

Abstract
During early development, γ-aminobutyric acid (GABA) depolarizes and excites neurons, contrary to its typical function in the mature nervous system. As a result, developing networks are hyperexcitable and experience a spontaneous network activity that is important for several aspects of development. GABA is depolarizing because chloride is accumulated beyond its passive distribution in these developing cells. Identifying all of the transporters that accumulate chloride in immature neurons has been elusive and it is unknown whether chloride levels are different at synaptic and extrasynaptic locations. We have therefore assessed intracellular chloride levels specifically at synaptic locations in embryonic motoneurons by measuring the GABAergic reversal potential (EGABA) for GABAA miniature postsynaptic currents. When whole cell patch solutions contained 17–52 mM chloride, we found that synaptic EGABA was around −30 mV. Because of the low HCO3− permeability of the GABAA receptor, this value of EGABA corresponds to approximately 50 mM intracellular chloride. It is likely that synaptic chloride is maintained at levels higher than the patch solution by chloride accumulators. We show that the Na+-K+-2Cl− cotransporter, NKCC1, is clearly involved in the accumulation of chloride in motoneurons because blocking this transporter hyperpolarized EGABA and reduced nerve potentials evoked by local application of a GABAA agonist. However, chloride accumulation following NKCC1 block was still clearly present. We find physiological evidence of chloride accumulation that is dependent on HCO3− and sensitive to an anion exchanger blocker. These results suggest that the anion exchanger, AE3, is also likely to contribute to chloride accumulation in embryonic motoneurons.
INTRODUCTION
A great deal of interest has recently been focused on the observation that neurons are depolarized by γ-aminobutyric acid (GABA) in early development in several different parts of the nervous system (Ben-Ari et al. 2007). In this developmental period, the reversal potential for GABAA receptor currents (referred to as EGABA) is more depolarized than the resting membrane potential because chloride, the main carrier of the GABAA current, is accumulated in immature neurons. The depolarizing nature of GABA is important in driving the spontaneous network activity (SNA) that is observed in virtually all developing circuits (Ben-Ari et al. 2007; O'Donovan 1999). In the spinal cord, episodes of SNA recruit the majority of neurons, drive embryonic movements, and are important in muscle, joint, and motoneuron development (Ben-Ari et al. 2007; Casavant et al. 2004; Gonzalez-Islas and Wenner 2006; Hanson and Landmesser 2004, 2006; O'Donovan 1999).
The modulation of intracellular chloride appears to be critical for the normal expression of SNA (Chub and O'Donovan 1998, 2001; Chub et al. 2006; Fedirchuk et al. 1999; Marchetti et al. 2005). During an episode of SNA, intracellular chloride is depleted as it passes out of the cell through activated GABAA receptor channels, thus weakening the driving force for GABAergic synapses, leaving the cord relatively less excitable. Then in the quiescent interepisode interval, chloride is reaccumulated by transporters that are thought to be critical for strengthening excitatory GABAergic synapses and generating the next episode (Marchetti et al. 2005).
Several studies demonstrate that the Na+-K+-2Cl− cotransporter, NKCC1, accumulates chloride in developing neurons (Achilles et al. 2007; Chub et al. 2006; Delpy et al. 2008;Kakazu et al. 1999; Rocha-Gonzalez et al. 2008; Rohrbough and Spitzer 1996; Sipila et al. 2006; Yamada et al. 2004). However, most of these studies block NKCC1 and still observe some chloride accumulation and it appears that in some developing neurons NKCC1 is not involved at all (Balakrishnan et al. 2003; Zhang et al. 2007). In a recent study significant chloride accumulation was observed in embryonic spinal motoneurons after blocking both NKCC1 and the chloride extruder KCC2 with furosemide (Delpy et al. 2008). The molecule(s) responsible for NKCC1-independent chloride accumulation remains unknown. In the current study we test the role of NKCC1 and other transporters in accumulating chloride in motoneurons in the chick embryo. We assess chloride levels at GABAergic synapses, which are likely to exist out in the dendrites. This allows us to determine the chloride transporters that are most relevant to setting the driving force for GABAergic inputs.
In the present report we assess EGABA at synaptic sites using miniature postsynaptic currents (mPSCs) in chick embryo motoneurons. Whole cell recordings of GABAergic mPSCs were obtained at different voltage steps and current–voltage (I–V) plots were constructed, providing conductance measurements, as well as EGABA for the synaptic currents. Chloride accumulators appear to define the synaptic chloride concentrations out in the dendrites. We show that although NKCC1 contributes to chloride influx in embryonic motoneurons, chloride accumulation still occurs after NKCC1 blockade and is likely mediated by the anion exchanger, AE3.
METHODS
Dissection
White Leghorn chicken eggs were incubated in a circulated air incubator (GQF Manufacturing) at 38°C. Electrophysiological experiments were performed on isolated spinal cords of embryonic day 10–11 (E10–E11), stages 36–37 (Hamburger and Hamilton 1951) chick embryos. SNA has been extensively studied at these stages. The lumbosacral spinal cord region, with attached femorotibialis (Fem) or adductor (Add) nerves, was dissected under cooled (15°C) oxygenated Tyrode's solution containing (in mM): 139 NaCl, 12 d-glucose, 17 NaHCO3, 3 KCl, 1 MgCl2, and 3 CaCl2. Tyrode's solution was constantly bubbled with a mixture of 95% O2-5% CO2 to maintain a pH around 7.3. After dissection, the cord was allowed to recover overnight in Tyrode's at 18°C. The cord was then transferred to a recording chamber, continuously perfused with oxygenated Tyrode's solution that was slowly heated to recording temperature (28°C). The cord was allowed to establish stable SNA (constant frequency of bursts) before starting the experiment.
Electrophysiology: mPSCs
Patch-clamp recordings were used to acquire mPSCs in the whole cell mode. Miniature postsynaptic currents were recorded from motoneurons antidromically identified as Fem or Add motoneurons by the stimulation of their particular muscle nerves via tight-fitting suction electrodes connected to high-gain differential amplifiers (A-M Systems). Tight seals (>2 GΩ before breaking into whole cell mode) were obtained using patch electrodes pulled from thin-walled glass in two stages using a P-87 Flaming/Brown micropipette puller (Sutter Instruments) and having resistances between 5 and 10 MΩ. Series resistance during recording varied from 10 to 20 MΩ among different motoneurons and was not compensated. Recordings were terminated whenever significant increases in input resistance (>20%) occurred. Extracellular recording solution for mPSCs, unless otherwise declared, contained the following (in mM): NaCl, 139; KCl, 5; NaHCO3, 17; CaCl2, 3; MgCl2,1; d-glucose, 12; tetraethylammonium (TEA), 30; CsCl, 5; tetrodotoxin (TTX), 0.001; pH was adjusted to 7.3 with NaOH. 6-Cyano-7-nitroquinoxaline-2,3-dione (CNQX, 10 μM) and 2-amino-5-phosphonopentanoic acid (AP-5, 50 μM) were also added to the bath to isolate GABAergic mPSCs (Gonzalez-Islas and Wenner 2006). It was not necessary to block glycinergic mPSCs because they are not observed at these stages in embryonic chick motoneurons (Gonzalez-Islas and Wenner 2006). Different extracellular solutions (low Na+: 17 mM; low Cl−: 53 mM; 0 HCO3−) were obtained by substituting appropriate concentrations of choline-Cl, choline-HCO3, and Na-gluconate. Solutions were constantly bubbled with a mixture of 95% O2-5% CO2 to maintain a pH around 7.3, except for the 0 mM HCO3− solution, which contained 10 mM HEPES and was bubbled throughout the experiment with 100% O2.
