

Abstract
Purpose
To evaluate noninvasive molecular imaging methods as correlative biomarkers of therapeutic efficacy of cetuximab in human colorectal cancer cell line xenografts grown in athymic nude mice. The correlation between molecular imaging and immunohistochemical analysis to quantify epidermal growth factor (EGF) binding, apoptosis, and proliferation was evaluated in treated and untreated tumor-bearing cohorts.
Experimental Design
Optical imaging probes targeting EGF receptor (EGFR) expression (NIR800-EGF) and apoptosis (NIR700-Annexin V) were synthesized and evaluated in vitro and in vivo. Proliferation was assessed by 3′-[18F]fluoro-3′-deoxythymidine ([18F] FLT) positron emission tomography. Assessment of inhibition of EGFR signaling by cetuximab was accomplished by concomitant imaging of NIR800-EGF, NIR700-Annexin V, and [18F] FLT in cetuximab-sensitive (DiFi) and insensitive (HCT-116) human colorectal cancer cell line xenografts. Imaging results were validated by measurement of tumor size and immunohistochemical analysis of total and phosphorylated EGFR, caspase-3, and Ki-67 immediately following in vivo imaging.
Results
NIR800-EGF accumulation in tumors reflected relative EGFR expression and EGFR occupancy by cetuximab. NIR700-Annexin V accumulation correlated with cetuximab-induced apoptosis as assessed by immunohistochemical staining of caspase-3. No significant difference in tumor proliferation was noted between treated and untreated animals by [18F] FLT positron emission tomography or Ki-67 immunohistochemistry.
Conclusions
Molecular imaging can accurately assess EGF binding, proliferation, and apoptosis in human colorectal cancer xenografts. These imaging approaches may prove useful for serial, noninvasive monitoring of the biological effects of EGFR inhibition in preclinical studies. It is anticipated that these assays can be adapted for clinical use.
The epidermal growth factor receptor (EGFR, HER1) is frequently overexpressed in colorectal cancer. Extensive preclinical data support pharmacologic blockade of the EGFR as an effective therapeutic strategy in advanced colorectal cancer (1-3). As a result, molecularly targeted agents designed to inhibit various aspects of EGFR signaling have been developed and extensively studied in individuals with colorectal cancer (4-11). These agents include chimeric (e.g., cetuximab; refs. 1, 7) or fully human (e.g., panitumumab; refs. 12, 13) monoclonal antibodies (mAb) that block ligand engagement with the extracellular domain of EGFR as well as small molecules that block the catalytic domain of EGFR tyrosine kinase. Whereas EGFR-targeted tyrosine kinase inhibitors such as gefitinib (Iressa; refs. 8, 14) or erlotinib (Tarceva; refs. 15) have not shown clinical activity in colorectal cancer as monotherapy (9), mAbs including cetuximab (C225, Erbitux) and panitumumab (Vectibix) have consistently produced single-agent response rates of 10% to 11% as second- or third-line therapy in patients with metastatic colorectal cancer, and both are now Food and Drug Administration approved for this purpose (5, 10, 16).
Translational Relevance.
Noninvasive molecular imaging is capable of visualizing and quantifying cellular and physiologic processes in vivo. We have evaluated the concomitant use of three complementary noninvasive molecular imaging modalities as correlative biomarkers of therapeutic efficacy of cetuximab in human colorectal cancer cell line xenografts grown in athymic nude mice. The reported in vivo imaging approaches, which include metrics of EGF uptake, apoptosis, and proliferation, were evaluated within the context of a cetuximab-sensitive (wild-type KRAS) and a cetuximab-resistant (mutant KRAS) human colorectal cancer cell line. Noninvasive imaging results agreed closely with immunohistochemical analysis of tumor tissues collected immediately following in vivo imaging. Our data suggest that these imaging approaches may prove useful for serial, non-invasive monitoring of the biological effects of EGFR inhibition in preclinical studies. Furthermore, it is anticipated that these assays can be adapted for clinical use.
Despite numerous studies showing that EGFR-directed mAbs significantly improve the outcome of patients with advanced colorectal cancer (10, 16-19), noninvasive biomarkers suitable for indicating and predicting therapeutic response to these agents are lacking. Using tumor biopsy specimens, it has recently been shown that tumor expression of epiregulin and amphiregulin (20), as well as wild-type KRAS (20, 21), predict sensitivity to cetuximab and panitumumab in patients with colorectal cancer. However, the only noninvasive indicator of therapeutic response to this and similar EGFR-targeted agents remains a characteristic acneiform rash, and not all patients who develop rash obtain a clinical response (5, 10, 22). This underscores the critical need to develop and employ noninvasive biomarkers suitable for objective and early assessment of clinical response to this class of therapeutics.
