

Abstract
The unique monooxygenase activity of cytochrome P450cam has been attributed to coordination of a cysteine thiolate to the heme cofactor. To investigate this interaction, we replaced cysteine with the more electron-donating selenocysteine. Good yields of the selenoenzyme were obtained by bacterial expression of an engineered gene containing the requisite UGA codon for selenocysteine and a simplified yet functional selenocysteine insertion sequence (SECIS). The sulfur-to-selenium substitution subtly modulates the structural, electronic, and catalytic properties of the enzyme. Catalytic activity decreases only 2-fold, whereas substrate oxidation becomes partially uncoupled from electron transfer, implying a more complex role for the axial ligand than generally assumed.
Keywords: protein engineering, selenocysteine insertion sequence element, selenoenzyme, stop codon suppression, X-ray crystallography
Cytochrome P450 enzymes are versatile heme-dependent monooxygenases that catalyze the stereoselective hydroxylation of nonactivated hydrocarbons. They have been subject to intense mechanistic scrutiny because of their critical role in diverse biochemical processes, ranging from steroid and lipid biosynthesis to xenobiotic detoxification and drug metabolism, and because of their potential synthetic utility outside the cell. High-valent iron oxo species have been postulated to be key intermediates in oxygen transfer (Fig. 1), but precisely how molecular oxygen is activated in these systems is still an ongoing debate (1–3).
Fig. 1.

The reaction path of P450cam. Compound I, a ferryl-oxo π-cation porphyrin radical, is the putative oxidant that reacts directly with substrate. Dashed arrows indicate possible uncoupling processes.
The axial ligand to the heme iron, a cysteine thiolate, is generally believed to control P450 reactivity. It has been difficult, however, to modulate its properties as an electron donor without destroying the heme environment. For example, mutating the proximal cysteine to histidine, a residue often found in the corresponding position of non-P450-type heme proteins, inactivates P450s (4, 5). Consequently, more conservative replacement of cysteine by a nonstandard amino acid such as selenocysteine is attractive. Although selenium and sulfur are nearly isosteric, selenols differ from thiols in their pKa values and redox potentials (6). Thus, their incorporation into proteins can be mechanistically informative (7). QM/MM calculations on one of the best characterized enzymes in this superfamily, P450cam, suggest that the greater electron donating ability of a selenolate compared with a thiolate should speed up formation of compound I, a highly reactive ferryl-oxo π-cation porphyrin radical, but slow its subsequent reaction with substrate ≈100-fold (8). Such properties could facilitate observation of this elusive species.
Artificial selenoproteins have been prepared previously by (semi)synthesis (9), posttranslational modification (10), and reassignment of cysteine codons to selenocysteine in auxotrophic strains (11). None of these methods is particularly suited for the targeted replacement of the proximal heme ligand in P450cam, however, given the large size of the enzyme, the relative inaccessibility of the heme–thiolate, and the presence of multiple cysteine residues in the sequence. In nature, selenocysteine is incorporated cotranslationally into proteins as a 21st building block by selective suppression of the UGA stop codon (12–14). The bacterial biosynthetic machinery for selenocysteine insertion has been successfully adapted for heterologous production of some natural selenoproteins and their analogues (15–18) but, with the exception of a C-terminal GCUG tetrapeptide added as a purification tag (19), this approach has been difficult to extend to other proteins because reassignment of the opal stop codon requires an overcoding mechanism. In bacteria, UGA is read as selenocysteine only when immediately followed by a specific mRNA stem loop structure, called a selenocysteine insertion sequence (SECIS) (13). If, as is often the case, this ≈40-nt-long segment encodes part of the protein, it is a challenge to create a SECIS element that retains function while minimizing additional sequence changes (20).
Here, we show that the P450cam gene can be equipped with a simplified SECIS element that directs efficient biosynthesis of an enzyme variant possessing a selenocysteine at position 357 as the proximal heme ligand. Although selenocysteine insertion requires 2 additional amino acid changes, the structural integrity of the active site is maintained as demonstrated by X-ray crystallography and retention of significant monooxygenase activity. The subtle changes in electronic properties resulting from the sulfur-to-selenium substitution provide a unique window for probing the role of the axial ligand in P450 chemistry.
Results
SECIS Design.
We used the bacterial SECIS element from formate dehydrogenase (21) (Fig. 2A) as a model for redesigning the P450cam gene (Fig. 2B). Biochemical and structural experiments have shown that the boxed 17-nt-long stem loop structure is essential for efficient stop codon suppression (22, 23). This “minimal SECIS element” is optimally positioned 11 nt downstream of the UGA and contains a conserved U bulge in its upper stem and 4 conserved bases in the loop. The exact structure of the lower stem appears to be less critical and need not be double-stranded.
Fig. 2.

Design of a simplified SECIS element for P450cam. (A) Canonical SECIS element from E. coli formate dehydrogenase as encoded by the fdhH gene. The boxed region shows the minimal SECIS element with essential SelB-contacting bases circled. (B) Sequence of the P450cam gene immediately downstream of the codon for the proximal heme ligand, Cys-357. Sequence differences relative to the canonical SECIS element are highlighted in blue. (C) Simplified SECIS element for production of C357U P450cam*, with the mutations that were introduced shown in red. (D) Gene and amino acid sequences of WT P450cam, the control protein P450cam*, which has a proximal cysteine ligand in addition to the SECIS mutations, and C357U P450cam*. Mutations are shown in red.