To record mPSCs, throughout the study we used five different pipette solution concentrations of chloride that are referred to as 17, 32, 52, 77, and 157 mM Cl− patch solutions. Chloride concentration was varied by substituting NaCl and Na-gluconate, and KCl and K-gluconate, and are shown in Table 1. pH in all cases was adjusted to 7.2 with KOH. The junction potentials were determined for each solution (Table 1) and were corrected on-line. To block calcium-, sodium-, and potassium-voltage gated currents we added verapamil (0.1 mM), N-(2,6-dimethylphenyl carbamoylmethyl)triethylammonium bromide (QX-314, 10 mM), TEA (10 mM), and CsCl (5 mM) to the normal pipette solution, respectively. Motoneurons were antidromically identified before QX-314 completely blocked action potentials. Once a stable motoneuron recording was obtained, we added TTX to prevent episodes of SNA from occurring and therefore also eliminate the episode-induced modulation of GABAergic mPSC amplitude. After 10–20 min when mPSC amplitudes become stable, a voltage ramp was performed to establish that voltage-activated currents were blocked (Fig. 1A). Voltage steps were then given in 10 mV increments, usually from −80 to +50 mV, although in some cases we used 20 mV steps and/or skipped voltages around the reversal potential (see following text). Each step was held for 2 min while mPSCs were acquired; however, in certain cases mPSC frequency was high and it was not necessary to record the full 2 min to collect sufficient numbers of mPSCs. Average amplitude of GABAergic mPSCs at each step was then plotted against voltage (ImPSC–V plot). In some cases amplitudes near the reversal potential were slightly larger than predicted by our fitted line (average mPSCs were likely biased toward larger values because smaller mPSCs fall into the noise; average mPSC amplitudes of <6 pA were typically off the fitted line and were excluded). Generating mPSC I–V plots using whole cell recording should provide an accurate assessment of EGABA. Whole cell electrodes have low access resistance and recordings of embryonic motoneurons at this stage appear to provide a very good space clamp. As shown previously, mPSC amplitude and decay are not correlated as would be expected if we had a poor space clamp (Gonzalez-Islas and Wenner 2006); furthermore, we never saw inward and outward currents in the same voltage step, consistent with the idea that the entire length of the dendrite is clamped at the same voltage. Finally, at moderate and high intracellular patch chloride concentrations, the measured EGABA matched that predicted by the Nernst equation.
TABLE 1.
Ionic concentrations of pipette solutions (in mM)
| Solution | Cl− | Na+ | K+ | Gluconate− | Osmolarity, mOsm | Junction Potential, mV |
|---|---|---|---|---|---|---|
| “17” | 17.2 | 5.1 | 130 | 135 | 299 | −17 |
| “32” | 32 | 5.1 | 130 | 120 | 298 | −12 |
| “52” | 52.2 | 5.1 | 130 | 100 | 281 | −10 |
| “77” | 77.2 | 5.1 | 130 | 75 | 278 | −9 |
| “157” | 157.2 | 10.0 | 130 | 0 | 286 | −5 |
All solutions also included (in mM): MgCl2 (1), CaCl2 (0.1), HEPES (10), ATP-Na (1), GTP-Mg (0.1), BAPTA (10); and voltage-gated channel blockers (in mM): verapamil (0.1), QX-314 (10), TEA-Cl (10), and CsCl (5). Junction potential was adjusted on-line.
FIG. 1.

Current–voltage (I–V) plots of (GABAA) receptor-mediated miniature postsynaptic currents (mPSCs). A: voltage-clamp recording of a voltage ramp from −90 to 50 mV, at 70 mV/s (52 mM chloride in patch solution). B: mPSCs were then recorded for 2 min at voltage steps of 10-mV increments (only 20-mV steps shown). C: the average amplitude of GABAergic mPSCs at each step was then plotted against the step voltage (ImPSC–V plot). Error bars represent SE.
Whole cell currents were acquired using an EPC 8 amplifier (HEKA Elektronik), controlled by PatchMaster 2.03 software (HEKA Elektronik) via an LIH 1600 interface (HEKA Elektronik). Currents were filtered on-line at 5 kHz, digitized at 10 kHz, and analyzed using Minianalysis software (Synaptosoft). Average mPSC amplitudes were fitted to a linear function with Kaleidagraph plotting software (Synergy Software) to obtain EGABA and conductance. Theoretical EGABA was calculated using the Nernst equation for Cl−.
Verapamil was purchased from Calbiochem; TTX, AP-5, and CNQX, were purchased from Tocris Cookson; CsCl was purchased from Fisher Scientific; 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) was purchased from Fluka; HEPES from Acros Organics. All other chemicals and drugs were purchased from Sigma–Aldrich (St. Louis, MO). Drugs were dissolved in saline unless otherwise mentioned.
Isoguvacine puffs
Experiments measuring the evoked ventral root potentials following local application of a GABAA agonist (isoguvacine) involved six different bath solutions (Table 2). For local application, isoguvacine (100 μM) was dissolved in DPBS (Dulbecco's phosphate-buffered saline with Ca2+ and Mg2+) and pressure applied (26–32 psi, 20- to 30-s duration) through glass micropipettes (tip diameters 2.5–5 μm) by means of Pneumatic Pico Pump (PV 800; WPI) using nitrogen gas. For repetitive (every 90 s) isoguvacine application, the duration of pressure pulses was reduced to 0.5–1 s. Micropipettes were positioned close to the Fem motor pool and the amplitude of the isoguvacine-evoked motoneuron responses were recorded (DAM 50, differential amplifier, low-pass filter; WPI) from the Fem nerve as electrotonic potentials. Signals were amplified with Cyber Amp 380, digitized with Digidata 1322A, stored in the computer, and analyzed with pClamp 9 software (Molecular Devices). To measure cellular Cl− accumulation over time, intracellular Cl− was first depleted for 50–60 min of bath application in the “low Cl” solution (Table 2), which completely collapsed the isoguvacine-evoked responses in motoneurons. Following this, the “low Cl” solution was changed back to normal bath solution (Table 2) and the amplitude of the nerve potentials to repetitive application of isoguvacine were measured during the following 60 min. To evaluate the transporters involved in cellular Cl− accumulation we repeated the process in the presence of transporter blockers.
TABLE 2.
