all other transcripts tested (not shown), except for NGFi-A.
Based on the comparable reduction in transcripts in wild-type and R6/1 mice, we attributed the reduction in the steady-state levels of these striatal-specific transcripts to a siHUNT-2 shRNA-mediated “off-targeting” effect. Further documentation of this off-targeting finding is the subject of a separate study (E. Rodriguez et al., manuscript in preparation).
Analysis of R6/1 mice injected unilaterally with the control rAAV5-siRho-1 shRNA revealed no significant differences in the levels of ppEnk, DARPP-32, PDE10A, PDE1B, NGFi-A, or D2 receptor mRNA between the left uninjected and the right injected striatum (Figs. 5E and 5F). Additionally, there was no side-to-side difference in the steady-state levels of any of the transcripts analyzed in the striatum of wild-type mice injected unilaterally with rAAV5-siHUNT-1 (Figs. 5A and 5B). We conclude that rAAV5-siHUNT-1-mediated reduction of mHtt levels in the striatum of R6/1 mice resulted in a small but significant increase in the steady-state levels of striatal ppEnk and DARPP-32 mRNAs relative to the levels observed in the untreated contralateral striatum and that, despite its apparent efficacy at reducing mHtt mRNA and protein levels, rAAV5-siHUNT-2 produced deleterious off-target effects.
Long-Term Bilateral Striatal Expression of rAAV5-siHUNT-1 in the R6/1 Mouse is Associated with a Delay in the Clasping Phenotype
R6/1 mice display a progressive neurological phenotype that includes clasping of the hind limbs and dyskinesias [24,43,44]. Additionally, R6/1 mice fail to gain weight at a normal rate and show a progressive decrease in retention times in the rotarod compared to age-matched wild-type littermate mice. To examine whether lowering the striatal levels of mHtt would prevent the progression of these phenotypes, we injected 6- to 8-week-old R6/1 and wild-type mice bilaterally at two different striatal sites (four injections total) with rAAV5-siHUNT-1 or rAAV5-GFP control (Fig. 2B). We initially assessed the progression of the HD-like phenotype in the R6/1 mice by recording weights weekly beginning at 1 week postsurgery.
As previously reported, we observed that R6/1 mice did not increase total body weight at the same rate over time compared to age-matched wild-type littermate mice (Figs. 6A versus 6B). Examination of the mean weekly weights of uninjected wild-type mice and wild-type mice injected with rAAV5-GFP or rAAV5-siHUNT-1 revealed that long-term expression of neither viral vector led to a significant change in the rate of weight gain over the experimental period (Fig. 6A). Similarly, long-term expression of neither rAAV5-GFP nor rAAV5-siHUNT-1 in R6/1 mice resulted in a significant change in rate of weight gain compared to untreated R6/1 (Fig. 6B). In addition, we examined rotarod retention times of both wild-type and R6/1 mice 16 weeks postsurgery (22 weeks of age, Fig. 6C). As reported previously [25], R6/1 mice displayed a significant deficit in their ability to remain on the rotarod compared to wild-type controls [F(1,22) = 16.7, P = 0.0005]. However, there was no effect of rAAV5-siHUNT-1 injection on rotarod performance [F(2,22) = 3.1, P > 0.05].
FIG. 6. Bilateral long-term striatal expression of rAAV5-siHUNT-1 delays the clasping phenotype of R6/1 mice.
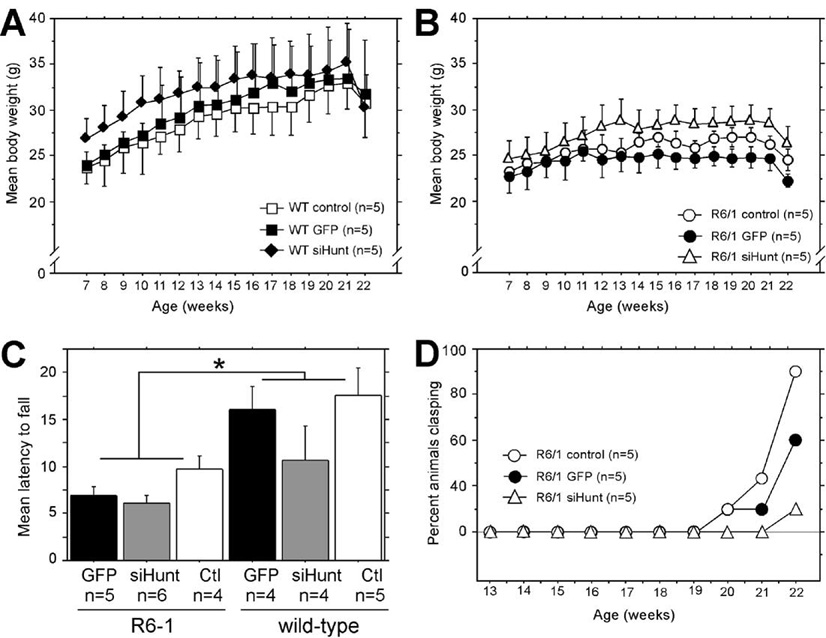
(A and B) Animal weight was recorded weekly starting at 1 week postsurgery (Fig. 2B). (A) The mean (±SEM) weight for wild-type littermate controls is shown. There was no effect of rAAV-siHUNT-1 treatment on weight regardless of genetic background or vector treatment, including rAAV5-GFP (rAAV repeated-measures ANOVA, F[2,22] = 0.63, P > 0.5). (B) The mean (±SEM) weight for R6/1 mice is shown. In general, R6/1 mice weighed less than age-matched littermate controls (n = 5) over the period of the experiment (F[1,22] = 4.7, P = 0.04 A vs B). (C) There was a significant difference in rotarod performance (mean latency to fall (s) +SEM) between R6/1 and wild-type mice but no difference among groups of R6/1 or wild-type mice treated with rAAV5-GFP or rAAV5-siHUNT-1 or untreated controls (*P = 0.0005). (D) Tail-suspension tests were performed weekly beginning at 6 weeks postsurgery (corresponding to 13 weeks of age) on age-matched control R6/1 mice (n = 5) and R6/1 mice injected bilaterally with rAAV5-siHUNT-1 (n = 5) or rAAV5-GFP (n = 5). Data are presented as percentage of animals scoring positive on the test as a function of time.