Based on this information and the inherent degeneracy of the genetic code, 9 nucleotide changes were introduced into the WT P450cam gene to construct a SECIS element immediately after the codon for residue 357 (Fig. 2C). Four substitutions ensure formation of the Watson–Crick base-paired upper stem at an appropriate distance from the engineered UGA, and 3 others optimize the nucleotides in the recognition loop. The final 2 changes adjust codon usage in the lower stem for efficient gene expression in Escherichia coli (24). Six of the 9 substitutions are silent; the other 3 result in coding changes for 2 residues, R365L near the surface of the protein and E366Q (Fig. 2D).† To assess the effect of these mutations, the gene encoding the double mutant (R365L/E366Q P450cam = P450cam*) was generated in parallel with the gene for the selenocysteine-containing variant (C357U P450cam*). Both constructs were cloned into expression plasmids under control of the salicylate promoter.
Enzyme Production.
Four gene products are required for efficient UGA suppression in bacteria (12–14). SelC is the suppressor tRNA with an anticodon complementary to UGA; SelA and SelD are the enzymes that load selenocysteine onto SelC; and SelB is a GTP-dependent elongation factor, homologous to EF-Tu, that recognizes the SECIS element and delivers the selenocysteinyl-tRNASec to the ribosomal active site. As high intracellular concentrations of SelA-C improve heterologous production of natural selenoproteins in bacteria (15), we produced C357U P450cam* in E. coli cotransformed with plasmid pSUABC (15), which directs expression of the selA-C genes. Cell cultures were supplemented with selenite as a selenium source and δ-aminolevulinic acid for heme biosynthesis. The control protein was produced by standard procedures in XL1 Blue cells. Both enzymes were isolated as previously described for P450cam (25) and purified to homogeneity by ion-exchange chromatography. One liter of cell culture typically afforded 5–10 mg of purified C357U P450cam*, which compares well with the yield of 20–30 mg of purified P450cam*. Although the selenoenzyme is produced in lower yield than the control protein, it is easier to purify because it is largely heme bound. The control protein is typically contaminated by large amounts of apoenzyme lacking the heme cofactor, so multiple chromatographic steps are needed to obtain pure holoenzyme. The proteins were characterized by SDS/PAGE and electrospray mass spectrometry (P450cam*: calculated 47561.9 Da, found 47561.6 Da; C357U P450cam*: calculated 47608.79 Da; found 47607.4 Da) [supporting information (SI) Fig. S1 A and B]. Selenocysteine incorporation in C357U P450cam* was additionally confirmed by X-ray fluorescence (Fig. S1C).
X-Ray Crystallographic Structure of the C357U P450cam* and P450cam* Camphor Complexes.
C357U P450cam* and P450cam* were crystallized in the presence of camphor, and their structures solved by molecular replacement (see Table S1 for details). Because the effects of the cysteine-to-selenocysteine exchange and the SECIS element mutations were expected to be small, the influence of crystal packing effects and refinement protocols was minimized by rerefining the native structures used for comparison with the same protocols as for the variants.
The structure of C357U P450cam*, including the active site (Fig. 3A), is very similar to that of WT P450cam and P450cam* (Table 1). The peptide loop, from which the axial heme ligand projects, adopts the same backbone conformation in all 3 proteins. Except for Leu-358, for which 2 rotameric conformations are observed in the selenoenzyme, the side chains in this region are also superimposable. The Se–Fe bond length is somewhat longer than the S–Fe bond length in WT P450cam structures determined from the same crystal forms (Table 1 and Table S2) and falls within the range of experimentally determined Se–Fe bond distances (2.39–2.49 Å) for synthetic iron–selenolate complexes (26–28) and nonheme enzymes (29). Despite the larger size of selenium, the network of hydrogen bonding interactions with the amide bonds of residues Leu-358, Gly-359, and Gln-360, which regulate the redox potential of the heme iron in the WT enzyme by stabilizing the heme–thiolate complex (30–32), is preserved in the selenoenzyme (see Table 1).
Fig. 3.

Structure of C357U P450cam* in the camphor-bound state. (A) Active site of the selenoenzyme (yellow) superimposed on that of P450cam* (green) and WT P450cam (1dz4, gray); all 3 structures were determined from the monoclinic crystal form. Although the axial cysteine ligand at position 357 was substituted by selenocysteine, the structural integrity of the enzyme is maintained. The increased Fe-Se distance may account for the 2 side chain conformations observed for Leu-358 in the selenoenzyme. The camphor-binding site is not affected by the mutations (see Fig. S2). The camphor molecule has been omitted for clarity. (B) View of the region around Gln-366 in C357U P450cam* (yellow), P450cam* (green), and the WT protein (gray). Mutating the native glutamate residue to glutamine induces displacement of the connected chain of ordered water molecules.
Table 1.