Noninvasive molecular imaging enables cellular and physiologic processes to be visualized and quantified in vivo. Widely recognized as a tool for cancer detection in most organ sites, the vast majority of clinical molecular imaging procedures employ the positron emission tomography (PET) tracer fluorodeoxyglucose. Noninvasive molecular imaging using a variety of probes offers great promise as a means to assess response to conventional and molecularly targeted therapeutic interventions (23-25). Given that EGFR serves as a mediator of cellular proliferation and apoptosis, quantitative assessment of these and other molecular events could potentially serve as biomarkers of response to EGFR-directed therapies. Conventional methods employed to assess tumor proliferation and apoptosis require invasive procurement of limited amounts of tissue with attendant risks and sampling errors due to tumor heterogeneity. Furthermore, serial tumor biopsies as are required to assess treatment response longitudinally are clinically impractical in many instances. Molecular imaging circumvents these limitations and offers potential advantages over traditional biopsy-based procedures to predict and/or assess treatment response. Our interest in developing noninvasive biomarkers to profile treatment response to molecularly targeted therapies in colorectal cancer led us to evaluate 3′-[18F] fluoro-3′-deoxythy-midine ([18F] FLT) PET imaging as a metric for assessing cellular proliferation (26-29) as well as near-infrared (NIR)-based imaging probes to assess EGF uptake (NIR800-EGF) and apoptosis (NIR700-Annexin V) within human colorectal cancer tumor xenografts. Whereas both Annexin V and [18F] FLT uptake are downstream markers for apoptosis and proliferation, respectively, EGF binding to its receptor is a specific, proximal molecular event, and its assessment provides a measure of EGFR occupancy by cetuximab. Data presented here suggest that these three imaging-based metrics have potential utility for determining the efficacy of EGFR-directed therapy.
Materials and Methods
Preparation of NIR800-EGF and NIR700-Annexin V imaging probes
NIR800-EGF and NIR700-Annexin V were prepared by reconstituting human recombinant EGF (Upstate) or human-derived Annexin V (Sigma) in ice-cold PBS (pH 7.4; 0.2 g/L). NIR dye (LI-COR 800CW or 700DX; LI-COR Biosciences) dissolved previously in DMSO was added to the protein solution with vortex mixing (NIR800-EGF, dye to protein stoichiometry, 3:1; NIR700-Annexin V, 9:1). The reaction vessel was protected from light and gently agitated for 2 h at 4°C. The progress of the labeling reaction was monitored by gel filtration chromatography (Superdex 200HR), eluting with 1 × PBS. Following conjugation, labeled probes were purified exhaustively by dialysis (1 × PBS, 4°C) using 3500 MWCO dialysis cassettes (Pierce). The purity of conjugates was assessed chromatographically, and dye/protein ratio (routinely 1:1) was quantified by spectrophotometry. Spectroscopic properties of the imaging probes were evaluated in 1 × PBS at room temperature. Absorbance of aqueous solutions was measured using a Shimadzu 1701 UV/Vis spectrophotometer and fluorescence emission measured using a PTI (QM-4ME) spectrofluorimeter.
Synthesis of [18F] FLT
[18F] FLT was prepared from [18F]fluoride in a two-step, one-pot reaction as described previously (27) using a GE TRACERlab FX-FN automated module. Aqueous [18F]fluoride from a H2[18O]O target was trapped by ion exchange (QMA; Waters) and then was eluted with Kryptofix-222 and K2CO3 in CH3CN/H2O into the reaction vessel. Three sequences of heating (110°C) with He(g) flow resulted in dry [18F]fluoride/Kryptofix-222/K2CO3. The cyclic precursor 2,3′-anhydro-5′-O-benzoyl-2′-deoxythymidine (ABX Advanced Biochemical Compounds) was added in DMSO and reacted for 10 min at 160°C. The benzoyl-protecting group was removed from the labeled intermediate by basic hydrolysis (0.25 mol/L NaOH, 50°C, 10 min). The reaction mixture was purified on a semipreparative C-18 high-performance liquid chromatography column eluting with 10% ethanol/10 mmol/L sodium phosphate buffer and sterilized by 0.2 μm membrane filtration. Radiochemical identity, purity, and specific activity were determined by analytic high-performance liquid chromatography. Product was obtained with average radiochemical purity of 98.3% and specific activity of 3,480 Ci/mmol.
In vitro NIR800-EGF binding assays
The in vitro specificity of NIR800-EGF was evaluated via competitive binding assays between the labeled probe and the unlabeled (cold) EGF in cell lines with known EGFR expression profiles (A431 and DiFi, both ~2 × 106 to 4 × 106 receptors per cell; refs. 30, 31). Cells were propagated in DMEM supplemented with 10% fetal bovine serum and 1 μg/mL gentamicin sulfate at 37°C, 5% CO2. For assay, cells were seeded at a density of 4 × 105 per well into 96-well optical bottom plates (Nunc) and allowed to adhere for 24 h. Cells were then incubated with 56 ng/mL NIR-EGF and either 0, 5, 50, 500, or 5000 ng/mL unlabeled EGF for 20 min at 37°C, 5% CO2. Following incubation, monolayers were rinsed three times with fresh culture medium, and cellular uptake of the imaging probe was assessed using an Odyssey plate reader.
