

Abstract
Self-splicing group II introns catalyze their own excision from pre-RNAs, thereby joining the flanking exons. The introns can be released in a lariat or linear form. Lariat introns have been shown to reverse the splicing reaction; in contrast, linear introns are generally believed to perform no or only poor reverse splicing. Here, we show that a linear group II intron derived from ai5γ can reverse the second step of splicing with unexpectedly high efficiency and precision. Moreover, the linear intron generates dramatically more reverse-splicing product than its lariat equivalent. The finding that linear group II introns can readily undergo the critical first step of mobility by catalyzing efficient reverse splicing into complementary target molecules demonstrates their innate potential for mobility and transposition and raises the possibility that reverse splicing by linear group II introns may have played a significant role in certain forms of intron mobility and lateral gene transfer during evolution.
Keywords: ribozyme, group II intron, reverse splicing, pH-dependent rate constants, 3′end heterogeneity, intron mobility
INTRODUCTION
Group II introns are self-splicing RNAs that catalyze their own excision from a pre-RNA. They are found in organellar genomes of higher plants and fungi, as well as in many bacteria, proteobacteria, blue algae, and more recently, in a primitive metazoan (Vallès et al. 2008). Group II introns share a common secondary structure consisting of six stem–loops, termed Domains D1–D6, which are arranged around a central wheel (Pyle 2008). Several tertiary interactions arrange the six domains in the natively folded structure. Domain D1 provides a scaffold for folding of the intron (Su et al. 2005). It contains the two exon binding sites EBS1 and EBS2, which recognize complementary sequences, termed intron binding sites IBS1 and IBS2, on the 5′-exon, determining the 5′-splice site. Domain D3 is a catalytic effector and removal of this domain reduces the catalytic rate dramatically (Koch et al. 1992). In contrast, Domain D4 is not required for catalysis and can be removed without affecting intron function (Jarrell et al. 1988). Domain D5 is the catalytic center of the intron and absolutely required for catalysis. Domain D6 provides the branch point adenosine, a bulged nucleotide that is located close to the 3′-terminus of the intron (vide infra).
Self-splicing of group II introns is a two-step reaction and can follow two different pathways (Fig. 1; Daniels et al. 1996). In the first step of the branching pathway, the 2′-OH group of the branch point adenosine nucleophilically attacks the phosphate at the 5′-splice site. The 5′-exon is released and the attacking adenosine adopts a 2′,5′-branched structure that gives the intron a lariat form. In the alternative hydrolytic pathway, a water molecule acts as the nucleophile in the first step and cleaves the 5′-exon from the intron without forming a branched structure. The second step of splicing is the same for both pathways; the 3′-terminal OH group of the 5′-exon attacks the phosphate at the 3′-splice site, thereby splicing the exons and releasing the lariat or linear intron.
FIGURE 1.

Pathways of group II intron splicing. Linear and lariat introns are depicted as solid lines or circles, respectively. Exon sequences are shown as solid (5′-exon) or open (3′-exon) boxes.
The lariat bI1 intron, which is a group II intron found in the COB gene in Saccharomyces cerevisiae mitochondria, can reinsert itself into its original location between the spliced exons by reversing both steps of splicing. However, the reaction was reported to proceed with very low efficiency (Augustin et al. 1990). Reverse splicing is a very important feature of group II introns because it is the obligate first step of intron mobility, through which group II introns invade duplex DNA. This process may have resulted in the propagation of ancestral introns and have pushed their evolution into modern forms (Pyle 2008). The actual process of intron mobility requires more than reverse splicing by intron RNA: it depends upon the action of an intron encoded maturase or host proteins, which provide endonuclease and reverse transcription activities. This process is known as target-primed reverse transcription (TPRT) (Pyle and Lambowitz 2006).
The linear intron is generally believed to be inefficient or unable to perform reverse splicing (Dème et al. 1999; Mohr et al. 2006; Gordon et al. 2007). Although a linear form of the bI1 group II intron was shown to perform reversal of the second step of splicing (Mörl and Schmelzer 1990), the reaction was inefficient and generated only traces of reverse-spliced RNA. In addition, the intron originated from a splicing reaction and was isolated by denaturing gel electrophoresis. This method bears the risk of contaminating the linear intron molecules with broken lariats, which accumulate significantly during the forward-splicing reaction and comigrate with linear intron during gel electrophoresis (Daniels et al. 1996). Thus, it cannot be ruled out that broken lariats were responsible for the observed appearance of reverse-splicing products.
We set out to characterize the reactivity of pure linear intron RNA molecules and establish their potential for transesterification and mobility. Here, we demonstrate that the linear group II intron ai5γ from the mitochondrial genome of S. cerevisiae readily reverses the second step of splicing with high yield and precision. In striking contrast, only trace amounts of reverse-splicing product were formed by lariat intron, suggesting that the covalently branched structure locks the majority of lariat molecules in a state that is incapable of performing reverse splicing. Using extensive kinetic investigations, including pH-rate analyses, we show that the reverse-splicing rate constant for the linear intron is limited by the deprotonation of the attacking nucleophile in the slightly acidic pH range. Likewise, we show that nucleophile deprotonation limits the rate constant for the second step of forward splicing and the spliced exon reopening (SER) reaction. The results have important implications for the chemical mechanism of reverse splicing and for the role of linear introns in evolutionary history.
RESULTS
Generation of linear intron with controlled 3′-end homogeneity
For mechanistic investigations of reverse splicing, intron molecules needed to be fully active and appropriately terminated with a 3′-hydroxyl group. The ai5γ intron construct employed here retained all functional domains, although Domain 4 (D4) was replaced by a hairpin.