Comparison of the active site geometry of camphor complexes of P450cam, P450cam*, and C357U P450cam* determined from monoclinic crystals using the same refinement procedure
| Complex (PDB code) | Space group Unit cell (a, b, c) ÅMolecules per asymmetric unit | Resolution, Å | RMSD */native, Å | Coord. error†, Å | Distance, Å |
||||
|---|---|---|---|---|---|---|---|---|---|
| a | b | c | d | ||||||
| Native ferric P450cam (1dz4) | P2167.4, 62.7, 95.5, β = 90.65° | 1.60 | na | 0.09 | 2.38 | 3.5 | 3.3 | 3.3 | 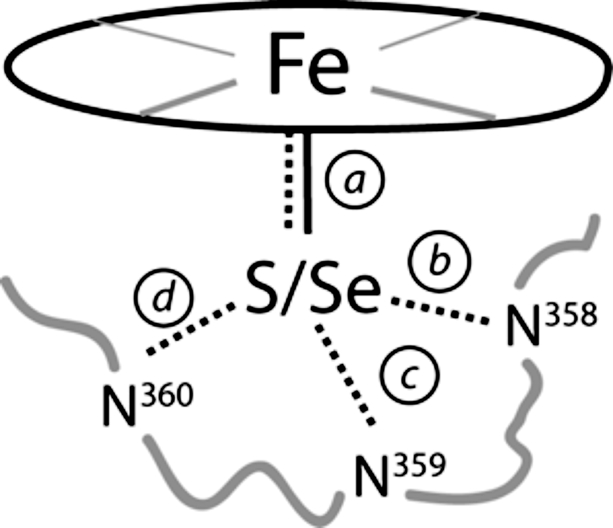 |
| 2.34 | 3.5 | 3.3 | 3.3 | ||||||
| 2 | |||||||||
| P450cam*(3fwg) | P21 67.2, 62.8, 95.6, β = 90.53° | 1.55 | 0.18 | 0.09 | 2.39 | 3.5 | 3.2 | 3.3 | |
| 0.17 | 2.34 | 3.5 | 3.2 | 3.4 | |||||
| 2 | |||||||||
| C357U P450cam*(3fwf) | P21 67.5, 61.9, 94.5, β = 90.80° | 1.83 | 0.26 | 0.13 | 2.47 | 3.5 | 3.3 | 3.6 | |
| 0.26 | 2.47 | 3.5 | 3.3 | 3.6 | |||||
| 2 | |||||||||
na, not applicable.
†Estimated coordinate (Coord.) error based on R-/Rfree value.
The additional R365L and E366Q mutations derived from the SECIS element lead to more pronounced structural changes than the selenium-for-sulfur swap. For example, Glu-128, which is adjacent to Arg-365, moves to fill the void created by the substitution of the latter with the smaller leucine (ΔCα = 0.8 Å), causing adjustments in nearby Val-124 (ΔCα = 0.8 Å) and residues in the neighboring C′ and D-helices (Fig. S2) as well as in the surrounding water structure. In contrast, mutation of Glu-366 to glutamine leads primarily to changes in the network of ordered water molecules near the active site. As the result of a ≈30° rotation of the amide side chain relative to the original carboxylate, Wat523, Wat566, and Wat687 are displaced by ≈1.5, ≈0.5, and ≈0.2 Å, respectively, from their positions in the WT enzyme (Fig. 3B). Because these waters have been postulated to facilitate proton transfer to the hydrophobic active site during catalytic turnover (33, 34), the observed changes could be functionally significant. Contrasting WT and control proteins enables this effect to be estimated. The consequences of replacing the thiolate heme ligand with the more electron donating selenolate can then be assessed in the absence of other confounding structural changes by comparing C357U P450cam* and P450cam*, which share the SECIS mutations and possess identical active site water structures (Fig. 3B).
Biophysical Characterization.
C357U P450cam* exhibits typical optical absorption spectra for cytochromes, although the maxima are red shifted relative to the WT and control proteins. For example, the ferric form of the selenoenzyme has an absorption maximum at 395 nm in the presence of D-(+)-camphor, rather than at 391 nm as seen for P450cam and P450cam* (Fig. 4A). Similarly, the Soret band for the ferrous CO form is shifted from 446 nm for the control protein to 453 nm for the selenocysteine-containing variant (Fig. 4B). Complexes of WT P450cam with benzeneselenol show a similar red shift relative to the corresponding thiophenol complexes (35), consistent with the lower ionization energy of selenium.
Fig. 4.

Characterization of C357U P450cam* (red) and the control protein P450cam* (black). (A) UV-vis absorption spectra of the ferric resting state with bound camphor. (B) Ferrous CO form of the enzymes. (C) EPR spectra at 15 K. (D) Transformation of camphor by C357U P450cam* at 25 °C as a function of time.