Binding variables Kd and Bmax were assessed in live DiFi cells. Cells were seeded in 96-well optical bottom plates as described above. Next, the cells were incubated with serial concentrations (half-log) of NIR800-EGF (0-5 × 10-6 mol/L; 13 total dilutions) for 20 min at 37°C, 5% CO2. Following incubation, cells were rinsed with fresh culture medium, and uptake was measured using an Odyssey system. Nonspecific binding of NIR800-EGF was assessed by incubating cells at the above concentrations concomitantly with excess (2 × 10-6 mol/L) unlabeled EGF and subsequently measuring agent uptake. Specific binding data were analyzed to determine Kd using GraphPad Prism 4.0. Bmax for NIR800-EGF in DiFi cells was determined by correlating the molar quantity of NIR800-EGF with plate reader-measured fluorescence intensities. A calibration curve for NIR800-EGF in PBS was found to be linear over the concentration range 0 to 100 pmol/L, thus enabling estimation of cellular bound imaging agent. Fluorescence intensity values recorded from the binding assay were converted to fmol agent/ mg protein using the fluorescence calibration data and total cellular protein was assayed per 4 × 105 DiFi cells (Bradford assay).
Cellular imaging of NIR800-EGF in DiFi cells
Intracellular uptake and distribution of NIR800-EGF was visualized by fluorescence microscopy in DiFi cells. Cells were propagated to 50% confluency in MatTek dishes and treated with 10 nmol/L NIR800-EGF for 20 min at 37°C, 5% CO2. Control populations were treated with NIR800-EGF plus excess unlabeled EGF or vehicle (sham). After incubation, monolayers were rinsed with fresh culture medium and imaged directly without fixation. The microscope (Nikon TE2000) was configured with mercury illumination, 775 × 50 nm excitation filter, 810 nm long pass dichroic mirror, 845 × 55 nm emission filter, and a Q-Imaging camera.
In vitro validation of NIR700-Annexin V
NIR700-Annexin V was validated in DiFi cells following 24 h treatment with mAb-C225 [cetuximab, 0 (control), 0.3, 3, and 30 μg/mL]. Control and treated cells were incubated with NIR700-Annexin V for 30 min. Following incubation, monolayers were rinsed three times with fresh culture medium, and resultant probe binding was measured using an Odyssey system. In parallel, apoptosis was independently evaluated in identically treated populations of control and treated cells using an apoptosis kit measuring caspase-3/7 activity (Caspase-Glo; Promega).
Cellular response to cetuximab in vitro
DiFi cells, propagated to near confluence, were treated with serial concentrations of cetuximab for 24 h. Apoptosis was assessed in treated and untreated cells by measuring caspase-3/7 activity (Caspase-Glo; Promega). Additionally, treated cells were stained with propidium iodide, and cell cycle analysis was done via flow cytometry (FACSCalibur; BD Biosciences).
In vivo imaging
All studies involving animals were conducted in compliance with federal and institutional guidelines. DiFi, HCT-116, and SW620 xenografts were generated in athymic nude mice (Harlan Sprague-Dawley) following subcutaneous injection of 2 × 106 to 4 × 106 cells. Palpable tumors were detected within 2 to 4 weeks. For treatment studies, tumor-bearing mice (0.5-1.0 cm longest dimension) were administered cetuximab (40 mg/kg) or saline vehicle intraperitoneally every 3 days for 1 week (three total injections). Following anesthesia (2% isoflurane in O2 at 2 L/min), in vivo optical images were recorded pre- and post-imaging agent administration at longitudinal time points using either a Xenogen IVIS 200 (Caliper Life Sciences) or CRI Maestro system. Single or multiple (NIR800-EGF and NIR700-Annexin V) optical imaging agents were administered intravenously (each agent 0.5 × 10-9 mol/animal) via a single 100 μL retro-orbital injection. For PET imaging, animals were administered 180 to 200 μCi [18F] FLT via intravenous injection before imaging on a Concorde Microsystems microPET Focus 220 (Siemens Preclinical Solutions). Animals remained conscious and were allowed free access to food and water during a 1 h uptake period. For PET scanning, mice were anesthetized (2% isoflurane in O2 at 2 L/min) and imaged in the prone position for 15 min. Body temperature was maintained before and during imaging using a thermostat controlled circulating warm water pad. For multimodality imaging, final optical (24 h) and PET images were acquired < 30 min apart. Immediately following imaging, both treated and untreated mice were sacrificed, and tissues were collected for histopathologic analysis for validation purposes.