It was also important that the population of linear intron molecules was free of broken lariat molecules. To avoid contamination by broken lariats, the linear intron was generated directly by run-off transcription, rather than isolating it from a forward-splicing reaction. A complication with the direct transcriptional approach is that T7 RNA polymerase adds one or more untemplated nucleotides to the 3′-end of most transcripts (Milligan et al. 1987). Since intron molecules with additional nucleotides at the 3′-end are expected to be incapable of reverse splicing (vide infra), we have adapted a previously reported method for the transcription of short RNAs with reduced 3′-end heterogeneity (Kao et al. 1999, 2001). The original method uses synthetic template DNA that contains C2′-methoxy (2′-OMe) groups on the penultimate or last 2 nucleotides (nt). For the transcription of the 769-nt-long ai5γ intron construct, we have generated the DNA template by PCR using a synthetic antisense primer that introduced the required 2′-OMe modifications. Analysis of the resulting transcription products revealed that 96% of the RNA molecules were correctly terminated; in contrast, only 18% of the RNA molecules that were transcribed from linearized plasmid DNA were correctly terminated. To distinguish between these two intron preparations, in this paper they will be referred to as homo-RNA (for homogeneous termini) and hetero-RNA (for heterogeneous termini), respectively.
Assaying reverse-splicing capabilities of the linear ai5γ intron
Our primary aim was to determine whether the linear intron has the capability to undergo reverse splicing. Our basic experiment involved incubation of linear intron with a short 3′-end-labeled RNA substrate (Fig. 2A). This substrate consisted of 24 nt. The first 17 nt corresponded to the terminal 17 nt of the natural 5′-exon and contained the two intron binding sites, IBS1 and IBS2. The terminal 7 nt of the substrate corresponded to the sequence of the natural 3′-exon (Fig. 2B, RS1). Effects caused by substrate binding and release were minimized by using single turnover conditions, i.e., reacting traces of labeled substrate with excess linear intron (Fedorova et al. 2002). The reactions were performed under slightly acidic conditions (pH = 6) in two common high salt buffer systems containing 100 mM MgCl2 and 500 mM KCl or (NH4)2SO4 (Daniels et al. 1996).
FIGURE 2.

Single turnover reverse-splicing assay. (A) Schematic of the reverse-splicing reaction. (B) Substrate RS1. 5′- And 3′-exonic sections, as well as intron binding sites IBS1 and IBS2, are indicated. (C) Time course (0–30 min) of the reaction between linear intron and 3′-end-labeled RNA substrate RS1. Observable species are indicated on the left side. (D) Time course (0–30 min) of the reaction between 5′-triphosphorylated linear intron and 5′-end-labeled RNA substrate RS1. (E) Time course (0–300 min) of the reaction between linear intron and 3′-end-labeled DNA substrate DS1.
Immediately after combining intron and 3′-end-labeled RNA substrate, a long labeled product evolves, which indicates that reversal of the second step of splicing has taken place (Fig. 2C). At long times, ∼40% of the substrate is converted into the putative reverse-splicing product. In addition, the appearance of a short 3′-end-labeled product indicates that the spliced exon reopening (SER) reaction also occurs. This hydrolytic cleavage of the original splice site frequently is observed in splicing assays (Jarrell et al. 1988; Daniels et al. 1996) and is related mechanistically to reversal of the second splicing step (Podar et al. 1995).
The short substrate facilitated straightforward investigation of the reverse-splicing reaction. However, the natural substrate for reverse splicing might be a relatively long mRNA. In order to determine whether the linear intron can also react with a long substrate, experiments were conducted with a 615-nt RNA substrate (RS2) in the manner described above. The splice site junction sequence was placed in the center of this substrate, resulting in a 293-nt upstream exon and a 322-nt downstream exon. The long substrate was converted into the corresponding reverse-splicing product with a somewhat lower efficiency than the short substrate. Experiments employing a twofold substrate excess over intron concentration indicated, however, that about 50% of the intron molecules are converted into reverse-splicing products (Supplemental Fig. S1).
The lariat intron is known to undergo both steps of reverse splicing (Augustin et al. 1990). Complete reverse splicing proceeds through two energetically neutral transesterifications. In the final step of this process, the 5′-exon is ligated to the intron by formation of a regular 3′,5′ linkage, while the 2′,5′ linkage at the branch point adenosine is opened. By contrast, the 2′,5′ linkage is missing in the linear intron; instead it is terminated by a 5′-monophosphate. Thus, it appears unlikely that linear intron could reverse the first step of splicing (Lehmann and Schmidt, 2003). However, if a suitable leaving group were placed at the 5′-end of the intron, e.g., a triphosphate or an m7G-cap, it might be possible to reverse the first step of splicing with a linear intron (Mörl et al. 1992). To test this hypothesis, intron constructs were capped with a 5′-triphosphate and reacted with 5′-end-labeled RS1 (Fig. 2D). However, a long product resulting from a complete reversal of splicing was not observed. The same results were obtained with the m7G-capped intron and, as expected, with 5′-monophosphorylated intron (not shown). These experiments suggest that the linear ai5γ intron is unable to reverse the first step of splicing, even if a potential leaving group is placed at the 5′-end.
To test if linear ai5γ will react with single-stranded DNA substrates, an analogous experiment was conducted using a short 3′-end-labeled DNA substrate (DS1) with the same sequence as the short RNA substrate RS1. In this experiment, neither SER nor reverse-splicing products were formed (Fig. 2E), which indicates that ai5γ cannot react with all-DNA substrates, and which agrees with previous cleavage experiments employing chimeric RNA/DNA substrates (Griffin et al. 1995).
Fidelity of the reverse-splicing reaction
We next aimed to investigate the accuracy and sequence selectivity of splice site selection. The reverse-splicing product was isolated by denaturing PAGE and the intron/3′-exon junction was sequenced by primer extension. For these experiments, we used a substrate containing an extended, 30-nt-long 3′-exon (RS3) in order to provide a primer binding site with sufficient distance from the splice junction. Primer extension revealed that the sequence of the resulting intron-exon junction boundary is an exact match for the expected 3′-splice site sequence (Fig. 3). No indication for errors in splice site selection could be detected, thereby demonstrating that the linear intron performs reverse splicing with high precision.
FIGURE 3.

Reverse-splicing precision. Reverse-splicing product was isolated and the 3′-splice site was sequenced by primer extension (right). Control experiments sequencing the splice sites of substrate RS3 (left) and of a linear ai5γ transcript with flanking exons bearing the expected splice site junction (center) were performed with the same primer.