The sulfur-to-selenium substitution significantly alters the electron paramagnetic resonance (EPR) spectrum of the camphor complex (Fig. 4C). As shown previously for WT P450cam, the control protein exists as a 60:40 mixture of high- (S = 5/2) and low- (S = 1/2) spin states at 15 K. The g values for both spin fractions are within the range of published values (36) (see Table S3). In contrast, >90% of the C357U P450cam* sample is in the low-spin state. These results are consistent with the larger field effect expected for the selenolate compared with the thiolate. However, because the g values for the low-spin fraction (gx = 2.00, gy = 2.27, and gz = 2.47) are only 1.5–2% larger than those for the corresponding low-spin state of P450cam*, large changes in the ligand geometry and environment can be excluded.
The redox potential of the Fe(III)/Fe(II) couple was investigated by Osteryoung-type square wave voltammetry using enzymes that were immobilized in a didodecyldimethylammonium bromide (DDAB) film on an edge-plane graphite electrode (37). The selenoprotein has a 48 mV more negative peak potential (2 ± 6 mV vs. NHE) than the control protein (50 ± 4 mV) (Fig. S3), again consistent with the stronger electron-donating effect of selenium compared with sulfur. The peak potential for WT P450cam (45 ± 2 mV) is similar to that of the control protein, and decreases to −46 ± 5 mV under denaturing conditions. As seen in other studies of P450 proteins in surfactant films (37), these values are ≈200–300 mV more positive than redox potentials determined with nonimmobilized enzymes.
Selenolate ligands like benzeneselenol, bound to the proximal cavity of a mutant P450-like enzyme lacking a distal electron-donating substituent, have been shown to reduce the heme iron to the ferrous state (35). In contrast, the ferric form of C357U P450cam* is stable under aerobic conditions. Despite the highly reducing nature of selenols compared with thiols (6), the relatively low redox potential of the Fe(III)/Fe(II) couple in the selenoenzyme, coupled with conserved hydrogen-bonding interactions with the axial ligand, presumably stabilize the selenolate.
Catalytic Properties.
Catalytic turnover in P450cam requires the enzymes putidaredoxin (Pdx) and putidaredoxin reductase (Pdr), which mediate electron transfer from NADH to the heme. If this process is tightly coupled to substrate hydroxylation, as is the case for WT P450cam (38), the rate of NADH oxidation reflects the rate of enzyme turnover. The consumption of NADH was therefore monitored spectroscopically at 25 °C in the presence of substrate for each variant (Table 2). The specific activity of the selenoenzyme is approximately half that of the control protein, which, in turn, is 2-fold less active than native P450. The latter effect can be ascribed to the SECIS mutations. In addition to modulating active-site water structure, they appear to perturb the putidaredoxin-binding site directly, because higher putidaredoxin concentrations are needed to saturate P450cam* and C357U P450cam* than the WT enzyme (Fig. S4). The further reduction in catalytic efficiency attributable to the modified electronic properties of the heme–selenolate cofactor is surprisingly modest.
Table 2.
Catalytic properties of C357U P450cam*, P450cam*, and WT P450cam
Because P450 mutations often redirect reducing equivalents into abortive side reactions, such as the production of superoxide and hydrogen peroxide (Fig. 1) (2), assessing the extent of uncoupling in the individual variants is important. Indeed, the ferrous oxygenated forms of P450cam* and the selenoenzyme autooxidize (k = 0.23 min−1 and 0.31 min−1, respectively) 7- to 10-times more rapidly than WT P450 (k = 0.033 min−1) (39). Disruption of the network of distal active-site waters, which has been previously invoked to explain strong uncoupling in the T252A mutant (33, 40), presumably accounts for this observation. Nevertheless, autooxidation does not appear to compete significantly with substrate hydroxylation, because formation of exo-5-hydroxycamphor, monitored by GC (Fig. 4D), remains coupled to NADH consumption for all 3 proteins. The initial rate of appearance of the hydroxylated products was within experimental error of the independently measured rates of NADH consumption for P450cam and P450cam* and only somewhat slower for the selenoenzyme (Table 2). These results were confirmed by direct quantification of hydrogen peroxide by an iron thiocyanate assay (38). Comparison with the amount of NADH consumed gave <2% uncoupling for WT P450cam and P450cam* but 6–10% for the selenoenzyme. The enhanced production of H2O2 by the selenoenzyme compared with the control cannot be rationalized by disruption of the active-site water structure, because C357U P450cam* and P450cam* have identical distal environments (Fig. 3).
Discussion
Selenium extends the properties of many natural proteins (13, 14), providing novel prospects for redox activity and metal binding. More generally, it represents a valuable spectroscopic and mechanistic probe (7) and has been used to create artificial catalysts with tailored hydrolytic and redox properties (9–11, 41–44). A robust method for placing selenocysteine at any site, in any protein would greatly enhance its utility. Our results show that the normal biosynthetic machinery of E. coli can be successfully exploited for targeted insertion of selenocysteine into large and complex proteins like P450cam, provided that a functional SECIS element can be engineered into the encoding gene.
Practical constraints on cotranslational selenocysteine insertion derive from the need for a productive interaction between the elongation factor SelB and the mRNA SECIS element (45). In the case of P450cam, this overcoding mechanism necessitated making 2 mutations in addition to the desired C357U. However, these substitutions could be introduced at positions in the protein where they are readily tolerated. Subtle changes in water structure at the active site give rise to minor differences in specific activity and coupling efficiency. Nonetheless, WT P450cam and the control protein exhibit similar spectroscopic and catalytic properties overall. Because the basic chemistry of the system is preserved, the effect of the axial selenolate ligand on the redox properties of the heme cofactor can be readily discerned by comparison of the selenoenzyme with the P450cam* control.