Composition of bath solutions (in mM)
| Solution | NaCl | Na-Priopionate | NaHCO3 | Tris-Cl | Tris-OH | Choline-HCO3 | HEPES | NaOH | |
|---|---|---|---|---|---|---|---|---|---|
| 95% O2-5% CO2 | “Normal” | 134 | 0 | 21 | 0 | 0 | 0 | 0 | 0 |
| “Low Cl” | 0 | 134 | 21 | 0 | 0 | 0 | 0 | 0 | |
| “0 Na” | 0 | 0 | 0 | 129 | 21 | 21 | 0 | 0 | |
| 100% O2 | “0 HCO3−” | 134 | 0 | 0 | 0 | 0 | 0 | 10 | 6 |
| “0 Na+ and 0 HCO3−” | 0 | 0 | 0 | 134 | 23 | 0 | 0 | 0 | |
| “Low Cl− and 0 HCO3−” | 0 | 134 | 0 | 0 | 0 | 0 | 10 | 10 |
All solutions also included (in mM): KCl (5). NaH2PO4 (0.58), MgCl2 (1), CaCl2 (3), d-glucose (12), and TTX (0.0025–0.005). Osmolarity was adjusted with sucrose to 345 mOsm when needed.
Statistics
Data are expressed as means ± SE. Most statistical analysis was performed using two-tailed Student's t-tests (paired and unpaired) unless mentioned otherwise. GraphPad Instat software was used for statistical analysis.
RESULTS
Determining the reversal potential at GABAergic synapses
To assess GABAergic synaptic reversal potentials, we used whole cell recordings of GABAA mPSCs to generate I–V plots (Jarolimek et al. 1999). One concern was that our patch solution would dialyze the cell and set the intracellular chloride concentration and therefore EGABA. To test this we isolated spinal cords and obtained whole cell recordings of antidromically identified spinal motoneurons (Fem and Add) from E10 chicks. Voltage-gated channels and glutamatergic transmission were blocked to isolate GABAergic mPSCs (Gonzalez-Islas and Wenner 2006), which were typically recorded in voltage steps of 10 mV (Fig. 1B). The average peak amplitude of GABAergic mPSCs at each step was then plotted against voltage (ImPSC–V plot), as shown for a motoneuron in Fig. 1C. The average EGABA and conductance were, respectively, −29.5 mV and 247 pS (Table 3, 52 mM chloride patch solution). To directly test the influence of the patch solution on intracellular chloride concentrations for GABAergic mPSCs we used patch electrodes with several different chloride concentrations in the pipette solution (17, 32, 52, 77, and 157 mM; Table 1, Fig. 2). If patch chloride concentration had set intracellular chloride, then Nernstian predictions for EGABA would match the measured EGABA. As shown in Fig. 2 and Table 3, the measured reversal potential for GABAergic mPSCs was Nernstian at higher patch chloride concentrations (52, 77, and 157 mM). However, EGABA for lower patch chloride (17 and 32 mM) was not Nerstian, but rather suggested that intracellular chloride was maintained at about 50 mM (EGABA: −30 mV), significantly higher than that predicted for 17 mM (EGABA: −59 mV, P < 0.0001) or 32 mM (EGABA: −43 mV, P < 0.005) patch chloride. When we reduced extracellular chloride from the normal 174 to 53 mM, while using 52 mM patch chloride, we found that measured EGABA was no different from the Nernstian prediction (−1.2 vs. −0.5 mV; Table 3). The results showed that extracellular chloride and higher patch chloride concentrations determine the reversal potential for GABAergic currents. On the other hand, patch solutions with lower chloride concentrations did not noticeably influence the reversal potential. Therefore we used low chloride (17 mM) patch solution for the remainder of the study to measure the GABAergic mPSC reversal potential while examining various chloride transporter inhibitors.
TABLE 3.
EGABA and GABA-mPSC conductance
| Recording Condition | EGABA, mV | Conductance, pS |
|---|---|---|
| 17 mM Cl− patch (16) | −30.7 ± 1.1 | 336 ± 36 |
| 17 mM Cl− patch/0 mM HCO3− bath (4) | −33.9 ± 2.2 | 302 ± 42 |
| 17 mM Cl− patch/10 M bumetanide bath (8) | −39.1 ± 1.0 | 254 ± 20 |
| 17 mM Cl− patch/17 mM Na+ bath (7) | −38.5 ± 1.5 | 224 ± 17 |
| 32 mM Cl− patch (5) | −24.3 ± 2.7 | 270 ± 14 |
| 52 mM Cl− patch (7) | −29.5 ± 2.4 | 247 ± 20 |
| 77 mM Cl− patch (3) | −25.3 ± 1.9 | 309 ± 25 |
| 157 mM Cl− patch (3) | −0.6 ± 3.3 | 316 ± 64 |
| 52 mM Cl− patch/53 mM Cl− bath (3) | −1.2 ± 5.6 | 349 ± 48 |
Number of motoneurons recorded in a given condition is in parentheses.
FIG. 2.

Low chloride patch solution does not perturb intracellular chloride concentrations. GABAergic reversal potential (EGABA) is plotted from ImPSC–V plots for individual cells with patch electrodes of varying chloride concentrations (circles: 17 mM; diamonds: 32 mM; square: 52 mM; downward triangle: 77 mM; upward triangle: 157 mM; horizontal lines represent averages for each concentration). The Nernstian predicted (gray circles) and measured EGABA was only different at low patch chloride (17 and 32 mM).
Chloride is the main carrier of GABAergic currents
Measurements of EGABA with 17 mM chloride patch electrodes suggested that either intracellular chloride was accumulated beyond 17 mM or that another ion could be involved. Since GABAA receptor channels are slightly permeable to bicarbonate ions (Ben-Ari et al. 2007; Farrant and Kaila 2007), it was possible that HCO3− played a significant role in determing EGABA when we used 17 mM chloride patch electrodes, and therefore HCO3− maintained the reversal potential at about −30 mV. The Goldman–Hodgkin–Katz equation predicts that bicarbonate ions do not influence EGABA when intracellular chloride is 17 mM (or higher), assuming the HCO3− to Cl− permeability ratio for GABAA receptor channels at 0.2–0.4 (Farrant and Kaila 2007; Kaila 1994). However, it remained possible that EGABA was maintained at −30 mV because GABAA receptor channels in chick embryo spinal motoneurons were more permeable to HCO3− than in other systems. Therefore we tested this possibility by depleting extracellular HCO3−. GABAergic mPSCs were acquired as described earlier to make mPSC I–V plots and derive EGABA in identified motoneurons. First, EGABA was determined in extracellular solution containing 17 mM HCO3− and then again after replacing the extracellular solution with a 0 mM HCO3− solution while bubbling with 100% oxygen (17 mM chloride in the pipette). Although EGABA did become slightly hyperpolarized 15–45 min after depleting HCO3− (control −30.3 ± 2.2 mV vs. 0 HCO3− −33.9 ± 2.2 mV, P < 0.05 paired t-test, n = 4), it was far from the −59-mV reversal potential predicted by the 17 mM chloride patch solution. Figure 3 shows the motoneuron that exhibited the most significant change in EGABA following perfusion of the 0 HCO3− solution. In the absence of HCO3− the measured reversal potential suggested that intracellular Cl− was about 45 mM. Further, in some cases EGABA appeared to recover if the cells were left in the 0 HCO3− solution for >60 min (not shown). Consistent with other developing systems, the results suggest that chloride is the predominant carrier of the GABAergic current in E10 chick motoneurons.
FIG. 3.

Depleting extracellular HCO3− hyperpolarizes EGABA. ImPSC–V plot from a motoneuron with 17 mM chloride patch solution before and 20 and 60 min after depleting extracellular HCO3−. EGABA slowly shifts to a more hyperpolarized level. Error bars represent SE.