This result is especially striking because the experiment was carried out with hetero-RNA (<20% correctly terminated RNAs) intron molecules. The absence of products containing additional nucleotides between the normal intron terminus and the 3′-exon suggests that intron molecules with additional nucleotides at the 3′-end cannot perform reverse splicing. This indicates that reverse splicing is extremely specific for the exact sequence at the downstream intron terminus.
A kinetic framework for the reverse-splicing reaction
While the previous experiments established that reverse splicing by linear intron is a robust and highly accurate reaction, kinetic analyses were required to assess the efficiency of reverse splicing and to obtain the rate constants necessary for comparative mechanistic studies. To this end, the kinetics of reverse splicing and the competing SER reaction were monitored and quantified in parallel under a variety of reaction conditions.
As a first step, it was important to evaluate the impact of SER on reaction products observed for both homo- and hetero-RNA. The product sequencing experiments described above indicate that intron molecules with additional nucleotides cannot perform reverse splicing, but they may be able to perform SER. Parallel time courses were conducted with homo- and hetero-RNA to answer this question (Fig. 4B). We found that, while both intron preparations have similar rate constants for reverse splicing and SER (Supplemental Table S1), homo-RNA produces about 4 times more reverse-splicing product, whereas hetero-RNA accumulates the free 3′-exon significantly faster. Therefore, we conclude that intron molecules with additional nucleotides are capable of performing SER, but unable to undergo reverse splicing.
FIGURE 4.

Kinetic investigations. (A) The proposed kinetic model used for data analysis. Linear intron is depicted as a straight line. Intron molecules with untemplated nucleotides at the 3′-end are indicated as a straight line with a stretch of “A.” Full and open boxes show the 5′- and 3′-exons as defined in Figure 1. (B) Kinetic traces from two single turnover experiments, one of which was conducted using homo-RNA (filled symbols, solid lines), the other one with hetero-RNA (open symbols, dashed lines). Symbols show experimental data, the lines represent least-square fits according to the model shown in (A). (C) Progress of reaction by linear intron (filled symbols, solid lines) and lariat intron (open symbols, dashed lines) with 3′-end-labeled substrate RS1. Interpolated lines connect the data points for better visibility. (D) Logarithmic rates obtained for the KCl buffer system as a function of pH. Symbols indicate experimental data, the lines represent best fits using Equation 1. Parameters obtained from the fits are shown in Table 2. (E) Logarithmic rates obtained for the (NH4)2SO4 buffer system as a function of pH.
The kinetic traces (Fig. 4B) show rapid development of reverse-splicing product. After reaching a peak, it starts to decrease, while free 3′-exon continues to form even though only trace amounts of substrate RS1 remain (Fig. 4B, see solid curves representing homo-RNA). This indicates that the initial intron–substrate complex is continuously re-formed from the reverse-splicing product by forward splicing. Since SER is irreversible, the chemical equilibrium between linear intron and reverse-splicing product eventually leads to complete depletion of substrate and reverse-splicing product.
None of the kinetic traces starts with a lag phase suggesting that substrate binding is fast compared to the chemical rates and can be excluded from the kinetic scheme. The kinetic framework derived from these observations includes three individual reactions. The kinetic parameters for these reactions are the reverse-splicing rate constant k rev, the forward-splicing rate constant k for, and the SER rate constant k SER (Fig. 4A). The resulting set of differential equations cannot be solved without major simplifications, and therefore curve fitting was done using kinetic simulation software (Kuzmic 1996). The solid lines in Figure 4B represent the best fit to the data points using the proposed kinetic framework. The fractions of correctly terminated intron molecules were experimentally determined (see Supplemental Fig. S4; see Materials and Methods) and included as fixed parameters into the model. The resulting rate constants k rev, k for, and k SER at pH 5.9 are shown in Table 1, while rate constants for other pH values are listed in Supplemental Table S1.
TABLE 1.
Rate constants of linear and lariat intron at pH 5.9 in the KCl- and (NH4)2SO4-buffer systems

Comparing linear and lariat intron reverse splicing
To compare linear and lariat intron activity in reverse splicing, we have generated lariat intron by forward splicing followed by isolation using denaturing gel electrophoresis (Dème et al. 1999). Single turnover experiments were conducted with 3′-end-labeled substrate RS1 as described above for the linear intron. The resulting gels show two different reverse-splicing products resulting from reversal of the second-splicing step and full reverse splicing. The fraction of reverse-splicing products decreases slowly after reaching a peak (Fig. 4C; Supplemental Fig S2, representative gel). An important difference is that the amplitude of reverse-splicing products is substantially lower for the lariat intron (<5%) than for the linear intron (>40%). Interestingly, the rate of SER is essentially the same for linear and lariat intron (Table 1), suggesting that it is not a general refolding problem of the lariat that causes this observation.
Since reverse and forward splicing form a chemical equilibrium, the higher product amplitude observed with the linear intron generally may be a result of perturbed forward splicing of this species. This view is supported by previous work, which has shown a dramatically slower second step forward-splicing rate constant for the linear intron compared to that of the lariat (Podar et al. 1998b; Dème et al. 1999). However, the reaction progress of the lariat intron observed here is not consistent with an extremely fast forward-splicing rate. This is apparent from the comparably late product peak and the slow decrease of this product caused by forward splicing und successive substrate hydrolysis. Instead, the reaction fits well to a model in which most (>90%) of the lariat intron molecules are unable to perform reverse splicing. Although the low reaction amplitude makes it impossible to determine accurate rate constants for the transesterifications, we can conclude from kinetic simulations that the forward- and reverse-splicing rates are in the same order of magnitude as those determined for the linear intron (Supplemental Fig. S2). The previously reported results might be different due to the variations in experimental setup, including use of chemically modified substrates, differences in exon lengths, intron to substrate ratios, and reaction temperatures.