X-ray crystallographic analysis confirms that replacing the heme–thiolate with a heme–selenolate constitutes a highly conservative substitution. Despite the slightly larger van der Waals radius of selenium compared with sulfur, the geometry and environment of the heme ligand are fully preserved in the mutant. However, the greater electron-donating ability of the selenolate compared with a thiolate causes distinct changes in the electronic and chemical properties of the enzyme, including diagnostic redshifts of the absorbtion maxima, perturbation of the spin equilibrium in favor of the low-spin resting state, and a lowered Fe(III)/Fe(II) redox potential. The rate of camphor hydroxylation is also reduced 2-fold compared with the control enzyme, consistent with the mutant's lowered redox potential. Although detailed stopped-flow and rapid-quench experiments will be needed to assess how the selenolate ligand influences individual steps in the catalytic cycle, this effect is considerably smaller than the 100-fold decrease in hydroxylation rate predicted by QM/MM calculations (8), suggesting that downstream chemistry is still very fast compared with the initial electron transfer steps. Nevertheless, the modest increase in H2O2 production observed for the selenoenzyme compared with the control protein is at odds with the general view (5, 30, 31) that a more electron-donating axial heme ligand should enhance coupling of NADH consumption with substrate oxidation by facilitating cleavage of the dioxygen bond. As a consequence, a detailed experimental and theoretical reexamination of the postulated role(s) of the axial heme ligand in P450 chemistry may be warranted.
Because selenols are versatile functional groups, with valuable nucleophilic, radical, and redox properties (6), the ability to produce proteins containing selenocysteine recombinantly should foster their broader application as sensitive probes in studies of enzyme mechanism and as prosthetic groups in tailor-made catalysts. By coevolving SECIS elements and cognate SelB elongation factors, it may be possible to further generalize this approach, simultaneously minimizing undesired sequence changes and expanding the recognition properties of SelB to new amino acids. Exploiting the basic bacterial overcoding strategy in this way to enhance stop codon suppression should also profitably extend ongoing efforts to expand the genetic code (46).
Methods
Site-Directed Mutagenesis.
The gene encoding cytochrome P450cam was subcloned into vector pMG211 (4,690 bp) (47) by standard overlap-extension PCR to attach a C-terminal Leu-Glu-His6 tag. The primers wtP450sense (5′-GGGAATTCCATATGACGACTGAAACC, NdeI site in italics) and wtP450anti (5′-GCCGCTCGAGTACCGCTTTGGTAGT, XhoI site in italics) were used for amplification. The resulting plasmid, pMG211_P450cam, served as the template to construct plasmids pMG211_P450cam* and pMG211_C357UP450cam*, encoding R365L/E366Q P450cam (P450cam*) and C357U P450cam*, respectively. Mutations were introduced by PCR mutagenesis using primers P450* sense (5′-TGCCTCGGCCAACATCTTGCACGTCTGCAGATCATCGTCACCCT); P450*anti (5′-CAGACGTGCAAGATGTTGGCCGAGGCACAGATGGCTGCCGTGG); C357U*sense (5′-TGACTCGGCCAACATCTTGCACGTCTGCAGATCATCGTCACCCT); and C357U*anti (5‘-CAGACGTGCAAGATGTTGGCCGAGTCACAGATGGCTGCCGTGG). The mutations are shown in bold type. All constructs were confirmed by DNA sequence analysis.
Protein Production and Purification.
C357U P450cam* was produced in E. coli XL1-Blue cells (Stratagene) cotransformed with plasmid pSUABC (15). Cells were grown to OD600 ≈1.8 in Terrific-Broth (TB) medium supplemented with 100 mg/L ampicillin, 34 mg/L chloramphenicol, and 0.6 mM l-cysteine. After induction with 200 μM salicylate, the broth was supplemented with 5 μM sodium selenite and 1 mM δ-aminolevulinic acid and incubated for ≈20 h at 30 °C. P450cam* and WT P450cam were produced in E. coli XL1-Blue cells by standard procedures. The proteins were purified by Ni–NTA affinity chromatography in the presence of D-(+)-camphor, followed by repeated ion exchange chromatography on Q-Sepharose until A391/A280 was ≥1.4 for enzyme used for kinetic measurements and ≥1.1 for EPR studies. The purity of all protein samples was assessed by SDS/PAGE. Masses were determined by ESI-MS analysis. Protein concentrations were determined spectroscopically at 391 nm (WT and P450cam*) and 395 nm (C357U P450cam*) by using a molar absorption coefficient of 102 mM−1 cm−1. The extinction coefficient of the C357U P450cam* Soret band was determined to be the same as for P450cam and P450cam* by normalizing all 3 samples to the same absorbance at the peak maximum, followed by measurement of protein and heme concentrations by using Bradford protein assay and the QuantiChrom Heme Chrome Assay kit (BioAssay Systems), respectively.