We also analyzed the progression of the clasping phenotype in uninjected and rAAV5-GFP- or rAAV5-siHUNT-1-injected R6/1 mice by performing a weekly tail-suspension test beginning at 13 weeks of age. Uninjected and rAAV5-GFP-injected R6/1 mice began to display clasping of the hind limbs at 20 weeks of age (Fig. 6D). In contrast, R6/1 mice expressing rAAV5-siHUNT-1 did not show evidence of clasping until 22 weeks of age, when only 20% overtly displayed the phenotype, in comparison with up to 80% of uninjected and rAAV5-GFP-injected R6/1 mice, which overtly displayed the hind limb clasping phenotype at this same time point.
We conclude that 6- to 8-week-old mice expressing rAAV5-siHUNT-1 bilaterally in the striatum for up to 14 weeks did not display any overt toxicity associated with our gene transfer approach. Furthermore, long-term expression of siHUNT-1 anti-mHtt shRNA resulted in a mild behavioral improvement of the R6/1 phenotype with respect to clasping behavior but did not affect body weight abnormalities or muscle-dependent activity such as rotarod retention.
DISCUSSION
The expression of one expanded mHtt allele initiates a cascade of events that leads to the neuronal dysfunction and pathology observed in HD [1]. It has been hypothesized that ablating the expression of mHtt might protect against or even reverse the effects associated with pQ-expanded Htt protein [18,29,45]. In this study, we assessed the molecular and behavioral effects of long-term intrastriatal rAAV5-mediated expression of shRNAs designed to suppress the expression of the human mHtt transgene that is present in R6/1 mice [24]. Our results demonstrate that posttranscriptional gene silencing of striatal mHtt in adult R6/1 mice by one of two shRNAs tested, siHUNT-1, has a mild positive effect on the rapid cellular and behavioral pathological changes that occur in this model. Improvement occurred in only two striatal-specific transcripts and this amelioration was found only in the striatal subregion of rAAV5-mediated transduction. These data, therefore, suggest that greater transduction efficiency in the rapidly progressing R6/1 mouse model could improve the functional effects obtained here.
Intrastriatal injections of rAAV5 vectors resulted in widespread transduction of the mouse striatum especially compared to previously reported levels of transduction in mice [46,47] but were very comparable to rAAV1-mediated striatal transduction in HD-N171-82Q mice [30]. Equivalency between striatal transduction efficiency for rAAV1 and rAAV5 has been previously reported in rats [35].
Long-term expression of rAAV5-siHUNT-1 in the R6/1 mouse striatum resulted in mRNA levels that were reduced by 75% compared to controls, while reduction in protein aggregates ranged from 25 to 38%. These data compare favorably to the 55% reduction of striatal mHtt mRNA reported after rAAV1-delivered shRNA [30].
Aggregated, but not soluble, mHtt was detected using our Western blot protocol, which may have contributed to the discrepancy between the magnitude of reduction of mRNA and mHtt protein. Indeed, discrepancies between levels of mRNA reduction and protein levels has been reported previously for siRNA and ribozyme experiments [48–50]. In fact, even though we tested several published antibodies and a variety of extraction protocols, we were unable to detect measurable levels of soluble mHtt N-terminal fragments in total striatal protein samples from untreated adult R6/1 mice.
In agreement with the data obtained from Hum-1 immunoblots indicating reduced striatal mHtt in transduced regions, Hum-1 immunohistochemical analysis of sections from rAAV5-siHUNT-1-treated R6/1 mice revealed a marked decrease in the size and number of NIIs within the area of viral transduction. Thus, since aggregation of soluble mHtt fragments leads to the formation of inclusions in vivo, our data suggest that expression of rAAV5-siHUNT-1 significantly diminished the available cellular pool of soluble mHtt N-terminal fragments in the striatum of the R6/1 mouse. Similar reductions of NIIs were reported in rAAV1-mediated shRNA-treated HD-N171-82Q mice [30].
The functional significance of NIIs in HD mouse models is currently controversial. NIIs have been hypothesized to be a protective mechanism in cells by functioning to sequester detrimental toxic forms of Htt and preventing them from damaging the cell [51,52]. Arrasate et al. [52] showed that cells often die without forming NIIs, which agrees with the idea that mHtt fragments that are pre-NII formation may be toxic. This observation suggests that intervention earlier in R6/1 mice may reduce the level of exposure of striatal cells to these more toxic mHtt fragments and therefore lead to a better functional outcome. In agreement with this idea, Harper et al. [30] treated the more slowly progressing HD-N171-82Q mice with shRNA starting at 4 weeks of age, as opposed to the treatment of the highly aggressive R6/1 HD model starting at 7 weeks of age as we have done here.