Reaction rates are limited by the chemical step
To shed more light on the chemical mechanisms of the underlying reactions, a series of experiments was conducted at varying pH values and reaction rate constants were determined. When the logarithm of the rate constants are plotted as a function of pH, one observes linear profiles with a slope of almost 1 in the acidic pH range and a decreasing slope in the neutral and slightly alkaline pH range for all of the observed reactions. The strict pH dependence is not unexpected, since the attacking nucleophile is created by deprotonation of the terminal 3′-OH group during the transesterification reactions or a water molecule during SER. The shape of the curves resemble that of a general acid–base catalyzed reaction, which is limited by deprotonation of the requisite base and can be described by (Herschlag and Khosla 1994; Knitt and Herschlag 1996)
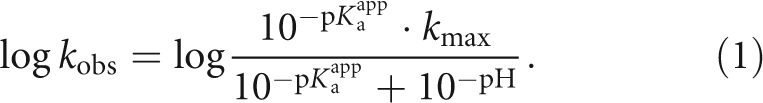 |
In this equation, k obs is the observed rate constant at a given pH value. As will be discussed below, the acidity constant in this equation is unlikely to reflect the true pK a of the attacking nucleophile and is therefore termed the apparent acidity constant pKappa. This value might represent the pH value at which an event other than chemistry becomes rate limiting, and the rate constant of this event is equivalent to the maximum rate constant for the reaction (k max).
The experimental data from each of the three observed reactions is well described by Equation 1 (Fig. 4D,E; Table 2), demonstrating that they are limited by chemistry in the slightly acidic pH range. It is striking that for all three reactions the resulting apparent pKappa values as well as the maximum reaction rate constants fall within a comparably narrow range. In the KCl-buffer system, the pKappa values are 6.7–6.9 and the maximum reaction rate constants are 0.12–0.47 min−1. Similarly, in the (NH4)2SO4-buffer system the pKappa values are 6.4–6.9 and the maximum rate constants are 0.15–0.21 min−1. The similar pKappa values and rate constants for the three reactions suggest that the same event is rate limiting in each case.
TABLE 2.
Parameters of the least-square fits using Equation 1 of the pH-dependent logarithmic rate constant shown in Figure 4, D and E

Interestingly, the lowest possible pH value at which reverse splicing was observed varied between the two investigated buffer systems; the KCl-buffer system appeared to be more robust and promoted reaction at a pH as low as 4.8, whereas the (NH4)2SO4-buffer system required a minimum pH of 5.4. The reduction in reactivity at lower pH values may result from protonation of adenine and cytosine nucleobases, which may disrupt important molecular interactions. The difference in the minimum pH requirements may indicate that KCl stabilizes the intron structure better than (NH4)2SO4.
Activity of the ΔG588, ΔA589 mutant
The reverse-splicing reaction is expected to be a stereochemical inversion of the second step of forward splicing, having the same transition state involving identical atomic interactions (Podar et al. 1995). The facile reversibility of the reaction, i.e., the observation of second step forward splicing, supports this view. To test whether the reverse-splicing reaction by linear intron is actually related to the second step of splicing, we created an intron mutant where the consecutive nucleotides G588 and A589, located in the region linking Domains D2 and D3 of ai5γ, were deleted (ai5γ-ΔG588,ΔA589). It was shown previously with a full length forward-splicing construct that this double mutant is almost unable to catalyze the second step of splicing (Mikheeva et al. 2000). The concept of microscopic reversibility requires that this mutation will also affect the true reversal of the second splicing step (Ricard 1978).
The ai5γ-ΔG588,ΔA589 mutant showed dramatically decreased reverse-splicing activity (Supplemental Fig. S3). In the KCl-buffer system, the reverse-splicing rate constant was about 250 times slower than that observed for the wild type intron and the SER rate constant was four times slower. The forward-splicing rate constant for the mutant could not be measured because it forms only traces of reverse-splicing product. Experiments in the (NH4)2SO4-buffer system showed an even more dramatic mutational effect, with reverse splicing more than 40,000 and SER 150 times slower than wild-type. These results reinforce the assumption that the observed reverse-splicing reaction is a true inversion of the second step of forward splicing. Since the two deleted nucleotides support assembly of the catalytic core (Toor et al. 2008), the less dramatic effect in the KCl buffer supports the view that KCl provides better structural stabilization than (NH4)2SO4.
DISCUSSION
Here, we show that, in contrast to prevailing opinion, the linear form of the ai5γ intron catalyzes reverse splicing efficiently and with high precision. This finding raises the possibility that reverse splicing by linear group II introns may have played a significant role in certain forms of intron mobility and lateral gene transfer. It is now clear that certain types of group II introns splice only through the hydrolytic pathway, exclusively releasing linear intron (Granlund et al. 2001; Vogel and Börner 2002). Indeed, even group II introns that splice predominantly through a branching pathway (such as ai5γ) still can splice efficiently through hydrolysis in vivo (Podar et al. 1998a). Given the prevalence of linear group II introns, their innate potential for mobility and transposition has become an important question. While we have not yet shown that linear group II introns can migrate into new genomic locations, we have shown that linear group II introns can readily undergo the critical first step of mobility by catalyzing efficient reverse splicing into complementary target molecules.
In striking contrast with the efficiency of the linear intron, reverse splicing by lariat intron is surprisingly unfavorable. Importantly, SER occurs with the same rate constant for both linear and lariat intron, indicating that the lariat active-site is intact and fully competent for chemical catalysis. Although it was not possible to precisely measure forward- and reverse-splicing rates of the lariat intron, we found that neither a fast forward-splicing rate nor a slow reverse-splicing rate can account for the paucity of reverse-splicing products. Instead, our data indicate that linear and lariat introns reverse splice with similar rates, but the majority of lariat intron molecules are locked in a conformation that is intrinsically deficient in reverse splicing. This finding suggests that maturase proteins might be required to activate the intron lariats, transforming them into a state that readily participates in both steps of reverse splicing.