Crystallization and Structure Determination of the C357U P450cam* and P450cam* Camphor Complexes.
The proteins were crystallized as described previously (48) with minor adjustments in the concentrations of DTE and camphor (2 mM each), and their structures solved by molecular replacement. See SI Methods for detailed procedures and a summary of crystal parameters, data collection, and refinement statistics.
Biophysical Characterization and Catalytic Properties.
For full experimental details, see SI Methods. EPR spectra of samples containing 10% glycerol were recorded at 15 K on a multipurpose Bruker E680 spectrometer equipped with an Oxford CF935 helium cryostat and EN4118X-MD-4-W1 probe head (≈9.73 GHz). Redox potentials were measured by square wave voltammetry at 4 °C in 50 mM potassium phosphate (pH 7.4), as previously described (37, 49). Catalytic activities were determined at 25 °C by monitoring the rate of NADH oxidation spectroscopically or by quantitating the formation of 5-exo-hydroxycamphor by gas chromatography (30). The rate of Fe(II)-O2 decay was measured at 4 °C as previously described (39), and hydrogen peroxide concentrations were determined by a colorimetric assay (38).
Supplementary Material
Acknowledgments.
We are grateful to Dr. Christiane Jung for the plasmids with the Pdr and Pdx genes and Dr. Elias Arnér for plasmid pSUABC. Diffraction data were collected at the Swiss Light Source, beamline X10SA, Paul Scherrer Institute, Villigen, Switzerland. We thank the Dortmund–Heidelberg team for data collection and the X10SA beamline staff for their support in setting up the beamline. We are grateful to Ingrid Vetter for support of the crystallographic software. This work was supported by the ETH Zurich. This project was carried out as a collaboration with Prof. Arthur Schweiger. It is dedicated in his memory.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 3FWG, 3FWF, 3FWJ, and 3FWI).
This article contains supporting information online at www.pnas.org/cgi/content/full/0810503106/DCSupplemental.
A web-based server for designing SECIS elements within coding sequences has been described (ref. 20) that is particularly useful for optimizing the sequence of the lower stem. However, because the program fixes the canonical bases in the SECIS hairpin loop, 3 mutations (R364G, R365L, and E366Q) would be required to replace the axial heme ligand of P450cam with selenocysteine rather than 2.
References
- 1.Dawson JH. Probing structure-function relations in heme-containing oxygenases and peroxidases. Science. 1988;240:433–439. doi: 10.1126/science.3358128. [DOI] [PubMed] [Google Scholar]
- 2.Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and chemistry of cytochrome P450. Chem Rev. 2005;105:2253–2278. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 3.Ortiz de Montellano PR. Cytochrome P450: Structure, Mechanism, and Biochemistry. New York: Kluwer Academic/Plenum; 2005. [Google Scholar]
- 4.Yoshioka S, Takahashi S, Hori H, Ishimori K, Morishima I. Proximal cysteine residue is essential for the enzymatic activities of cytochrome P450cam. Eur J Biochem. 2001;268:252–259. doi: 10.1046/j.1432-1033.2001.01872.x. [DOI] [PubMed] [Google Scholar]
- 5.Auclair K, Moënne-Loccoz P, Ortiz de Montellano PR. Roles of the proximal heme thiolate ligand in cytochrome P450cam. J Am Chem Soc. 2001;123:4877–4885. doi: 10.1021/ja0040262. [DOI] [PubMed] [Google Scholar]
- 6.Patai S, Rappoport Z. The Chemistry of Organic Selenium and Tellurium Compounds. Chichester, UK: Wiley; 1986. [Google Scholar]
- 7.Moroder L. Isosteric replacement of sulfur with other chalcogens in peptides and proteins. J Pept Sci. 2005;11:187–214. doi: 10.1002/psc.654. [DOI] [PubMed] [Google Scholar]
- 8.Cohen S, Kumar D, Shaik S. In silico design of a mutant of cytochrome P450 containing selenocysteine. J Am Chem Soc. 2006;128:2649–2653. doi: 10.1021/ja056586c. [DOI] [PubMed] [Google Scholar]
- 9.Hondal RJ, Raines RT. Semisynthesis of proteins containing selenocysteine. Methods Enzymol. 2002;347:70–83. doi: 10.1016/s0076-6879(02)47009-7. [DOI] [PubMed] [Google Scholar]
- 10.Wu Z-P, Hilvert D. Conversion of a protease into an acyl transferase: Selenolsubtilisin. J Am Chem Soc. 1989;111:4513–4514. [Google Scholar]
- 11.Müller S, et al. The formation of diselenide bridges in proteins by incorporation of selenocysteine residues: Biosynthesis and characterization of (Se)2-thioredoxin. Biochemistry. 1994;33:3404–3412. doi: 10.1021/bi00177a034. [DOI] [PubMed] [Google Scholar]