Taken together, these results suggest that at low patch chloride concentrations, chloride accumulators maintain higher chloride levels and resist the dialysis of the patch chloride. This is consistent with the possibility that the vast majority of GABAergic synaptic inputs, from which the mPSCs arise, are located out in the dendrites, further away from the influence of the patch electrode.
Chloride accumulation is partly mediated by NKCC1
Current studies suggest that in many developing neurons NKCC1 contributes to the chloride accumulation that gives rise to the depolarizing GABAergic currents. To assess NKCC1's role in chloride accumulation in E10 chick motoneurons we used several techniques, each offering slightly different information and, together, providing greater confidence than that provided by any single technique. We first tested chloride accumulation in a cell-wide fashion. Muscle nerve recordings were made while a GABAA agonist, isoguvacine, was locally applied to motoneurons (Fig. 4A, schematic). The isoguvacine-evoked nerve potential was then assessed as a monitor of the chloride-mediated response (TTX, a voltage-gated Na+ channel blocker was added to bath to isolate the GABAA-mediated responses). We then bath applied a “0 Na” solution (Table 2) to completely block any Na+-dependent transporter (e.g., NKCC1) and observed that this reduced the isoguvacine-evoked response to 44.7 ± 2.8% of the control (n = 3, Fig. 4). By 30 min after the reintroduction of normal Na+ solution the amplitude recovered to 80.6 ± 6.9% of the control. These findings suggested that there was a significant chloride accumulation that is mediated by a Na-dependent process.
FIG. 4.
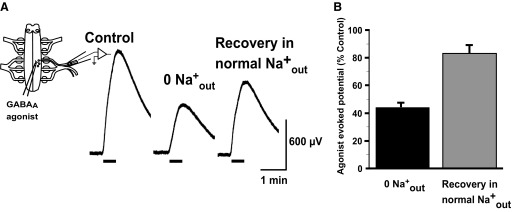
Na+-dependent process contributes to part of the chloride accumulation in embryonic motoneurons. A: isoguvacine is locally applied to the motor column while muscle nerve potentials are recorded (schematic of procedure shown on left). Traces show that 0 Na+ extracellular solution significantly reduces response (P < 0.001), which then recovers on reintroduction of normal Na+ levels. B: average responses to isoguvacine are shown in bar chart.
To assess the progressive accumulation of chloride we first depleted intracellular chloride by bath perfusion of a low chloride extracellular solution (13 mM, Table 2) for about 50 min, followed by bath perfusion of a normal chloride solution (158 mM, Table 2) while locally applying a GABAA agonist (100 μM isoguvacine, 0.5 s, 15-psi picospritzer) to the motor column and again recording muscle nerve potentials (Fig. 5A). This provided a measure of cell-wide chloride accumulation in the motoneuron population because these GABAergic currents grow stronger with the accumulation of chloride. Isoguvacine puffs were delivered every 90 s (Fig. 5A, lines below traces) and the isoguvacine-induced potentials increased and stabilized to predepletion levels (288 ± 50 μV, n = 4) in about 35–40 min. Chloride accumulation in the order of minutes has been described previously (Achilles et al. 2007), although small perturbations in intracellular chloride can recover more quickly in other systems (Brumback and Staley 2008). The procedure was then repeated while blocking NKCC1 with bumetanide (10 μM, added to both “low Cl” and subsequent normal chloride solutions). Isoguvacine-induced nerve potentials did begin to recover, but stabilized at a value of 34.9 ± 1.4% of that in the absence of bumetanide (25–30 min, Fig. 5B). Figure 5 shows that the initial phase of reaccumulation (phase 1) was less dependent on NKCC1 than later stages of the accumulation (phase 2). These findings suggested that NKCC1 moved chloride into the cell beyond a passive distribution of the ion, but suggested that another accumulator must exist, which might be important at lower intracellular chloride concentrations (phase 1).
FIG. 5.

Na+-K+-2Cl− cotransporter (NKCC1) contributes to chloride accumulation in embryonic chick motoneurons. A: procedure is shown for intracellular chloride depletion and reaccumulation. Isoguvacine puffs were applied every 90 s, 0.5-s duration (horizontal line with vertical marks) and potentials were recorded from the Fem muscle nerve. As “low Cl” solution is superfused onto the cord, isoguvacine-evoked potentials were abolished within 15–20 min, indicating a collapse of the chloride gradient. As normal chloride solution was reintroduced, potentials increased to predepletion levels within 40–50 min (bottom traces are first positive potentials from above, shown at an expanded timescale). This suggested our “low Cl” solution had no long-term effects. This process was then repeated in the presence of bumetanide (right). B: average of the recovery in nerve potential amplitudes is shown before adding bumetanide (control) and in the presence of bumetanide (n = 4). Data were normalized to control values (determined as mean of 5 last responses of the 50-min record in normal bath solution). The recovery showed 2 phases: phase 1, where a largely NKCC1-independent chloride accumulation occurs, and phase 2, where a largely NKCC1-dependent accumulation occurs. Error bars represent SE.
We next tested chloride accumulation specifically at synaptic sites, rather than in a cell-wide manner, as before. In the chick embryo, episodes of SNA lead to significant GABAergic transmission, which depletes intracellular chloride. Although this weakens GABAergic currents, the currents recover as chloride is reaccumulated in the interepisode interval (Chub et al. 2006; Fedirchuk et al. 1999; Marchetti et al. 2005). We can monitor this process of chloride regulation by stimulating motoneurons projecting in one ventral root and making extracellular recordings in the adjacent ventral root. Ventral root stimulation activates R-interneurons, which then make direct synaptic projections back to motoneurons, and these GABAergic responses can be recorded in the adjacent ventral root (Wenner and O'Donovan 1999). Ventral root responses are weakened after an episode of SNA and then become progressively stronger in the interval between episodes (Fig. 6). The ventral root responses were then reduced after adding bumetanide to block NKCC1. The response was eliminated when a GABAA antagonist was applied, demonstrating the response was GABAergic (Fig. 6A). When responses were binned in consecutive 2-min intervals after an episode, we saw that the average response of the fourth bin (6–8th min) was slightly, but significantly, reduced in bumetanide (Fig. 6B). Although the ventral root response was reduced, it continued to modulate, suggesting chloride was still accumulated in the absence of NKCC1 function. These findings are consistent with the idea that NKCC1 is important in chloride accumulation at synaptic sites, but that NKCC1 was not solely responsible for the accumulation.
FIG. 6.
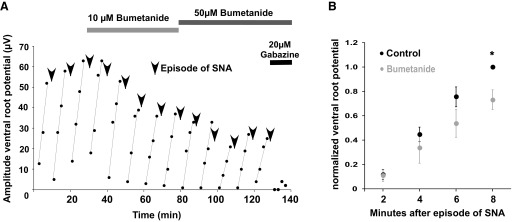
GABAergic synaptic potentials are reduced but continue to modulate following NKCC1 block. A: ventral root-evoked ventral root response is shown modulating over time. After each episode of spontaneous network activity (SNA, arrowhead) this GABAergic ventral root response is reduced but then increases in the interval between episodes (lines). When the NKCC1 blocker bumetanide was added to the bath, responses were reduced, although the modulation still occurred even when a higher concentration of bumetanide was added. The response was knocked out when GABAergic transmission was blocked by gabazine. B: the average amplitude of the ventral root-evoked ventral root response is shown in the first 8 min after an episode of SNA, broken into 2-min bins (0–2, >2–4, >4–6, >6–8 min; n = 4 experiments). After adding bumetanide (waiting ≥20 min) the amplitude of the response had decreased, but still showed modulation. Responses in the 4th bin (6–8th min) were significantly reduced following NKCC1 block (*P < 0.05, one sample t-test). Error bars represent SE.