mHtt has been shown to affect the cellular localization and function of several regulators of transcription known to be critical for proper neuronal function, which results in the specific down-regulation of a subset of striatal mRNAs [53]. Expression of rAAV5-siHUNT-1 in the striatum of the R6/1 mouse resulted in a significant increase in the steady-state mRNA levels of both ppEnk and DARPP-32 mRNAs as demonstrated by in situ hybridization analysis. No significant changes were observed in either the dopamine type 2 receptor or NGFi-A mRNA levels. Additionally, expression of rAAV5-GFP or the inactive control shRNA, rAAV5-siRho-1, did not result in any detectable changes in the levels of any of the transcripts analyzed. The promoter for ppEnk is regulated by the cAMP-responsive element (CRE) transcriptional pathway [54]. The CRE transcriptional pathway is extensively involved in the regulation of genes needed for neuronal function and survival and expression of mHtt has been shown to interfere with this pathway [55–57]. CRE-mediated transcription is modulated by TAFII130, and the pQ domain of soluble mHtt N-terminal fragments can sequester TAFII130 into aggregates. This aberrant protein–protein interaction is thought to affect CRE-regulated transcription negatively by affecting the proper localization and function of TAFII130 [56]. Therefore, reduced levels of striatal mHtt might have a direct effect on the activity of CRE-mediated transcription. In this study, while the frequency and size of NIIs were reduced and disease-affected striatal transcripts were improved by administration of siHUNT-1, the relationship between NIIs and disease progression and the exact role of mHtt in transcriptional dysregulation remain poorly understood.
We have observed that the levels for both ppEnk and DARPP-32 mRNA begin to decrease at ~4–5 weeks of age and reach a minimum steady-state level around 12 weeks of age (E. M. Denovan-Wright, unpublished data). In this study, we initiated shRNA treatment to reduce striatal mHtt expression in R6/1 mice that were between 6 and 8 weeks of age. Since we did not fully restore ppENK and DARPP-32 mRNAs to wild-type levels, our data suggest that lowering the levels of striatal mHtt affected the rate of loss in steady-state mRNA levels. Alternatively, R6/1 striatal neurons expressing low levels of mHtt protein may have maintained an increased level of transcriptional activity compared to striatal neurons expressing normal levels of mHtt.
Finally, we investigated whether long-term expression of rAAV5-siHUNT-1 would affect the behavioral phenotype displayed in the R6/1 mouse. In this study, we evaluated several physiological and behavioral tests, including effect on weight loss, clasping phenotype, and performance on the rotarod apparatus. There was no difference in weight among the groups of R6/1 mice bilaterally treated with rAAV5-siHUNT-1 or rAAV5-GFP R6/1 and uninjected age-matched R6/1 controls. Analysis of retention times in the rotarod apparatus demonstrated a clear difference between genotype; however, we observed no differences among the three different R6/1 treatment groups. In contrast, there was a delayed onset of the clasping phenotype on R6/1 mice that were bilaterally injected with rAAV5-siHUNT-1 compared to uninjected or rAAV5-GFP-injected R6/1 mice. This mild behavioral improvement may suggest that suppression of mHtt expression in other regions of the brain, i.e., cortex, may be required to achieve a more explicit positive effect on behavior. Also, some of the behavioral phenotypes observed in the R6/1 mouse may be affected by dysfunction of other systems outside the CNS. In fact, problems with metabolism and muscle wasting have been observed in the R6 lines [58]. These systemic abnormalities would not be expected to improve after suppression of striatal mHtt expression. In contrast, Harper et al. [30] obtained substantial phenotypic correction after striatal-specific rAAV1-mediated anti-mHtt shRNA in the HD-N171-82Q mouse. Striatal transduction efficiencies and knockdown of mHtt mRNA levels were strikingly similar in this study compared to those of Harper et al. [30]. Therefore, in addition to the timing of vector injection, differences in the mouse model may account for the greater behavioral effects found in Harper et al. [30]. For example, the HDN171-82Q mouse does not display significant weight loss or muscle wasting [59], which may allow for better striatal-mediated improvement in coordinated movement as measured by the rotarod paradigm in the Harper et al. study [30].
In addition to the observation that siHUNT-1 reduced mHtt levels, reduced NIIs, and partially normalized levels of two mHtt-affected transcripts and had a positive effect on the behavioral phenotype of R6/1 mice, we observed that the other active anti-mHtt shRNA tested, siHUNT-2, reduced levels of striatal-specific transcripts in wild-type and R6/1 mice, indicating that this molecule, despite its apparent efficacy at reducing mHtt mRNA and protein levels in HEK293 cells and in vivo, had deleterious effects that were independent of mHtt knockdown. As shown by these results, extreme caution should be taken when interpreting data from shRNAs in vivo, and detailed analysis of cellular gene expression can detect off-targeting effects associated with the intracellular expression of shRNAs.
In conclusion, long-term rAAV5-mediated striatal expression of an anti-mutant Htt shRNA was well tolerated in both wild-type and R6/1 transgenic mice and led to changes that are consistent with reduced expression levels of mHtt protein. Moreover, improved striatal transduction, use of a slower progressing HD mouse model [30], transduction in additional critical brain regions such as cerebral cortex, intervention earlier in the time course of pathology [30], or a combination of these factors may lead to better shRNA-mediated striatal mHtt knockdown effects. Nevertheless, we have demonstrated that reduced levels of striatal mHtt can be achieved through the use of RNAi and that this treatment results in a mildly improved cellular and behavioral phenotype in the R6/1 line of HD. Since polymorphisms associated with the mHtt allele have been described [60–62], these results suggest that mutant allele-specific gene silencing may be a clinically viable approach once remaining efficiency and safety issues are resolved [29,45].
MATERIALS AND METHODS
rAAV-shRNA plasmid construction
Two complementary DNA oligonucleotides (Invitrogen, San Diego, CA, USA) were allowed to anneal to produce a double-stranded DNA fragment coding for the sense strand, 9-nucleotide loop. and antisense strand of both anti-huntingtin shRNAs tested. siHUNT-1 targeted nucleotides 262–281 (5′-GCCGCGAGTCGGCCCGAGGC-3′, Fig. 1B) and siHUNT-2 targeted nucleotides 342–363 (5′-GGCCTTCGAGTCCCTCAAGTCC-3′, Fig. 1B) of the human HD mRNA (GenBank Accession No. L12392). Double-stranded DNA fragments were ligated into the BglII/HindIII sites of the rAAV vector pSOFF-H1p-hrGFP. This vector contains the human RNase P H1 promoter downstream of the rAAV serotype-5 ITR and a cDNA encoding the humanized Renilla reniformis GFP (Stratagene, La Jolla, CA, USA) protein under the control of the herpes simplex virus thymidine kinase promoter.