- 12.Böck A, Forchhammer K, Heider J, Baron C. Selenoprotein synthesis: An expansion of the genetic code. Trends Biochem Sci. 1991;16:463–467. doi: 10.1016/0968-0004(91)90180-4. [DOI] [PubMed] [Google Scholar]
- 13.Stadtman TC. Selenocysteine. Annu Rev Biochem. 1996;65:83–100. doi: 10.1146/annurev.bi.65.070196.000503. [DOI] [PubMed] [Google Scholar]
- 14.Johansson L, Gafvelin G, Arnér ESJ. Selenocysteine in proteins—Properties and biotechnological use. Biochim Biophys Acta. 2005;1726:1–13. doi: 10.1016/j.bbagen.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 15.Arnér ESJ, Sarioglu H, Lottspeich F, Holmgren A, Böck A. High-level expression of Escherichia coli of selenocysteine-containing rat thioredoxin reductase utilizing gene fusions with engineered bacterial-type SECIS elements and co-expression with the selA, selB and selC genes. J Mol Biol. 1999;292:1003–1016. doi: 10.1006/jmbi.1999.3085. [DOI] [PubMed] [Google Scholar]
- 16.Hazebrouck S, Camoin L, Faltin Z, Strosberg AD, Eshdat Y. Substituting selenocysteine for catalytic cysteine 41 enhances enzymatic activity of plant phospholipid hydroperoxide glutathione peroxidase expressed in Escherichia coli. J Biol Chem. 2000;275:28715–28721. doi: 10.1074/jbc.M004985200. [DOI] [PubMed] [Google Scholar]
- 17.Bar-Noy S, Moskovitz J. Mouse methionine sulfoxide reductase B: Effect of selenocysteine incorporation on its activity and expression of the seleno-containing enzyme in bacterial and mammalian cells. Biochem Biophys Res Commun. 2002;297:956–961. doi: 10.1016/s0006-291x(02)02314-8. [DOI] [PubMed] [Google Scholar]
- 18.Jiang Z, et al. Expression of selenocysteine-containing glutathione S-transferase in Escherichia coli. Biochem Biophys Res Commun. 2004;321:94–101. doi: 10.1016/j.bbrc.2004.06.110. [DOI] [PubMed] [Google Scholar]
- 19.Johansson L, et al. Exploiting the 21st amino acid—Purifying and labeling proteins by selenolate targeting. Nat Methods. 2004;1:61–66. doi: 10.1038/nmeth707. [DOI] [PubMed] [Google Scholar]
- 20.Busch A, Will S, Backofen R. SECISDesign: A server to design SECIS-elements within the coding sequence. Bioinformatics. 2005;21:3312–3313. doi: 10.1093/bioinformatics/bti507. [DOI] [PubMed] [Google Scholar]
- 21.Zinoni F, Heider J, Böck A. Features of the formate dehydrogenase mRNA necessary for decoding of the UGA codon as selenocysteine. Proc Natl Acad Sci USA. 1990;87:4660–4664. doi: 10.1073/pnas.87.12.4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Z, Reches M, Groisman I, Engelberg-Kulka H. The nature of the minimal “selenocysteine insertion sequence” (SECIS) in Escherichia coli. Nucleic Acids Res. 1998;26:896–902. doi: 10.1093/nar/26.4.896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sandman KE, Tardiff DF, Neely LA, Noren CJ. Revised Escherichia coli selenocysteine insertion requirements determined by in vivo screening of combinatorial libraries of SECIS variants. Nucleic Acids Res. 2003;31:2234–2241. doi: 10.1093/nar/gkg304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharp PM, et al. Codon usage patterns in Escherichia coli, Bacillus subtilis, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Drosophila melanogaster and Homo sapiens; A review of the considerable within-species diversity. Nucleic Acids Res. 1988;16:8207–8211. doi: 10.1093/nar/16.17.8207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shimoji M, Yin H, Higgins L, Jones JP. Design of a novel P450: A functional bacterial–human cytochrome P450 chimera. Biochemistry. 1998;37:8848–8852. doi: 10.1021/bi972775z. [DOI] [PubMed] [Google Scholar]
- 26.Song L-C, Sun Y, Hu Q-M, Liu Y. Synthesis of bridging carbyne butterfly Fe/Se cluster complexes via reaction of complex anions [(μ-RSe)(μ-CO)Fe2(CO)6]−: Crystal structure of (μ-EtOC)(μ-p-MeC6H4Se)Fe2(CO)6. J Organomet Chem. 2003;676:80–84. [Google Scholar]
- 27.El-Khateeb M. Iron Se-bonded mono-selenocarbonates CpFe(CO)2SeCO2R: The first selenocarbonate complexes. Inorg Chim Acta. 2004;357:4341–4344. [Google Scholar]
- 28.Mathur P, et al. Synthesis and structural characterisation of a novel tris-methylene bridged compound (NO) 4Fe2 Se(μ-CH2)3. J Organomet Chem. 1997;540:165–168. [Google Scholar]
- 29.Garcin E, et al. The crystal structure of a reduced [NiFeSe] hydrogenase provides an image of the activated catalytic center. Structure (London) 1999;7:557–566. doi: 10.1016/s0969-2126(99)80072-0. [DOI] [PubMed] [Google Scholar]