To more directly test chloride accumulation at synaptic sites we next used whole cell recordings and made mPSC I–V plots to determine the effect of blocking NKCC1 on EGABA, a measure of intracellular chloride accumulation. If NKCC1 were solely responsible for the accumulation of high chloride at GABAergic synaptic sites (∼50 mM) then we reasoned that blocking this transporter would shift EGABA toward the value predicted for the 17 mM patch chloride (−59 mV) because the patch solution dialyzed the cell (Jarolimek et al. 1996, 1999). We found that inhibiting NKCC1 with 10 μM bumetanide hyperpolarized EGABA by only 8.4 mV to −39.1 mV (Fig. 7, A and B) and this change occurred within 10 min. We also noticed that conductance was slightly reduced in the presence of the blocker (Fig. 7, A and C, Table 3). This finding suggested the possibility that bumetanide had a slight effect on GABAA receptor channel conductance in the chick embryo. We also inhibited sodium-dependent chloride transporters by effectively collapsing the Na+ gradient (low Naout+ solution, 17 mM). This maneuver hyperpolarized EGABA by −7.8 mV (P < 0.001, n = 7 cells, Table 3) in the first 2 h after perfusing with the low Na+ solution (Fig. 7D). We noticed that EGABA appeared to recover to control values if longer incubations were used (not shown). Therefore using a different means of inhibiting inward Cl− movement through NKCC1, we again determined that this cotransporter was involved in chloride accumulation at synaptic sites. Because the hyperpolarization of EGABA never reached −59 mV predicted by the Nernst equation (given a 17 mM chloride patch solution), there must be another chloride accumulator. Furthermore, the apparent recovery of EGABA is consistent with the idea that a Na+-independent accumulator could compensate for the loss of NKCC1 function.
FIG. 7.

NKCC1 blockade hyperpolarizes EGABA. A: ImPSC–V plots from a motoneuron before and after bath application of 10 μM bumetanide to block NKCC1 (17 mM Cl− patch solution). EGABA shifted to a more hyperpolarized level and conductance was also slightly reduced. B: average EGABA is shown from all motoneurons in control conditions and in the presence of bumetanide. EGABA was significantly hyperpolarized when NKCC1 was blocked. In certain cells we were able to measure the reversal potential before and after adding bumetanide in the same cell (n = 3, lines). C: GABAergic mPSC conductance was slightly reduced in the presence of bumetanide. P values were determined using Mann–Whitney test and were significant even when the 800-pS data point was removed. D: example of an ImPSC–V plot before and after reducing the Na+ gradient in one motoneuron. EGABA is shifted in the negative direction. Error bars represent SE.
NKCC1-independent chloride accumulation
The anion exchanger AE3 moves chloride into the cell as it expels bicarbonate. AE3 is up-regulated in the embryonic mouse spinal cord during the period SNA is expressed (Hentschke et al. 2006). We therefore tested the possibility that chloride was accumulated by AE3 in developing chick motoneurons. We repeated the experiments described earlier, but instead of NKCC1, we tested the anion exchanger by blocking it pharmacologically with 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS). We used the technique of locally applying a GABAA agonist while recording the potential produced in the muscle nerve and saw that application of DIDS reduced the response to 53.6 ± 4.7% (Fig. 8, A and B). The remaining response was largely abolished when we then added bumetanide (8.1 ± 2.9%) and this response could be partly recovered on washout to 43.0 ± 4.9% (60 min, n = 5). Because we were concerned that DIDS could have nonspecific effects (Cabantchik and Greger 1992), we repeated these experiments, blocking the exchanger in a different way, with a 0 HCO3− extracellular solution. Solutions were bubbled with 100% oxygen and HEPES was added to maintain the pH at about 7.3. Very similar responses to that of DIDS were produced when blocking with “0 HCO3−” solution (Fig. 8, C and D). The “0 HCO3−” solution significantly reduced the isoguvacine response to 67.9 ± 3.1% and this was further decreased to 4.4 ± 1.5% in a “0 HCO3− and 0 Na” bath solution. The response recovered to 88.5 ± 4.7% after a 45-min washout period (n = 5).
FIG. 8.
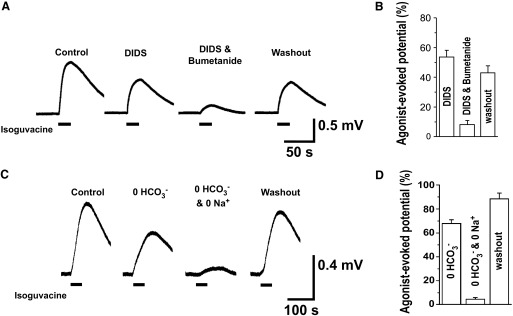
NKCC1-independent chloride accumulation is sensitive to 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS) and dependent on HCO3−. Isoguvacine was puffed (100 μM, 20-s duration, every 15 min) onto the motor column while the evoked potentials were recorded in the Fem nerve. A: the Cl−/HCO3− exchanger inhibitor, DIDS (50 μM), was added to the bath (60 min), which significantly reduced the evoked potential. This response was then nearly abolished on subsequent addition of 10 μM bumetanide (45 min). The response partially recovered after drug washout (45 min). B: averages are shown in the bar chart on the right (n = 5, P < 0.001). C: a “0 HCO3−” extracellular solution (45 min) also reduced the evoked potential, which is then nearly abolished with the perfusion of “0 Na and 0 HCO3−” extracellular solution (45 min). The response recovers after normal solution is restored. D: averages are shown in the bar chart on the right (n = 3, P < 0.001). Error bars represent SE.
In the next set of experiments we depleted and reintroduced chloride and monitored chloride reaccumulation in the presence of DIDS. We saw that the reaccumulation was inhibited compared with control values, 51.7 ± 1.1% (Fig. 9A, 50 min). When NKCC1 was also blocked with bumetanide, chloride accumulation was nearly prevented (7.1 ± 0.3% in 50 min). In a separate set of experiments, we used a “0 HCO3−” solution to block the anion exchanger and we observed the recovery was reduced to 65.3 ± 5.5% of the control (Fig. 9B, 50 min). Blocking AE3 with either method demonstrated that chloride accumulation was reduced. Together these findings suggest that NKCC1 significantly contributes to chloride accumulation in embryonic chick motoneurons, but that additional accumulation likely occurs through the bicarbonate-dependent anion exchanger AE3.
FIG. 9.