Testing of shRNA efficacy in cultured cells
A portion of the mHtt transgene was PCR amplified from R6/1 mouse genomic DNA using forward (5′-AGGGCTGTCAATCATGCTGG-3′) and reverse (5′-TCTGGGTTGCTGGGTCACTCTGTCTCTGCGGAGCCGGGGG-3′) primers. The resulting product was cloned into pCR 2.1-TOPO TA (Invitrogen), sequenced, and subcloned into the HindIII/NsiI sites present in the pRC/CMV (Invitrogen) eukaryotic expression plasmid. The resultant plasmid (pCMV-R6/1) expressed part of the 5′ UTR, the coding region within exon 1 with ~115 contiguous CAG repeats, and part of intron 1 of the mHtt transgene under the control of the minimal cytomegalovirus (CMV) promoter.
Human embryonic kidney 293T (HEK293) cells (ATCC, Manassas, VA, USA) were transfected using Lipofectamine and Plus reagents (Invitrogen) with pCMV-R6/1 or pCMV-R6/1 and siHUNT-1, siHUNT-2, or siRho-1 constructs. Transfection efficiency was determined by analysis of GFP-positive cells and ranged from 70 to 80%. Cells were harvested 48 h posttransfection for Northern and Western analysis. Total RNA was isolated from transfected HEK293 cells using TRIzol reagent (Invitrogen). Northern blot analysis of 10 µg of total RNA was performed using standard techniques [63]. A 150-bp HindIII/AgeI fragment of pCMV-R6/1 spanning the 5′ UTR and ATG initiation codon of the mHtt transgene was radiolabeled using [α-32P]dCTP (3000 mCi/ml; MP Biochemicals, Irvine, CA, USA) and used as the hybridization probe. RNA loading was normalized by removing the mHtt-specific probe and reprobing the membrane with a radiolabeled fragment complementary to nucleotides 150–270 of the human β-actin mRNA (GenBank Accession No. BC002409). Blots were exposed to phosphoimager screens and the intensity of labeled bands was determined using a Molecular Dynamics Phosphoimager.
Western blot analysis using Hum-1 antibody
Hum-1 polyclonal antibody was raised in New Zealand White rabbits against the human Htt synthetic peptide Ac-PQLPQPPPQAQPLLPQPQC-OH and affinity-purified (New England Peptide, Gardner, MA, USA). The peptide corresponded to the region of human Htt immediately downstream of the polyglutamine repeat. Total protein from transfected cells was solubilized in RIPA buffer [0.35 M Tris (pH 6.8), 10.28% SDS, 36% glycerol, 0.6 M dithiothreitol, 0.012% bromophenol blue]. Protein samples (40 µg) were boiled for 3 min, fractionated by SDS–PAGE, and subjected to Western blot analysis as described in [33] using a 1:500 dilution of affinity-purified Hum-1 and 1:2000 dilution of peroxidase-labeled goat anti-rabbit IgG secondary antibody (1:2000; Vector Laboratories, Burlingame, CA, USA).
rAAV vector production
All rAAV vectors used in this study were produced by the University of Florida Powell Gene Therapy Center Vector Core Facility using the method described in [64]. The rAAV5 vector consisted of rAAV5 capsids and AAV5 ITRs and is, therefore, not a pseudotyped vector [65]. A standard triple-transfection method using the helper DNA pDG [66] for required adenoviral proteins and the plasmids containing shRNAs and pAAV5.2, which has rep and cap from AAV5, was employed. Crude rAAV5 virus was then purified by iodixanol step gradients and Sepharose Q column chromatography as previously described [67]. The final products were concentrated to final titers of 1 to 5 × 1013 genome copies per milliliter.
Intrastriatal injection of shRNA-expressing AAV vectors
All animal care, handling, and surgical protocols were in accordance with the guidelines established by the Canadian Council on Animal Care and were approved by the Carleton Animal Care Committee at Dalhousie University. Breeding, care, and genotyping have been described previously [33]. Stereotaxic administration of AAV vectors were performed on 6- to 8- week-old wild-type and R6/1 mice under isoflurane anesthesia. The anterior–posterior (AP) and medial–lateral (ML) stereotaxic coordinates for injection were calculated from bregma and the dorsal–ventral (DV) coordinates were calculated from the dural surface. Mice received intra-striatal injections of rAAV5 expressing siHUNT-1, siHUNT-2, siRho-1, or TRUF11 (rAAV5-hrGFP, see [35]) suspended in phosphate-buffered saline (PBS) at a dose of 2 µl/site and an infusion rate of 0.5 µl/min using a continuous infusion pump attached to a 10-µl Hamilton microsyringe fitted with a glass micropipette with an outer diameter of 60–80 µm [68]. One minute after the cessation of the infusion, the micropipette was retracted an additional 1 mm, allowed to remain at this position for 4 min, and then slowly retracted from the brain. The stereotaxic coordinates used for the two injections within the same striatum were, site 1, AP +1.0, ML ±1.8, DV −3.3; site 2, AP +0.4, ML ±2.1, DV −3.4. All coordinates were measured with an empirically determined flat skull.
The effect of shRNA and control vectors on mHtt mRNA and protein levels was assessed in a group of animals that received unilateral intrastriatal injections of rAAV vectors. In these experiments, the contralateral hemisphere was used as an internal control. Ten-weeks after the intrastriatal infusion of rAAV, mice were deeply anesthetized with sodium pentobarbital (65 mg/kg ip) and decapitated. The brains were removed and stored at −70°C. Tissue sections (14 µm) were cut using a Micron cryostat through the rostral–caudal axis of the striatum, thaw-mounted onto Fisher SuperFrost slides, and stored at −70°C. For each animal, five coronal brain sections, each separated by approximately 350 µm, were placed on a single slide. This distribution of tissue was used to ensure that each slide contained sections taken throughout the transduced region of the striatum.