- 30.Yoshioka S, Takahashi S, Ishimori K, Morishima I. Roles of the axial push effect in cytochrome P450cam studied with the site-directed mutagenesis at the heme proximal site. J Inorg Biochem. 2000;81:141–151. doi: 10.1016/s0162-0134(00)00097-0. [DOI] [PubMed] [Google Scholar]
- 31.Yoshioka S, et al. Roles of the proximal hydrogen bonding network in cytochrome P450cam-catalyzed oxygenation. J Am Chem Soc. 2002;124:14571–14579. doi: 10.1021/ja0265409. [DOI] [PubMed] [Google Scholar]
- 32.Schöneboom JC, et al. The elusive oxidant species of cytochrome P450 enzymes: Characterization by combined quantum mechanical/molecular mechanical (QM/MM) calculations. J Am Chem Soc. 2002;124:8142–8151. doi: 10.1021/ja026279w. [DOI] [PubMed] [Google Scholar]
- 33.Harris DL, Loew GH. A role for Thr252 in cytochrome P450cam oxygen activation. J Am Chem Soc. 1994;116:11671–11674. doi: 10.1021/ja00105a007. [DOI] [PubMed] [Google Scholar]
- 34.Kumar D, et al. New features in the catalytic cycle of cytochrome P450 during the formation of compound I from compound 0. J Phys Chem B. 2005;109:19946–19951. doi: 10.1021/jp054754h. [DOI] [PubMed] [Google Scholar]
- 35.Jiang Y, Ortiz de Montellano PR. Selenolate complexes of CYP101 and the heme-bound hHO-1/H25A proximal cavity mutant. Inorg Chem. 2008;47:3480–3482. doi: 10.1021/ic800148a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lipscomb JD. Electron-paramagnetic resonance detectable states of cytochrome P-450cam. Biochemistry. 1980;19:3590–3599. doi: 10.1021/bi00556a027. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Z, Nassar A-EF, Lu Z, Schenkman JB, Rusling JF. Direct electron injection from electrodes to cytochrome P450cam in biomembrane-like films. J Chem Soc Faraday Trans. 1997;93:1769–1774. [Google Scholar]
- 38.Atkins WM, Sligar SG. Deuterium isotope effects in norcamphor metabolism by cytochrome P-450cam: Kinetic evidence for the two-electron reduction of a high-valent iron-oxo intermediate. Biochemistry. 1988;27:1610–1616. doi: 10.1021/bi00405a033. [DOI] [PubMed] [Google Scholar]
- 39.Sligar SG, Lipscomb JD, Debrunner PG, Gunsalus IC. Superoxide anion production by the autooxidation of cytochrome P450cam. Biochem Biophys Res Commun. 1974;61:290–296. doi: 10.1016/0006-291x(74)90565-8. [DOI] [PubMed] [Google Scholar]
- 40.Imai M, et al. Uncoupling of the cytochrome P-450cam monooxygenase reaction by a single mutation, threonine-252 to alanine or valine: A possible role of the hydroxy amino acid in oxygen activation. Proc Natl Acad Sci USA. 1989;86:7823–7827. doi: 10.1073/pnas.86.20.7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu Z-P, Hilvert D. Selenosubtilisin as a glutathione peroxidase mimic. J Am Chem Soc. 1990;112:5647–5648. [Google Scholar]
- 42.Casi G, Roelfes G, Hilvert D. Selenoglutaredoxin as a glutathione peroxidase mimic. ChemBioChem. 2008;9:1623–1631. doi: 10.1002/cbic.200700745. [DOI] [PubMed] [Google Scholar]
- 43.Berry SM, Gieselman MD, Nilges MJ, van der Donk WA, Lu Y. An engineered azurin variant containing a selenocysteine copper ligand. J Am Chem Soc. 2002;124:2084–2085. doi: 10.1021/ja0169163. [DOI] [PubMed] [Google Scholar]
- 44.Sarangi R, et al. Spectroscopic and density functional theory studies of the blue-copper site in M121SeM and C112SeC azurin: Cu-Se versus Cu-S bonding. J Am Chem Soc. 2008;130:3866–3877. doi: 10.1021/ja076495a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoshizawa S, et al. Structural basis for mRNA recognition by elongation factor SelB. Nat Struct Mol Biol. 2005;12:198–203. doi: 10.1038/nsmb890. [DOI] [PubMed] [Google Scholar]
- 46.Wang L, Schultz PG. Expanding the genetic code. Chem Commun. 2002:1–11. doi: 10.1039/b108185n. [DOI] [PubMed] [Google Scholar]
- 47.Sasso S, Ramakrishnan C, Gamper M, Hilvert D, Kast P. A secreted chorismate mutase from the pathogen Mycobacterium tuberculosis. FEBS J. 2005;272:375–389. doi: 10.1111/j.1742-4658.2004.04478.x. [DOI] [PubMed] [Google Scholar]
- 48.Schlichting I, et al. The catalytic pathway of cytochrome p450cam at atomic resolution. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 49.Shumyantseva VV, et al. A new format of electrodes for the electrochemical reduction of cytochromes P450. J Inorg Biochem. 2006;100:1353–1357. doi: 10.1016/j.jinorgbio.2006.03.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.