Reverse splicing by linear intron is not only biologically interesting, it is also useful. For example, the reverse-splicing reaction enabled us to investigate three reactions simultaneously: the second step of splicing, the reversal of this reaction, and SER. Despite the substantive differences between these reactions, all three of them are strictly limited by chemistry in the slightly acidic pH range. Under these conditions, the reverse-splicing reaction can be used for mechanistic studies on the second step of splicing and can reveal intron strategies for transition-state stabilization. It will now be possible to further investigate the role of metal ions (Gordon et al. 2007) and catalytic nucleobases during catalysis. A second useful attribute of the reverse-splicing reaction is that it covalently links the intron with an RNA substrate. The resulting products can be isolated through the incorporation of affinity tags, or through amplification. As in early studies of directed molecular evolution on group I introns, the reverse-splicing reaction can be used as a selection step for chemogenetic analysis of intron structural features using methods such as NAIM and NAIS (Boudvillain and Pyle 1998; Waldsich and Pyle 2007), or for the development of introns with new properties (Joyce 2007).
A rate limiting event at neutral pH
The pH-rate profiles indicate that deprotonation of the attacking nucleophile is rate limiting at pH values below the pKappa value. The pKappa values mark the pH values at which the rates lose strict pH dependency. This behavior could be explained by one of the following scenarios (Knitt and Herschlag 1996): (1) The pKappa reflects the actual pK a of the attacking nucleophile. (2) There is a change in the rate-limiting step at pH ∼7.0
The first scenario assumes that the attacking nucleophile becomes almost completely deprotonated once the pH exceeds the pK a of the nucleophile and the observed rate will thus approach the theoretical maximum rate constant, k max. In the present case, it is unlikely that the pKappa represents the actual pK a of the nucleophile. The pK a value of a free terminal 3′-OH group is around 12.8 (Johnson et al. 1988; Åström et al. 2004). It was shown recently that a Mg2+ ion binds directly to the attacking nucleophile in group II introns (Sontheimer et al. 1999; Gordon et al. 2000, 2007) which would be expected to lower the pK a of this group by up to three units (Karbstein et al. 2002) to 9.8. Even if additional interactions within the intron would contribute to further acidification of the nucleophile, it is unlikely to reach the observed pKappa of <7. A second consideration is that the pKappa observed for SER is essentially the same as that for the transesterifications, although in this case the nucleophile is H2O. Since the pK a of H2O is 15.7, which is almost three units higher than that of the terminal 3′-OH group, a similar difference would be expected for the observed pKappa values. Given these issues, it is very unlikely that the first scenario applies.
In the second scenario, the plateau in the pH-rate profile is caused by a change in the rate-limiting step near neutral pH. At higher pH values, a deprotonation event might disrupt important structural features of the ribozyme. In this case, all reactions of the ribozyme would be affected at the same pH value, resulting in similar pKappa values. Indeed, all the pKappa values fall within the narrow range between 6.4 and 6.9, which is consistent with this type of model. Within the folded core of certain ribozymes, it has recently been shown that important nucleobases can undergo shifts in acidity, such that their pK a values fall in the neutral range (Ryder et al. 2001; Oyelere et al. 2002; Wilson et al. 2007). It is therefore possible that the pH-rate profiles are influenced by a pK a shifted nucleobase, which is important for the structural integrity of the molecule.
Alternatively, a pH-independent event that is intrinsically important for reaction could become rate limiting when the chemical rate constant exceeds a certain value. If this pH-independent event is the same for all observed reactions, their maximum apparent rate constants would be similar. This might be applicable here, since all maximum reaction rate constants fall into the comparably small range of 0.12–0.47 min−1 in the KCl buffer and 0.15–0.21 min−1 in the (NH4)2SO4 buffer. Potentially rate-limiting events include substrate binding, product release, or a structural rearrangement associated with the chemical step. The reactions were conducted under single-turnover, pre-steady-state conditions, so they will not be affected by the efficiency of substrate binding, or by events that occur after the chemical reaction. Under such conditions, however, it remains possible that the E–S complex undergoes a rate-limiting conformational change prior to, or concomitant with the chemical reaction.
Biological significance
In addition to the implications for mobility by linear introns (vide supra), it is significant that reverse splicing by linear intron is fast, with a rate constant that is comparable to that of other biologically relevant ribozymes (Table 2). The short half-life of RNA in vivo requires that ribozyme reactions are fast enough to occur in a biologically significant context. Here, we show that this is possible for reverse splicing by linear intron, provided that the rate constants remain comparable in a physiological environment. If linear intron undergoes reverse splicing with high efficiency in vivo, it would be expected to influence the concentration of spliced mRNA, leading to downregulation of the encoded protein. In addition, reverse splicing might extend the half-life of excised introns by slowing down exonuclease digestion from the 3′-end.
It is highly significant that linear intron was not observed to undergo both steps of reverse splicing, even when a cleavable leaving group was introduced at the 5′-terminus. This suggests that any mobility reaction involving linear intron would be restricted to mechanisms that involve a single step of target cleavage with attachment of the intron to downstream sequences. Since mobility involves TPRT reactions that can occur on partially incorporated intron molecules, it has been hypothesized that DNA repair processes might complete the process of cDNA integration (Zimmerly et al. 1995). However, it is also possible that the intron recruits proteins in vivo to complete the process of reverse splicing. Indeed, forward splicing of ai5γ under physiological conditions requires proteins and the mechanism by which these proteins act remains unknown to date (Huang et al. 2005; Solem et al. 2006; Fedorova and Zingler 2007). The ai5γ intron is not known to be a mobile genetic element, but it is important to keep in mind that even a single incident can introduce a retroelement permanently into a genome. Candidate proteins that could have mobilized linear ai5γ molecules are those associated with mobility of the group II introns ai1 and ai2, and which are located in the same pre-mRNA as ai5γ. The strikingly high efficiency and precision of the observed reverse splicing make such a scenario appear likely.
MATERIALS AND METHODS
All substrates, primers, DNAzymes and transcripts were gel purified prior to further use and stored in 10 mM MOPS pH 6.0 and 1 mM EDTA. RNA 3′-end labeling was carried out using [32P]pCp and T4 RNA ligase as described (England and Uhlenbeck 1978). 5′-end labeling was performed using [32P]γ-ATP and polynucleotide kinase (NEB) according to the manufacturers protocol. All labeled nucleic acids were gel purified prior to further use.