To determine the levels of mHtt RNA and protein, striatal tissue was isolated from frozen sections that had been thaw-mounted on slides. Cortical tissue was removed using a razor blade and discarded. To collect tissue mainly from the transduction area, sections were first examined under epifluorescence to visualize the transduction area. The striatal tissue from the right and left sides of five brain sections per animal were then manually scraped from each slide taking only the approximate area of GFP positivity.
RNA was extracted using Trizol (Invitrogen), quantified spectrophotometrically, and used as the substrate for reverse-transcriptase (RT) reactions to generate single-stranded cDNA. The reaction was optimized to reverse transcribe the 5′ end of the human Htt transgene. Briefly, 1 µg of total RNA and 0.75 µg of random hexamers were incubated with 1 µl of 5× Q solution (Qiagen, Valencia, CA, USA) in a total volume of 5.75 µl at 70°C for 3 min, mixed, and placed on ice for 5 min. M-MLV RT buffer and dNTPs were added to a final concentration of 1× and 1.25 µM, respectively, in a final reaction volume of 10 µl. Twenty units of RNasin (Promega, Madison, WI, USA) and 100 units of M-MLV RT (Promega) were added and the reaction was allowed to proceed at 48°C for 60 min. The reaction was terminated by heating at 70°C for 10 min. −RT reactions differed from +RT reactions by the substitution of H2O for RT in the reactions. Quantitative PCR (Lightcycler; Roche) was used to amplify a 115-bp region of mHtt cDNA from striatal tissue. The PCRs contained 1× QuantiTect SYBR Green PCR Master Mix (Qiagen) and 500 nM each sense (5′-AGAGCCCCATTCATTGCC-3′) and antisense (5′-GGACTTGAGGGACTCGAA-3′) primer. Cycling conditions included a denaturation step of 95°C for 15 min, followed by 45 cycles of 94°C for 15 s, 58°C for 20 s, and 72°C for 20 s, and ending with a melting step from 65 to 99°C over 30 s. Total SYBR green fluorescence was measured at the end of each PCR cycle and continuously through the melting step. Known quantities of mHtt cDNA were simultaneously amplified with experimental samples and used to create a standard curve. The levels of cDNA in each sample were normalized to the levels of hypoxanthine ribosyl transferase [41].
For Western blot analysis, striatal tissue from four or five sections per animal were isolated and homogenized in 0.32 M sucrose, quantified using the BCA protein determination assay (Pierce), fractionated on SDS–PAGE gels using standard protocols [42], and subjected to the immunoblotting conditions described for analysis of mHtt protein levels in transiently transfected HEK293 cells.
For analysis of the distribution of immunoreactive inclusion bodies, fresh-frozen coronal brain sections on SuperFrost slides were allowed to come to room temperature and rinsed 3 × 10 min with PBS (100 mM phosphate, pH 7.4, and 0.9% NaCl) and the tissue was fixed in 4% (v/v) paraformaldehyde for 15 min. The tissue was incubated in 0.1% (v/v) H2O2, 10% methanol, and PBS for 10 min and rinsed 3 × 10 min in PBS. Nonspecific binding sites were blocked in a solution of 5% normal goat serum in 0.25% (v/v) Triton X-100/0.01 M PBS for 1 h at room temperature. Immunostaining was performed using a 1:500 dilution of affinity-purified Hum-1 or 1:4000 dilution of polyclonal rabbit anti-ubiquitin (IgG) antibody (DakoCytomation, Carpinteria, CA, USA) in 1% (v/v) normal goat serum, 0.25% (v/v) Triton X-100/PBS. Tissues were incubated overnight at 4°C. A 1:500 dilution of goat anti-rabbit biotin-labeled secondary antibody (Vector Laboratories) was prepared in 1% (v/v) normal goat serum, 0.25% (v/v) Triton X-100/0.01 M PBS. Slides were incubated in secondary antibody solution for 2 h at room temperature and then rinsed 3 × 10 min in 0.01 M PBS. Interaction between the primary and the secondary antibodies was detected using the Vectastain Elite ABC Kit (Vector Laboratories) and DAB using the company’s standard protocol.
The size and distribution of Hum-1-immunoreactive NIIs were determined by using the Analyze Particle function in the Image J NIH software (http://rsb.info.nih.gov/ij/). The relative size (µm2) and number of NIIs were determined by analyzing coded digital images of four different regions (left motor cortex, right motor cortex, left medial striatum, right medial striatum) in coronal sections that were subjected to the immunohistochemical procedure described above. This method was employed because 14-µm sections were too thin for unbiased stereological estimates of NII number and the NII diameter was too small to use the rotator method of volume estimation [69]. The Image J software (NIH) was configured to detect particles of a specified size range (0.03–0.5 µm2) and a constant upper and lower threshold value was used for all grayscale images from all areas analyzed. The mean relative size area (µm2) of particles (NIIs) as well as the mean number of particle counts within the specified size range was calculated by a blinded operator for each of the four different regions in all of the unilaterally injected R6/1 mice and reported as percentage of the size and number of NIIs present in the left motor cortex. Since absolute numbers or areas are not reported and internal section controls were used to determine percentage controls, this method of quantification of NIIs is valid.
In situ hybridization analysis
In situ hybridization was performed on coronal mouse brain sections (bregma +1.70 to −0.50 [70]) using radiolabeled antisense gene-specific oligonucleotide probes (Table 1). The methods employed for in situ hybridization and quantification of the hybridization signal have been described previously [40,42,71,72]. Representative sections shown in Fig. 5 were scanned and false-colored in Adobe PhotoShop 7.0 by changing the image to RGB color and applying a gradient map. No other manipulations were performed on these sections.