Chloride accumulation following chloride depletion demonstrate a DIDS-sensitive, bicarbonate-dependent process. A: plot shows that isoguvacine-evoked potentials (100 μM, 0.5-s duration, every 90 s) increase over time in control solution following chloride depletion, but less so during reaccumulation with DIDS (50 μM); reaccumulation was nearly abolished with application of DIDS and bumetanide (10 μM, n = 4). B: plot shows that isoguvacine-evoked potentials during reaccumulation were similarly inhibited on the perfusion of a “0 HCO3−” extracellular solution (100% O2, n = 4). Error bars represent SE.
DISCUSSION
In this study we examined chloride accumulation in embryonic motoneurons using several different approaches. We determined the reversal potential and conductance of GABAA synaptic currents, using whole cell recordings of GABAergic mPSCs. Synaptic chloride accumulates to about 50 mM and this is mediated in part by the chloride cotransporter NKCC1. We also show evidence that NKCC1 is involved in cell-wide chloride accumulation, as shown by muscle nerve responses to local application of a GABAA agonist. Although reduced, synaptic and cell-wide chloride accumulation continued after NKCC1 was blocked, suggesting an additional accumulator must exist. We show evidence that this residual chloride accumulation is dependent on HCO3− and is sensitive to DIDS, suggesting that, at least in embryonic motoneurons, the additional accumulator is likely to be AE3.
Measuring synaptic chloride through mPSC I–V plots
During development, GABAergic synapses are depolarizing and can be excitatory, leading to the expression of spontaneous network activity. Later in development GABAergic synapses become hyperpolarizing and SNA abates. To understand how the driving force for chloride changes and influences GABAergic synaptic strength during this period, it may be important to assess GABAergic reversal potential where the synapses are located. GABAA reversal potential is often determined by local application of a GABAA agonist, while recording from a cell using the perforated-patch technique. This technique has the advantage that intracellular contents are minimally influenced by the pipette. On the other hand, the tested reversal potential will likely assess somal or cell-wide intracellular chloride (wherever synaptic and extrasynaptic GABAA receptor activations occur). Because extra- and synaptic GABAA receptors could have distinct domains of expression, it is possible that EGABA measured by mPSCs versus a cell-wide application of a GABA agonist will be different because chloride concentrations can vary within an individual neuron from soma to dendrites (Chub et al. 2006; Duebel et al. 2006; Gavrikov et al. 2006; Hara et al. 1992; Jarolimek et al. 1999; Li et al. 2008; Miller and Dacheux 1983; Vardi et al. 2000; Varela et al. 2005). We have used whole cell recordings of GABAergic mPSCs to assess synaptic EGABA, which may provide a more specific measure of synaptic chloride. We find that intracellular chloride is about 50 mM at GABAergic synaptic locations, which are likely to be dendritic. This is considerably higher than estimates in the same cells using perforated patch recordings while locally applying a GABAA agonist (∼30 mM, unpublished observations). These findings are consistent with the possibility that motoneuron dendrites have higher chloride concentrations than the soma.
Measured EGABA for mPSCs was more depolarized than would be predicted by the Nernst equation when chloride in the patch solution was <50 mM. This suggests that a mechanism exists to maintain synaptic chloride at about 50 mM when patch chloride is below this. It is likely that chloride transporters actively resist dialysis out in the dendrites. This is further supported by the observation that EGABA moved toward values predicted by the patch solution when we blocked chloride transporters. On the other hand, when patch chloride is high, the transporters did not effectively extrude cellular chloride. This may be due to the observation that high intracellular chloride can inhibit transporter function (Lytle and Forbush 1996). Together, the findings suggested that these embryonic spinal neurons tightly regulate synaptic chloride. Similar findings have been reported previously, where whole cell recordings show that EGABA is different in the soma and dendrites of relatively mature neurons and that chloride in the patch solution did not define dendritic chloride unless the KCC2 transporter was blocked (Jarolimek et al. 1996, 1999). Making I–V plots directly from the synaptic currents themselves could prove to be a useful technique in identifying mechanisms underlying changes in mPSC amplitude because it simultaneously provides measurements of both conductance and reversal potential.
NKCC1 contributes to chloride accumulation in embryonic spinal motoneurons
In the E10 chick embryo spinal cord the transporter that has been thought to be largely responsible for restoring high intracellular chloride in the interepisode interval is the NKCC1 cotransporter (Chub and O'Donovan 2001; Chub et al. 2006). NKCC1 has been shown to be important in maintaining high intracellular chloride in mature DRG cells and in many different classes of developing neuron, leading to depolarizing GABAergic currents (Alvarez-Leefmans et al. 1988; Ben-Ari et al. 2007; Payne et al. 2003; Yamada et al. 2004). In the chick spinal cord GABA appears to switch from depolarizing to hyperpolarizing shortly after E15 (Xu et al. 2005). This switch, during early development in spinal motoneurons, is thought to be mediated by a reduction in NKCC1 function and a concurrent up-regulation of KCC2 (Delpy et al. 2008; Hübner et al. 2001; Jean-Xavier et al. 2006; Li et al. 2002; Stein et al. 2004).
We have several results that confirm and extend the above-cited findings. We have used muscle nerve responses to local application of GABAA agonists to monitor chloride accumulation. We can eliminate these nerve responses by bath application of low Cl solutions. This is consistent with the idea that chloride is accumulated by secondarily active transporters dependent on a chloride gradient, such as NKCC1 and AE3. Further, these chloride-dependent nerve potentials were inhibited by reducing NKCC1 function with bumetanide, or a “0 Na” solution. We were also able to demonstrate that NKCC1 contributes to the chloride accumulation at GABAergic synaptic sites. First, we showed that currents from GABAergic interneuronal inputs to motoneurons were reduced following bumetanide bath application. Second, using GABAergic mPSC I–V plots we determined that EGABA was hyperpolarized by about 8 mV (Clin− reduced to ∼39 mM) following NKCC1 inhibition by either bumetanide or “low Na” extracellular solutions. These results confirm earlier reports of an NKCC1-dependent chloride accumulation in embryonic spinal motoneurons. Further, they extend previous findings by showing that other Na+-dependent transporters, like the Na+-Cl− transporter (NCC), are unlikely to contribute to chloride accumulation since low and 0 mM Na+ solutions had effects similar to those of bumetanide (similar reductions in EGABA and nerve potentials). In addition, the results show that NKCC1 is important in chloride accumulation at GABAergic synaptic sites, likely in the motoneuron dendrites.
Anion exchanger also appears to contribute to chloride accumulation in embryonic chick motoneurons
Although a contributor, NKCC1 did not appear to be solely responsible for chloride accumulation in embryonic chick motoneurons. This is also consistent with previous work in several different tissues (Delpy et al. 2008; Kakazu et al. 1999; Rocha-Gonzalez et al. 2008; Rohrbough and Spitzer 1996; Yamada et al. 2004). Blockade of chloride influx through NKCC1 (bumetanide, low/0 Naout−) did not prevent chloride accumulation in any of the experiments we performed (GABAA agonist-induced nerve potentials, ventral root-evoked nerve response, ImPSC–V). This suggests that another transporter exists that, like NKCC1, is dependent on the chloride gradient to drive chloride into the cell. As discussed earlier it is unlikely to be the Na+-Cl− transporter NCC, which is not thought to be expressed in the nervous system (Gamba et al. 1994). The potassium chloride cotransporter KCC2 typically extrudes chloride, but under some circumstances can accumulate chloride because KCC2 can be near its thermodynamic equilibrium in mature neurons (Jarolimek et al. 1999; Payne 1997). However, it is highly unlikely that KCC2 could load chloride in embryonic motoneurons where intracellular chloride is high (∼50 mM) and therefore the potassium gradient will strongly favor chloride extrusion.