Preparation of intron RNA
Linear ai5γ intron was generated by run-off transcription using T7 polymerase and EcoR V digested plasmid pGD45. To reduce addition of untemplated nucleotides to the 3′-end of the transcripts, we have generated template DNAs with 2′-OMe groups on the penultimate or the last 2 nt (Kao et al. 1999, 2001). The modified transcription templates were generated by PCR using sense primer SB (5′-GAATTCTAATACGACTCACTATAGAGCGGTCTGAAAG-3′) and the 2′-OMe modified antisense primers AS2B-MR1 (5′-ATCCCGATAGGTAGACCTTTACAAGTTTTC-3′) or AS2B-MR2 (5′-ATCCCGATAGGTAGACCTTTACAAGTTTTC-3′), respectively (2′-OMe modified nucleotides are printed in bold). The template for the transcription of the ai5γ-ΔG588,ΔA589 mutant was generated by PCR using primers SB and AS2B-MR1 and plasmid pGD52. 5′-Monophosphorylated intron was prepared by dephosphorylation of gel purified intron with alkaline phosphatase (Roche) followed by monophosphorylation with polynucleotide kinase (NEB), both according to the manufacturers protocol. 5′-Terminal m7G-capped intron was prepared using the ScriptCap m7G Capping System from Epicentre.
Analysis of 3′-end heterogeneity
Intron RNA was transcribed from the two 2′-OMe modified PCR templates and, for comparison, from two batches of linearized plasmid DNA. The four transcripts were 3′-end labeled and cleaved with two different site-directed DNAzymes in separate experiments (Pyle et al. 2000; Chu et al. 2001). DNAzyme 1 (5′-TCCCGATAGGGGCTAGCTACAACGAAGACCTTTACAAG-3′) and DNAzyme 2 (5′-AGACCTTTACAAGGGCTAGCTACAACGATTTCCCCC-3′) were designed to cleave 11 and 25 nt upstream of the 3′-end of the intron RNA, respectively. The resulting short RNA fragments were resolved on a 20% denaturing polyacrylamide gel (Supplemental Fig. S4) and the fractions of correctly sized molecules and molecules having up to an additional 5 nt were quantified; fractions of longer products were negligible. This analysis revealed that only 18% of the RNA molecules that were transcribed from linearized plasmid DNA were correctly terminated (hetero-RNA), while 96% of the RNAs transcribed from either of the two 2′-OMe modified templates were correctly terminated (homo-RNA). All experiments were conducted using homo-RNA, unless otherwise noted.
Preparation of nonintronic oligonucleotides
RNA substrates RS1 (5′-CGUGGUGGGACAUUUUC/ACUAUGU-3′) and RS3 (5′-CGUGGUGGGACAUUUUC/ACUAUGUAUUAUCAAUGGGUGCUAUUUUCU-3′), as well as the 2′-OMe modified primers AS2B-MR1 and AS2B-MR2 were synthesized and deprotected as described (Wincott et al. 1995). DNA substrate DS1, primers SB and 3ex30reverse, DNAzyme 1 and DNAzyme 2 were purchased from Invitrogen. Substrate RS2 was transcribed from Hind III digested plasmid pMR8 using T7 RNA polymerase.
Reverse-splicing assays and time courses
Two different buffer systems were used for reverse-splicing experiments. The KCl buffer system consisted of 40 mM MOPS-KOH or MES-KOH at varying pH values, 100 mM MgCl2 and 500 mM KCl; the (NH4)2SO4 buffer contained 40 mM MOPS-NH3 or MES-NH3, 100 mM MgCl2, and 500 mM (NH4)2SO4. The pH values of the MOPS or MES buffers were first adjusted with KOH or NH4OH at room temperature. For the pH-dependent rate determinations the buffers were diluted, mixed with salts and heated to 42°C to simulate the experimental conditions; the pH values of these mixtures were measured with a pH electrode calibrated for this temperature.
Single turnover experiments were carried out using 100 nM Intron and traces (2 nM) of the respective 3′- or 5′-end-labeled substrate at 42°C. Intron and substrate were separately denatured in the respective pH buffer for 1 min at 95°C and cooled 1 min to 42°C. The salts were added, intron and substrate were allowed to fold for 10 min before they were combined. Aliquots were taken after increasing reaction times, immediately mixed with four volumes of quench buffer (75% formamide and 50 mM EDTA). Products were separated on biphasic polyacrylamide gels (5% and 20%) and quantified by their radioactive counts. The reaction of linear ai5γ with the long RNA substrate RS2 was carried out in the KCl buffer in the same way as described for the single turnover experiments, except that 200 nM (195 nM cold and 5 nM 3′-end-labeled) substrate and 100 nM intron were used.
Calculation of reaction rates
All reaction rates were calculated using the kinetic simulation software package Dynafit 3 from BioKin, Ltd. (Kuzmic 1996). The simulations were based on the model shown in Figure 4A. A sample script and experimental values for the simulation of the reaction of homo-RNA shown in Figure 4B can be found in the Supplemental Material.
Determination of the reverse-splicing precision
Linear intron (5 μM) transcribed from EcoR V digested plasmid pGD45 was reacted with the same concentration of trace 3′-end-labeled RNA substrate RS3 in KCl/MES buffer (pH 5.5) at 42°C as described above. Products were separated by denaturing PAGE. The reverse-splicing product was visualized with an Instant Imager, excised and eluted from the gel. The isolated reverse-splicing product was annealed to 5′-end-labeled primer 3ex30reverse and reverse transcribed in the presence of the four respective ddNTPs with AMV-RT (Roche) following the manufacturers protocol.
SUPPLEMENTAL MATERIAL
Supplemental material can be found at http://www.rnajournal.org.
ACKNOWLEDGMENTS
M.R. thanks Amanda Solem, Nora Zingler, and Olga Fedorova for careful proofreading of the manuscript.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.1392009.