Effect of anti-mHtt shRNA on phenotype
The effect of anti-mHtt shRNA and control vectors on phenotype was assessed in animals that received bilateral injections of siHUNT-1, siHUNT-2, and control rAAV vectors. Mice were weighed weekly following injection of rAAV. Average weight, grouped by genotype and treatment, was determined for each week. The percentage of mice per group at each time point that exhibited clasping was recorded. Motor coordination was analyzed by placing the mice on a rotarod that increased from 0 to 40 rpm over 1 min. The time from placing the mouse on the rotarod until the mouse fell off was recorded for each of four trials and the average time that each mouse remained on the rotarod was recorded.
Statistics
ANOVA was used to evaluate the probability of differences between experimental groups. Where appropriate, one-way ANOVA was performed and individual post hoc differences between groups were assessed using Fisher’s PLSD (Statview). Post hoc differences in two-way ANOVA designs were assessed in a hierarchical fashion as described by Kirk [73] using simple main-effects analysis. The minimum probability accepted for significance (α level) was 0.05.
ACKNOWLEDGMENTS
This work was supported by grants from the Hereditary Disease Foundation (R.J.M.), the Canadian Institutes of Health Research (E.D-W.), and the Huntington Society of Canada through the Laura’s Hope Fund (E.D-W.). E.R. was supported by a National Institutes of Health Minority Predoctoral Fellowship (5 F31 NS 011182- 05). We thank M. Huang for technical assistance. We also thank Dr. Marina Gorbatyuk (Department of Molecular Genetics, University of Florida) for providing the siRho-1 vector.
REFERENCES
- 1.The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 2.Gauthier LR, et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 3.Ross CA. Huntington’s disease: new paths to pathogenesis. Cell. 2004;118:4–7. doi: 10.1016/j.cell.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 4.Bates G. Huntingtin aggregation and toxicity in Huntington’s disease. Lancet. 2003;361:1642–1644. doi: 10.1016/S0140-6736(03)13304-1. [DOI] [PubMed] [Google Scholar]
- 5.Sharp AH, et al. Widespread expression of Huntington’s disease gene (IT15) protein product. Neuron. 1995;14:1065–1074. doi: 10.1016/0896-6273(95)90345-3. [DOI] [PubMed] [Google Scholar]
- 6.Trottier Y, et al. Cellular localization of the Huntington’s disease protein and discrimination of the normal and mutated form. Nat. Genet. 1995;10:104–110. doi: 10.1038/ng0595-104. [DOI] [PubMed] [Google Scholar]
- 7.Cha JH. Transcriptional dysregulation in Huntington’s disease. Trends Neurosci. 2000;23:387–392. doi: 10.1016/s0166-2236(00)01609-x. [DOI] [PubMed] [Google Scholar]
- 8.Li SH, Li XJ. Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004;20:146–154. doi: 10.1016/j.tig.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 9.Petersen A, Mani K, Brundin P. Recent advances on the pathogenesis of Huntington’s disease. Exp. Neurol. 1999;157:1–18. doi: 10.1006/exnr.1998.7006. [DOI] [PubMed] [Google Scholar]
- 10.Zuccato C, et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- 11.Duyao MP, et al. Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science. 1995;269:407–410. doi: 10.1126/science.7618107. [DOI] [PubMed] [Google Scholar]
- 12.Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat. Genet. 1995;11:155–163. doi: 10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]
- 13.White JK, et al. Huntingtin is required for neurogenesis and is not impaired by the Huntington’s disease CAG expansion. Nat. Genet. 1997;17:404–410. doi: 10.1038/ng1297-404. [DOI] [PubMed] [Google Scholar]
- 14.Dragatsis I, Levine MS, Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat. Genet. 2000;26:300–306. doi: 10.1038/81593. [DOI] [PubMed] [Google Scholar]
- 15.Menalled LB, et al. Early motor dysfunction and striasomal distribution of huntingtin microaggregates in Huntington’s disease knock-in mice. J. Neurosci. 2002;22:8266–8276. doi: 10.1523/JNEUROSCI.22-18-08266.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin CH, et al. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum. Mol. Genet. 2001;10:137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- 17.Cattaneo E, et al. Loss of normal huntingtin function: new developments in Huntington’s disease research. Trends Neurosci. 2001;24:182–188. doi: 10.1016/s0166-2236(00)01721-5. [DOI] [PubMed] [Google Scholar]
- 18.Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- 19.Thakur AK, Yang W, Wetzel R. Inhibition of polyglutamine aggregate cytotoxicity by a structure-based elongation inhibitor. FASEB J. 2004;18:923–925. doi: 10.1096/fj.03-1238fje. [DOI] [PubMed] [Google Scholar]
- 20.Colby DW, et al. Potent inhibition of huntingtin aggregation and cytotoxicity by a disulfide bond-free single-domain intracellular antibody. Proc. Natl. Acad. Sci. USA. 2004;101:17616–17621. doi: 10.1073/pnas.0408134101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lecerf JM, et al. Human single-chain Fv intrabodies counteract in situ huntingtin aggregation in cellular models of Huntington’s disease. Proc. Natl. Acad. Sci. USA. 2001;98:4764–4769. doi: 10.1073/pnas.071058398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller TW, et al. A human single-chain Fv intrabody preferentially targets amino-terminal huntingtin fragments in striatal models of Huntington’s disease. Neurobiol. Aging. 2005;19:47–56. doi: 10.1016/j.nbd.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 23.Mangiarini L, et al. Instability of highly expanded CAG repeats in mice transgenic for the Huntington’s disease mutation. Nat. Genet. 1997;15:197–200. doi: 10.1038/ng0297-197. [DOI] [PubMed] [Google Scholar]