Alternatively, the anion exchanger AE3 has been suggested as one potential candidate for chloride accumulation (Balakrishnan et al. 2003; Rocha-Gonzalez et al. 2008). AE3 is expressed in the developing spinal cord at the stage that SNA is exhibited (Hentschke et al. 2006) and can accumulate chloride as it extrudes HCO3−, in a Na+-independent manner. We now show evidence supporting a role in chloride accumulation for AE3. The perfusion of 0 mM HCO3− solution did lead to a slight hyperpolarization of EGABA and this developed progressively over time (Fig. 3), which might be expected as chloride slowly leaks out of the cell. The 0 mM HCO3− solution reduces intracellular HCO3−, which is necessary for AE3 function, and therefore could potentially reduce accumulated chloride. Further, DIDS and 0 HCO3− solutions reduced isoguvacine-evoked nerve potentials (Fig. 8) and reduced the reaccumulation of chloride in the chloride-depletion experiments (Fig. 9). A very similar finding had been reported previously, where GABAA currents, indirectly measured by calcium imaging, were weakened by bumetanide and a 0 mM HCO3− extracellular solution in embryonic motoneurons (Kulik et al. 2000). In addition, blocking AE3 function with DIDS produced a result similar to that of the 0 mM HCO3− extracellular solution. We cannot rule out the possibility that our methods of blocking the anion exchanger alter pH, which could then change some other aspect of chloride regulation. However, we do not favor this possibility because DIDS and 0 mM HCO3− both reduce chloride accumulation, but would likely influence pH differently.
NKCC1 and AE3 appear to have distinct functions in the recovery of chloride levels following chloride depletion in embryonic motoneurons. NKCC1 seems to be responsible for approximately two thirds of the steady-state chloride accumulation, whereas AE3 is responsible for the remaining third. Although both accumulators contribute to intracellular chloride, they appear to function differently at different stages of the reaccumulation following intracellular chloride depletion. AE3 may be important at early stages of the reaccumulation when intracellular chloride is relatively low. This can be observed in Fig. 5B, where in the first 10 min (phase 1), the recovery of the evoked potential does not change significantly when NKCC1 is blocked. On the other hand, NKCC1 appears to be important in the continued recovery of the evoked potentials past the first 10 min (phase 2, Fig. 5B). Together, our results suggest that chloride accumulation throughout the cell and at GABAergic synaptic sites occurs through NKCC1, and likely through the anion exchanger as well.
GRANTS
This research was supported by National Institute of Neurological Disorders and Stroke (NINDS) Grant NS-046510 and National Science Foundation Grant 0616097 to P. Wenner and by the intramural program of the NINDS.
Acknowledgments
We thank Dr. Martin Pinter for helpful discussions.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- Achilles 2007.Achilles K, Okabe A, Ikeda M, Shimizu-Okabe C, Yamada J, Fukuda A, Luhmann HJ, Kilb W. Kinetic properties of Cl− uptake mediated by Na+-dependent K+-2Cl− cotransport in immature rat neocortical neurons. J Neurosci 27: 8616–8627, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Leefmans 1988.Alvarez-Leefmans FJ, Gamino SM, Giraldez F, Nogueron I. Intracellular chloride regulation in amphibian dorsal root ganglion neurones studied with ion-selective microelectrodes. J Physiol 406: 225–246, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan 2003.Balakrishnan V, Becker M, Lohrke S, Nothwang HG, Guresir E, Friauf E. Expression and function of chloride transporters during development of inhibitory neurotransmission in the auditory brainstem. J Neurosci 23: 4134–4145, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari 2007.Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev 87: 1215–1284, 2007. [DOI] [PubMed] [Google Scholar]
- Brumback 2008.Brumback AC, Staley KJ. Thermodynamic regulation of NKCC1-mediated Cl− cotransport underlies plasticity of GABA(A) signaling in neonatal neurons. J Neurosci 28: 1301–1312, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabantchik 1992.Cabantchik ZI, Greger R. Chemical probes for anion transporters of mammalian cell membranes. Am J Physiol Cell Physiol 262: C803–C827, 1992. [DOI] [PubMed] [Google Scholar]
- Casavant 2004.Casavant RH, Colbert CM, Dryer SE. A-current expression is regulated by activity but not by target tissues in developing lumbar motoneurons of the chick embryo. J Neurophysiol 92: 2644–2651, 2004. [DOI] [PubMed] [Google Scholar]
- Chub 2006.Chub N, Mentis GZ, O'Donovan MJ. Chloride-sensitive MEQ fluorescence in chick embryo motoneurons following manipulations of chloride and during spontaneous network activity. J Neurophysiol 95: 323–330, 2006. [DOI] [PubMed] [Google Scholar]
- Chub 1998.Chub N, O'Donovan MJ. Blockade and recovery of spontaneous rhythmic activity after application of neurotransmitter antagonists to spinal networks of the chick embryo. J Neurosci 18: 294–306, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chub 2001.Chub N, O'Donovan MJ. Post-episode depression of GABAergic transmission in spinal neurons of the chick embryo. J Neurophysiol 85: 2166–2176, 2001. [DOI] [PubMed] [Google Scholar]
- Delpy 2008.Delpy A, Allain AE, Meyrand P, Branchereau P. NKCC1 cotransporter inactivation underlies embryonic development of chloride-mediated inhibition in mouse spinal motoneuron. J Physiol 586: 1059–1075, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duebel 2006.Duebel J, Haverkamp S, Schleich W, Feng G, Augustine GJ, Kuner T, Euler T. Two-photon imaging reveals somatodendritic chloride gradient in retinal ON-type bipolar cells expressing the biosensor Clomeleon. Neuron 49: 81–94, 2006. [DOI] [PubMed] [Google Scholar]
- Farrant 2007.Farrant M, Kaila K. The cellular, molecular and ionic basis of GABA(A) receptor signalling. Prog Brain Res 160: 59–87, 2007. [DOI] [PubMed] [Google Scholar]
- Fedirchuk 1999.Fedirchuk B, Wenner P, Whelan PJ, Ho S, Tabak J, O'Donovan MJ. Spontaneous network activity transiently depresses synaptic transmission in the embryonic chick spinal cord. J Neurosci 19: 2102–2112, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamba 1994.Gamba G, Miyanoshita A, Lombardi M, Lytton J, Lee WS, Hediger MA, Hebert SC. Molecular cloning, primary structure, and characterization of two members of the mammalian electroneutral sodium-(potassium)-chloride cotransporter family expressed in kidney. J Biol Chem 269: 17713–17722, 1994. [PubMed] [Google Scholar]