REFERENCES
- Åström H., Limén E., Strömberg R. Acidity of secondary hydroxyls in ATP and adenosine analogues and the question of a 2′,3′-hydrogen bond in ribonucleosides. J. Am. Chem. Soc. 2004;126:14710–14711. doi: 10.1021/ja0477468. [DOI] [PubMed] [Google Scholar]
- Augustin S., Müller M.W., Schweyen R.J. Reverse self-splicing of group II intron RNAs in vitro. Nature. 1990;343:383–386. doi: 10.1038/343383a0. [DOI] [PubMed] [Google Scholar]
- Boudvillain M., Pyle A.M. Defining functional groups, core structural features and inter-domain tertiary contacts essential for group II intron self-splicing: A NAIM analysis. EMBO J. 1998;17:7091–7104. doi: 10.1093/emboj/17.23.7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu V.T., Adamidi C., Liu Q., Perlman P.S., Pyle A.M. Control of branch-site choice by a group II intron. EMBO J. 2001;20:6866–6876. doi: 10.1093/emboj/20.23.6866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels D.L., Michels W.J., Jr, Pyle A.M. Two competing pathways for self-splicing by group II introns: A quantitative analysis of in vitro reaction rates and products. J. Mol. Biol. 1996;256:31–49. doi: 10.1006/jmbi.1996.0066. [DOI] [PubMed] [Google Scholar]
- Dème E., Nolte A., Jacquier A. Unexpected metal ion requirements specific for catalysis of the branching reaction in a group II intron. Biochemistry. 1999;38:3157–3167. doi: 10.1021/bi982462j. [DOI] [PubMed] [Google Scholar]
- England T.E., Uhlenbeck O.C. 3′-Terminal labelling of RNA with T4 RNA ligase. Nature. 1978;275:560–561. doi: 10.1038/275560a0. [DOI] [PubMed] [Google Scholar]
- Fedorova O., Zingler N. Group II introns: Structure, folding, and splicing mechanism. Biol. Chem. 2007;388:665–678. doi: 10.1515/BC.2007.090. [DOI] [PubMed] [Google Scholar]
- Fedorova O., Su L.J., Pyle A.M. Group II introns: Highly specific endonucleases with modular structures and diverse catalytic functions. Methods. 2002;28:323–335. doi: 10.1016/s1046-2023(02)00239-6. [DOI] [PubMed] [Google Scholar]
- Gordon P.M., Sontheimer E.J., Piccirilli J.A. Metal ion catalysis during the exon-ligation step of nuclear pre-mRNA splicing: Extending the parallels between the spliceosome and group II introns. RNA. 2000;6:199–205. doi: 10.1017/s1355838200992069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon P.M., Fong R., Piccirilli J.A. A second divalent metal ion in the group II intron reaction center. Chem. Biol. 2007;14:607–612. doi: 10.1016/j.chembiol.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Granlund M., Michel F., Norgren M. Mutually exclusive distribution of IS1548 and GBSi1, an active group II intron identified in human isolates of group B streptococci. J. Bacteriol. 2001;183:2560–2569. doi: 10.1128/JB.183.8.2560-2569.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin E.A., Jr, Qin Z., Michels W.J., Jr, Pyle A.M. Group II intron ribozymes that cleave DNA and RNA linkages with similar efficiency, and lack contacts with substrate 2′-hydroxyl groups. Chem. Biol. 1995;2:761–770. doi: 10.1016/1074-5521(95)90104-3. [DOI] [PubMed] [Google Scholar]
- Herschlag D., Khosla M. Comparison of pH dependencies of the Tetrahymena ribozyme reactions with RNA 2′-substituted and phosphorothioate substrates reveals a rate-limiting conformational step. Biochemistry. 1994;33:5291–5297. doi: 10.1021/bi00183a036. [DOI] [PubMed] [Google Scholar]
- Huang H.R., Rowe C.E., Mohr S., Jiang Y., Lambowitz A.M., Perlman P.S. The splicing of yeast mitochondrial group I and group II introns requires a DEAD-box protein with RNA chaperone function. Proc. Natl. Acad. Sci. 2005;102:163–168. doi: 10.1073/pnas.0407896101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrell K.A., Dietrich R.C., Perlman P.S. Group II intron domain 5 facilitates a trans-splicing reaction. Mol. Cell. Biol. 1988;8:2361–2366. doi: 10.1128/mcb.8.6.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R.W., Marschner T.M., Oppenheimer N.J. Pyridine nucleotide chemistry. A new mechanism for the hydroxide-catalyzed hydrolysis of the nicotinamide-glycosyl bond. J. Am. Chem. Soc. 1988;110:2257–2263. [Google Scholar]
- Joyce G.F. Forty years of in vitro evolution. Angew. Chem. Int. Ed. 2007;46:6420–6436. doi: 10.1002/anie.200701369. [DOI] [PubMed] [Google Scholar]
- Kao C., Zheng M., Rüdisser S. A simple and efficient method to reduce nontemplated nucleotide addition at the 3 terminus of RNAs transcribed by T7 RNA polymerase. RNA. 1999;5:1268–1272. doi: 10.1017/s1355838299991033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao C., Rüdisser S., Zheng M. A simple and efficient method to transcribe RNAs with reduced 3′ heterogeneity. Methods. 2001;23:201–205. doi: 10.1006/meth.2000.1131. [DOI] [PubMed] [Google Scholar]
- Karbstein K., Carroll K.S., Herschlag D. Probing the Tetrahymena group I ribozyme reaction in both directions. Biochemistry. 2002;41:11171–11183. doi: 10.1021/bi0202631. [DOI] [PubMed] [Google Scholar]
- Knitt D.S., Herschlag D. pH dependencies of the Tetrahymena ribozyme reveal an unconventional origin of an apparent pK a . Biochemistry. 1996;35:1560–1570. doi: 10.1021/bi9521147. [DOI] [PubMed] [Google Scholar]
- Koch J.L., Boulanger S.C., Dib-Hajj S.D., Hebbar S.K., Perlman P.S. Group II introns deleted for multiple substructures retain self-splicing activity. Mol. Cell. Biol. 1992;12:1950–1958. doi: 10.1128/mcb.12.5.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmic P. Program DYNAFIT for the analysis of enzyme kinetic data: Application to HIV proteinase. Anal. Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- Lehmann K., Schmidt U. Group II introns: Structure and catalytic versatility of large natural ribozymes. Crit. Rev. Biochem. Mol. Biol. 2003;38:249–303. doi: 10.1080/713609236. [DOI] [PubMed] [Google Scholar]