- 24.Mangiarini L, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 25.Carter RJ, et al. Characterization of progressive motor deficits in mice transgenic for the human Huntington’s disease mutation. J. Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davies SW, et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 27.Petersen A, et al. Mice transgenic for exon 1 of the Huntington’s disease gene display reduced striatal sensitivity to neurotoxicity induced by dopamine and 6-hydroxydopamine. Eur. J. Neurosci. 2001;14:1425–1435. doi: 10.1046/j.0953-816x.2001.01765.x. [DOI] [PubMed] [Google Scholar]
- 28.Hansson O, et al. Resistance to NMDA toxicity correlates with appearance of nuclear inclusions, behavioural deficits and changes in calcium homeostasis in mice transgenic for exon 1 of the huntingtin gene. Eur. J. Neurosci. 2001;14:1492–1504. doi: 10.1046/j.0953-816x.2001.01767.x. [DOI] [PubMed] [Google Scholar]
- 29.Miller VM, et al. Allele-specific silencing of dominant disease genes. Proc. Natl. Acad. Sci. USA. 2003;100:7195–7200. doi: 10.1073/pnas.1231012100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harper SQ, et al. RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc. Natl. Acad. Sci. USA. 2005;102:5820–5825. doi: 10.1073/pnas.0501507102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raoul C, et al. Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat. Med. 2005;11:423–428. doi: 10.1038/nm1207. [DOI] [PubMed] [Google Scholar]
- 32.Ralph GS, et al. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat. Med. 2005;11:429–433. doi: 10.1038/nm1205. [DOI] [PubMed] [Google Scholar]
- 33.Ralph GS, Mazarakis ND, Azzouz M. Theraputic gene silencing in neurological disorders, using interfering RNA. J. Mol. Med. 2005;83:413–419. doi: 10.1007/s00109-005-0649-1. [DOI] [PubMed] [Google Scholar]
- 34.Xia H, et al. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat. Med. 2004;10:816–820. doi: 10.1038/nm1076. [DOI] [PubMed] [Google Scholar]
- 35.Burger C, et al. Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol. Ther. 2004;10:302–317. doi: 10.1016/j.ymthe.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 36.Bartlett JS, Samulski RJ, McCown TJ. Selective and rapid uptake of adeno-associated virus type 2 in brain. Hum Gene Ther. 1998;9:1181–1186. doi: 10.1089/hum.1998.9.8-1181. [DOI] [PubMed] [Google Scholar]
- 37.Mandel RJ, et al. Characterization of intrastriatal recombinant adeno-associated virus mediated gene transfer of human tyrosine hydroxylase and human GTP-cyclohydroxylase I in a rat model of Parkinson’s disease. J. Neurosci. 1998;18:4271–4284. doi: 10.1523/JNEUROSCI.18-11-04271.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cha JH, et al. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human Huntington disease gene. Proc. Natl. Acad. Sci. USA. 1998;95:6480–6485. doi: 10.1073/pnas.95.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luthi-Carter R, et al. Decreased expression of striatal signaling genes in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2000;9:1259–1271. doi: 10.1093/hmg/9.9.1259. [DOI] [PubMed] [Google Scholar]
- 40.Hu H, McCaw EA, Hebb AL, Gomez GT, Denovan-Wright EM. Mutant huntingtin affects the rate of transcription of striatum-specific isoforms of phosphodiesterase 10A. Eur. J. Neurosci. 2004;20:3351–3363. doi: 10.1111/j.1460-9568.2004.03796.x. [DOI] [PubMed] [Google Scholar]
- 41.McCaw EA, et al. Structure, expression and regulation of the cannabinoid receptor gene (CB1) in Huntington’s disease transgenic mice. Eur. J. Biochem. 2004;271:4909–4920. doi: 10.1111/j.1432-1033.2004.04460.x. [DOI] [PubMed] [Google Scholar]
- 42.Hebb AL, Robertson HA, Denovan-Wright EM. Striatal phosphodiesterase mRNA and protein levels are reduced in Huntington’s disease transgenic mice prior to the onset of motor symptoms. Neuroscience. 2004;123:967–981. doi: 10.1016/j.neuroscience.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 43.Petersen A, et al. Evidence for dysfunction of the nigrostriatal pathway in the R6/1 line of transgenic Huntington’s disease mice. Neurobiol. Dis. 2002;11:134–146. doi: 10.1006/nbdi.2002.0534. [DOI] [PubMed] [Google Scholar]
- 44.Naver B, et al. Molecular and behavioral analysis of the R6/1 Huntington’s disease transgenic mouse. Neuroscience. 2003;122:1049–1057. doi: 10.1016/j.neuroscience.2003.08.053. [DOI] [PubMed] [Google Scholar]
- 45.Davidson BL, Paulson HL. Molecular medicine for the brain: silencing of disease genes with RNA interference. Lancet Neurol. 2004;3:145–149. doi: 10.1016/S1474-4422(04)00678-7. [DOI] [PubMed] [Google Scholar]
- 46.Matalon R, et al. Adeno-associated virus-mediated aspartoacylase gene transfer to the brain of knockout mouse for canavan disease. Mol. Ther. 2003;7:580–587. doi: 10.1016/s1525-0016(03)00066-2. [DOI] [PubMed] [Google Scholar]
- 47.Szczypka MS, et al. Viral gene therapy selectively restores feeding and prevents lethality of dopamine-deficient mice. Neuron. 1999;22:167–178. doi: 10.1016/s0896-6273(00)80688-1. [DOI] [PubMed] [Google Scholar]