- Gavrikov 2006.Gavrikov KE, Nilson JE, Dmitriev AV, Zucker CL, Mangel SC. Dendritic compartmentalization of chloride cotransporters underlies directional responses of starburst amacrine cells in retina. Proc Natl Acad Sci USA 103: 18793–18798, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Islas 2006.Gonzalez-Islas C, Wenner P. Spontaneous network activity in the embryonic spinal cord regulates AMPAergic and GABAergic synaptic strength. Neuron 49: 563–575, 2006. [DOI] [PubMed] [Google Scholar]
- Hamburger 1951.Hamburger V, Hamilton HC. A series of normal stages in the development of the chick embryo. J Morphol 88: 49–92, 1951. [PubMed] [Google Scholar]
- Hanson 2004.Hanson MG, Landmesser LT. Normal patterns of spontaneous activity are required for correct motor axon guidance and the expression of specific guidance molecules. Neuron 43: 687–701, 2004. [DOI] [PubMed] [Google Scholar]
- Hanson 2006.Hanson MG, Landmesser LT. Increasing the frequency of spontaneous rhythmic activity disrupts pool-specific axon fasciculation and pathfinding of embryonic spinal motoneurons. J Neurosci 26: 12769–12780, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara 1992.Hara M, Inoue M, Yasukura T, Ohnishi S, Mikami Y, Inagaki C. Uneven distribution of intracellular Cl− in rat hippocampal neurons. Neurosci Lett 143: 135–138, 1992. [DOI] [PubMed] [Google Scholar]
- Hentschke 2006.Hentschke M, Wiemann M, Hentschke S, Kurth I, Hermans-Borgmeyer I, Seidenbecher T, Jentsch TJ, Gal A, Hübner CA. Mice with a targeted disruption of the Cl− HCO3− exchanger AE3 display a reduced seizure threshold. Mol Cell Biol 26: 182–191, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hübner 2001.Hübner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron 30: 515–524, 2001. [DOI] [PubMed] [Google Scholar]
- Jarolimek 1996.Jarolimek W, Brunner H, Lewen A, Misgeld U. Role of chloride-homeostasis in the inhibitory control of neuronal network oscillators. J Neurophysiol 75: 2654–2657, 1996. [DOI] [PubMed] [Google Scholar]
- Jarolimek 1999.Jarolimek W, Lewen A, Misgeld U. A furosemide-sensitive K+-Cl− cotransporter counteracts intracellular Cl− accumulation and depletion in cultured rat midbrain neurons. J Neurosci 19: 4695–4704, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean-Xavier 2006.Jean-Xavier C, Pflieger JF, Liabeuf S, Vinay L. Inhibitory postsynaptic potentials in lumbar motoneurons remain depolarizing after neonatal spinal cord transection in the rat. J Neurophysiol 96: 2274–2281, 2006. [DOI] [PubMed] [Google Scholar]
- Kaila 1994.Kaila K Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol 42: 489–537, 1994. [DOI] [PubMed] [Google Scholar]
- Kakazu 1999.Kakazu Y, Akaike N, Komiyama S, Nabekura J. Regulation of intracellular chloride by cotransporters in developing lateral superior olive neurons. J Neurosci 19: 2843–2851, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulik 2000.Kulik A, Nishimaru H, Ballanyi K. Role of bicarbonate and chloride in GABA- and glycine-induced depolarization and [Ca2+]i rise in fetal rat motoneurons in situ. J Neurosci 20: 7905–7913, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li 2008.Li B, McKernan K, Shen W. Spatial and temporal distribution patterns of Na-K-2Cl cotransporter in adult and developing mouse retinas. Vis Neurosci 25: 109–123, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li 2002.Li H, Tornberg J, Kaila K, Airaksinen MS, Rivera C. Patterns of cation-chloride cotransporter expression during embryonic rodent CNS development. Eur J Neurosci 16: 2358–2370, 2002. [DOI] [PubMed] [Google Scholar]
- Lytle 1996.Lytle C, Forbush B 3rd Regulatory phosphorylation of the secretory Na-K-Cl cotransporter: modulation by cytoplasmic Cl. Am J Physiol Cell Physiol 270: C437–C448, 1996. [DOI] [PubMed] [Google Scholar]
- Marchetti 2005.Marchetti C, Tabak J, Chub N, O'Donovan MJ, Rinzel J. Modeling spontaneous activity in the developing spinal cord using activity-dependent variations of intracellular chloride. J Neurosci 25: 3601–3612, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller 1983.Miller RF, Dacheux RF. Intracellular chloride in retinal neurons: measurement and meaning. Vision Res 23: 399–411, 1983. [DOI] [PubMed] [Google Scholar]
- O'Donovan 1999.O'Donovan MJ The origin of spontaneous activity in developing networks of the vertebrate nervous system. Curr Opin Neurobiol 9: 94–104, 1999. [DOI] [PubMed] [Google Scholar]
- Payne 1997.Payne JA Functional characterization of the neuronal-specific K-Cl cotransporter: implications for [K+]o regulation. Am J Physiol Cell Physiol 273: C1516–C1525, 1997. [DOI] [PubMed] [Google Scholar]
- Payne 2003.Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci 26: 199–206, 2003. [DOI] [PubMed] [Google Scholar]
- Rocha-Gonzalez 2008.Rocha-Gonzalez HI, Mao S, Alvarez-Leefmans FJ. Na+,K+,2Cl− cotransport and intracellular chloride regulation in rat primary sensory neurons: thermodynamic and kinetic aspects. J Neurophysiol 100: 169–184, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrbough 1996.Rohrbough J, Spitzer NC. Regulation of intracellular Cl− levels by Na(+)-dependent Cl− cotransport distinguishes depolarizing from hyperpolarizing GABAA receptor-mediated responses in spinal neurons. J Neurosci 16: 82–91, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipila 2006.Sipila ST, Schuchmann S, Voipio J, Yamada J, Kaila K. The cation-chloride cotransporter NKCC1 promotes sharp waves in the neonatal rat hippocampus. J Physiol 573: 765–773, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein 2004.Stein V, Hermans-Borgmeyer I, Jentsch TJ, Hübner CA. Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J Comp Neurol 468: 57–64, 2004. [DOI] [PubMed] [Google Scholar]
- Vardi 2000.Vardi N, Zhang LL, Payne JA, Sterling P. Evidence that different cation chloride cotransporters in retinal neurons allow opposite responses to GABA. J Neurosci 20: 7657–7663, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela 2005.Varela C, Blanco R, De la Villa P. Depolarizing effect of GABA in rod bipolar cells of the mouse retina. Vision Res 45: 2659–2667, 2005. [DOI] [PubMed] [Google Scholar]
- Wenner 1999.Wenner P, O'Donovan MJ. Identification of an interneuronal population that mediates recurrent inhibition of motoneurons in the developing chick spinal cord. J Neurosci 19: 7557–7567, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu 2005.Xu H, Whelan PJ, Wenner P. Development of an inhibitory interneuronal circuit in the embryonic spinal cord. J Neurophysiol 93: 2922–2933, 2005. [DOI] [PubMed] [Google Scholar]
- Yamada 2004.Yamada J, Okabe A, Toyoda H, Kilb W, Luhmann HJ, Fukuda A. Cl− uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J Physiol 557: 829–841, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang 2007.Zhang LL, Delpire E, Vardi N. NKCC1 does not accumulate chloride in developing retinal neurons. J Neurophysiol 98: 266–277, 2007. [DOI] [PubMed] [Google Scholar]