- Mikheeva S., Murray H.L., Zhou H., Turczyk B.M., Jarrell K.A. Deletion of a conserved dinucleotide inhibits the second step of group II intron splicing. RNA. 2000;6:1509–1515. doi: 10.1017/s1355838200000972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan J.F., Groebe D.R., Witherell G.W., Uhlenbeck O.C. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987;15:8783–8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr S., Matsuura M., Perlman P.S., Lambowitz A.M. A DEAD-box protein alone promotes group II intron splicing and reverse splicing by acting as an RNA chaperone. Proc. Natl. Acad. Sci. 2006;103:3569–3574. doi: 10.1073/pnas.0600332103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mörl M., Schmelzer C. Integration of group II intron bI1 into a foreign RNA by reversal of the self-splicing reaction in vitro. Cell. 1990;60:629–636. doi: 10.1016/0092-8674(90)90666-3. [DOI] [PubMed] [Google Scholar]
- Mörl M., Niemer I., Schmelzer C. New reactions catalyzed by a group II intron ribozyme with RNA and DNA substrates. Cell. 1992;70:803–810. doi: 10.1016/0092-8674(92)90313-2. [DOI] [PubMed] [Google Scholar]
- Oyelere A.K., Kardon J.R., Strobel S.A. pK a perturbation in genomic Hepatitis Delta Virus ribozyme catalysis evidenced by nucleotide analog interference mapping. Biochemistry. 2002;41:3667–3675. doi: 10.1021/bi011816v. [DOI] [PubMed] [Google Scholar]
- Podar M., Perlman P.S., Padgett R.A. Stereochemical selectivity of group II intron splicing, reverse splicing, and hydrolysis reactions. Mol. Cell. Biol. 1995;15:4466–4478. doi: 10.1128/mcb.15.8.4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podar M., Chu V.T., Pyle A.M., Perlman P.S. Group II intron splicing in vivo by first-step hydrolysis. Nature. 1998a;391:915–918. doi: 10.1038/36142. [DOI] [PubMed] [Google Scholar]
- Podar M., Perlman P.S., Padgett R.A. The two steps of group II intron self-splicing are mechanistically distinguishable. RNA. 1998b;4:890–900. doi: 10.1017/s1355838298971643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle A.M. Group II introns: Catalysts for splicing, genomic change and evolution. In: Lilley D.M.J., Eckstein F., editors. Ribozymes and RNA catalysis. RCS Publishing; Cambridge, U.K: 2008. pp. 201–228. [Google Scholar]
- Pyle A.M., Lambowitz A.M. Group II introns: Ribozymes that splice RNA and invade DNA. In: Gesteland R.F., et al., editors. The RNA world. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2006. pp. 469–505. [Google Scholar]
- Pyle A.M., Chu V.T., Jankowsky E., Boudvillain M. Using DNAzymes to cut, process, and map RNA molecules for structural studies or modification. Methods Enzymol. 2000;317:140–146. doi: 10.1016/s0076-6879(00)17012-0. [DOI] [PubMed] [Google Scholar]
- Ricard J. Generalized microscopic reversibility, kinetic co-operativity of enzymes and evolution. Biochem. J. 1978;175:779–791. doi: 10.1042/bj1750779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryder S.P., Oyelere A.K., Padilla J.L., Klostermeier D., Millar D.P., Strobel S.A. Investigation of adenosine base ionization in the hairpin ribozyme by nucleotide analog interference mapping. RNA. 2001;7:1454–1463. [PMC free article] [PubMed] [Google Scholar]
- Solem A., Zingler N., Pyle A.M. A DEAD protein that activates intron self-splicing without unwinding RNA. Mol. Cell. 2006;24:611–617. doi: 10.1016/j.molcel.2006.10.032. [DOI] [PubMed] [Google Scholar]
- Sontheimer E.J., Gordon P.M., Piccirilli J.A. Metal ion catalysis during group II intron self-splicing: Parallels with the spliceosome. Genes & Dev. 1999;13:1729–1741. doi: 10.1101/gad.13.13.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su L.J., Waldsich C., Pyle A.M. An obligate intermediate along the slow folding pathway of a group II intron ribozyme. Nucleic Acids Res. 2005;33:6674–6687. doi: 10.1093/nar/gki973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toor N., Keating K.S., Taylor S.D., Pyle A.M. Crystal structure of a self-spliced group II intron. Science. 2008;320:77–82. doi: 10.1126/science.1153803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallès Y., Halanych K.M., Boore J.L. Group II introns break new boundaries: Presence in a bilaterian's genome. PLoS One. 2008;3:e1488. doi: 10.1371/journal.pone.0001477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel J., Börner T. Lariat formation and a hydrolytic pathway in plant chloroplast group II intron splicing. EMBO J. 2002;21:3794–3803. doi: 10.1093/emboj/cdf359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldsich C., Pyle A.M. A folding control element for tertiary collapse of a group II intron ribozyme. Nat. Struct. Mol. Biol. 2007;14:37–44. doi: 10.1038/nsmb1181. [DOI] [PubMed] [Google Scholar]
- Wilson T.J., McLeod A.C., Lilley D.M. A guanine nucleobase important for catalysis by the VS ribozyme. EMBO J. 2007;26:2489–2500. doi: 10.1038/sj.emboj.7601698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wincott F., DiRenzo A., Shaffer C., Grimm S., Tracz D., Workman C., Sweedler D., Gonzalez C., Scaringe S., Usman N. Synthesis, deprotection, analysis and purification of RNA and ribozymes. Nucleic Acids Res. 1995;23:2677–2684. doi: 10.1093/nar/23.14.2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerly S., Guo H., Eskes R., Yang J., Perlman P.S., Lambowitz A.M. A group II intron RNA is a catalytic component of a DNA endonuclease involved in intron mobility. Cell. 1995;83:529–538. doi: 10.1016/0092-8674(95)90092-6. [DOI] [PubMed] [Google Scholar]