- 48.Millington-Ward S, et al. RNAi of COL1A1 in mesenchymal progenitor cells. Eur. J. Hum. Genet. 2004;12:864–866. doi: 10.1038/sj.ejhg.5201230. [DOI] [PubMed] [Google Scholar]
- 49.Blalock TD, Yuan R, Lewin AS, Schultz GS. Hammerhead ribozyme targeting connective tissue growth factor mRNA blocks transforming growth factor-beta mediated cell proliferation. Exp. Eye Res. 2004;78:1127–1136. doi: 10.1016/j.exer.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 50.Dawson PA, Marini JC. Hammerhead ribozymes selectively suppress mutant type I collagen mRNA in osteogenesis imperfecta fibroblasts. Nucleic Acids Res. 2000;28:4013–4020. doi: 10.1093/nar/28.20.4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- 52.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 53.Sugars KL, Brown R, Cook LJ, Swartz J, Rubinsztein DC. Decreased cAMP response element-mediated transcription: an early event in exon 1 and full-length cell models of Huntington’s disease that contributes to polyglutamine pathogenesis. J. Biol. Chem. 2004;279:4988–4999. doi: 10.1074/jbc.M310226200. [DOI] [PubMed] [Google Scholar]
- 54.Kobierski LA, Wong AE, Srivastava S, Borsook D, Hyman SE. Cyclic AMP-dependent activation of the proenkephalin gene requires phosphorylation of CREB at serine-133 and a Src-related kinase. J. Neurochem. 1999;73:129–138. doi: 10.1046/j.1471-4159.1999.0730129.x. [DOI] [PubMed] [Google Scholar]
- 55.Shimohata T, Onodera O, Tsuji S. Interaction of expanded polyglutamine stretches with nuclear transcription factors leads to aberrant transcriptional regulation in polyglutamine diseases. Neuropathology. 2000;20:326–333. doi: 10.1046/j.1440-1789.2000.00350.x. [DOI] [PubMed] [Google Scholar]
- 56.Shimohata T, et al. Expanded polyglutamine stretches interact with TAFII130, interfering with CREB-dependent transcription. Nat. Genet. 2000;26:29–36. doi: 10.1038/79139. [DOI] [PubMed] [Google Scholar]
- 57.Steffan JS, et al. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luthi-Carter R, et al. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum. Mol. Genet. 2002;11:1911–1926. doi: 10.1093/hmg/11.17.1911. [DOI] [PubMed] [Google Scholar]
- 59.Schilling G, et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum. Mol. Genet. 1999;8:397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- 60.Rubinsztein DC, Leggo J, Goodburn S, Barton DE, Ferguson-Smith MA. Haplotype analysis of the delta 2642 and (CAG)n polymorphisms in the Huntington’s disease (HD) gene provides an explanation for an apparent ‘founder’ HD haplotype. Hum. Mol. Genet. 1995;4:203–206. doi: 10.1093/hmg/4.2.203. [DOI] [PubMed] [Google Scholar]
- 61.Novelletto A, et al. Polymorphism analysis of the huntingtin gene in Italian families affected with Huntington disease. Hum. Mol. Genet. 1994;3:1129–1132. doi: 10.1093/hmg/3.7.1129. [DOI] [PubMed] [Google Scholar]
- 62.Lucotte G, Gerard N, Roubertoux P, Schmitt I, Riess O. Relationships of the 2642 deletion polymorphism (delta 2642) in the huntingtin gene with the CAG repeat expansion length and age at onset of the disease. Genet. Couns. 1996;7:297–302. [PubMed] [Google Scholar]
- 63.Ausubel FM, et al. Current Protocols in Molecular Biology. Indianapolis: Wiley; 1988. [Google Scholar]
- 64.Zolotukhin S, et al. Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods. 2002;28:158–167. doi: 10.1016/s1046-2023(02)00220-7. [DOI] [PubMed] [Google Scholar]
- 65.Chiorini JA, Kim F, Yang L, Kotin RM. Cloning and characterization of adeno-associated virus type 5. J. Virol. 1999;73:1309–1319. doi: 10.1128/jvi.73.2.1309-1319.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grimm D, Kern A, Rittner K, Kleinschmidt JA. Novel tools for production and purification of recombinant adenoassociated virus vectors. Hum. Gene Ther. 1998;9:2745–2760. doi: 10.1089/hum.1998.9.18-2745. [DOI] [PubMed] [Google Scholar]
- 67.Zolotukhin S, et al. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6:973–985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]
- 68.Mandel RJ, Hardy S, Snyder RO, Naldini L, Leff SE. Intracerebral gene transfer using viral vectors. In: Dunnett SB, Bolton A, Baker G, editors. NeuroMethods. Totowa, NJ: Humana Press; 1999. pp. 103–130. [Google Scholar]
- 69.Gundersen HJG, et al. The new stereological tools: disector, fractionator, nucleator and point sampled intercepts and their use in pathological research and diagnosis. Acta Pathol. Microbiol. Immunol. Scand. 1988;96:857–881. doi: 10.1111/j.1699-0463.1988.tb00954.x. [DOI] [PubMed] [Google Scholar]
- 70.Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. San Diego: Academic Press; 2001. 2001. [Google Scholar]
- 71.Denovan-Wright EM, Robertson HA. Cannabinoid receptor messenger RNA levels decrease in a subset of neurons of the lateral striatum, cortex and hippocampus of transgenic Huntington’s disease mice. Neuroscience. 2000;98:705–713. doi: 10.1016/s0306-4522(00)00157-3. [DOI] [PubMed] [Google Scholar]
- 72.Harris AS, Denovan-Wright EM, Hamilton LC, Robertson HA. Protein kinase c beta II mRNA levels decrease in the striatum and cortex of transgenic Huntington’s disease mice. J. Psychiatry Neurosci. 2001;26:117–122. [PMC free article] [PubMed] [Google Scholar]
- 73.Kirk RE. Experimental Design: Procedures for the Behavioral Sciences. Belmont, CA: Brooks/Cole; 1968. [Google Scholar]