Image analysis
Optical imaging data were processed using Living Imaging 2.50 (Xenogen), Matlab 7.2.0, or CRI supplied software (multispectral imaging) and consisted of region-of-interest analysis [average pixel intensity/unit area or total photon counts (for burden measurements only)]. Changes in the optical imaging variables in response to therapy were evaluated by comparing the difference between pretreatment (baseline) and post-treatment imaging data. PET data were reconstructed using maximum a posteriori algorithm software provided with the microPET scanner. The 18 maximum a posteriori iterations (β = 0.1) were preceded by 2 iterations of a three-dimensional ordered subset expectation maximization (OSEM3D) algorithm (9 subsets), and the final images consisted of 128 × 128 × 95 voxels at a zoom of 4. Decay and dead-time corrections were made as part of the image reconstruction, but no attenuation or scatter corrections were applied. Volumes of interest were defined about the tumor and normal muscle regions using AsiPro software (Siemens Preclinical Solutions) to determine the average pixel intensity in both volumes (T/M ratio). Statistical analyses were done using GraphPad Prism 4.0. The statistical significance of treatment response data was determined using the Wilcoxon sign-rank test for paired data.
High-resolution ultrasound imaging
Three-dimensional ultrasound tumor volumes were collected in treated and untreated mouse time-course experiments using a Visualsonics Vevo 770 high-resolution imaging system. For imaging, mice were anesthetized with 2% isoflurane in O2 at 2 L/min. Following imaging, three-dimensional data sets were imported into Amira 4.1 (Visage Imaging) and manually segmented to determine tumor volumes.
Tissue analysis
Animals were sacrificed <1 h following final imaging. Tumor tissues were collected, sectioned (5 μm thickness), and stained for tumor proliferation markers (mouse monoclonal anti-Ki-67 antibody from DakoCytomation), apoptosis (rabbit polyclonal anti-caspase-3 antibody from Cell Signaling Technology), EGFR (mouse monoclonal anti-EGFR from DakoCytomation), and phosphorylated EGFR (p-EGFR; rabbit monoclonal anti-p-EGFR from Cell Signaling). Immunohistochemistry was done using a DakoCytomation Envision System HRP Detection Kit and evaluated by a qualified pathologist (M.K.W.).
Results
In vitro cellular responses to cetuximab treatment
To determine the suitability of measuring tumor cell apoptosis and proliferation as in vivo biomarkers of response to cetuximab treatment, these physiologic readouts were initially assayed in vitro. As expected, cetuximab-treated DiFi cells exhibited concentration-dependent apoptosis, with a dose as little as 0.5 μg/mL (~3 nmol/L) being sufficient to induce quantifiable caspase-3/7 activity (Fig. 1A). Similarly, cell cycle analysis (propidium iodide and flow cytometry) of cetuximab-treated DiFi cells revealed a corresponding decrease in the G2-M and S fractions as shown in Fig. 1B and C, suggesting a decrease in proliferation. These in vitro observations in DiFi cells are in agreement with work of Wu et al. (1) and suggest that cellular apoptosis and proliferation are suitable readouts of response to EGFR axis blockade with cetuximab and other EGFR-directed mAbs.
Fig. 1.
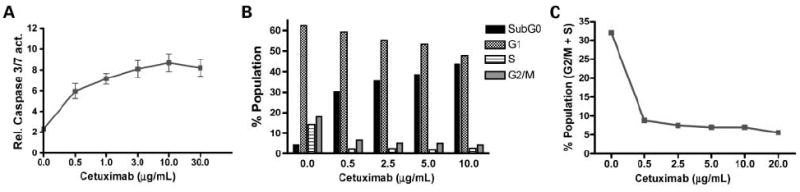
In vitro treatment of DiFi cells with cetuximab induces apoptosis and decreases proliferation. A, cetuximab-induced caspase-3/7 activity (relative bioluminescence intensity) in cultured DiFi cells. B, cell cycle analysis of cetuximab-treated DiFi cells showed a dose-dependent increase in sub-G0 phase (apoptotic cells) and a corresponding decrease in G2-M and S phase (proliferative cells) highlighted in C.
Imaging EGF uptake
Following synthesis and spectroscopic characterization of NIR800-EGF (Fig. 2A), we visualized uptake and localization of the probe by fluorescence microscopy in DiFi cells. Following a brief incubation with the agent, EGF binding to DiFi cells could be easily visualized by fluorescence microscopy. Furthermore, cellular uptake of the imaging probe could be inhibited by pretreatment with unlabeled EGF, indicating EGFR specificity (Fig. 2B-G).
Fig. 2.
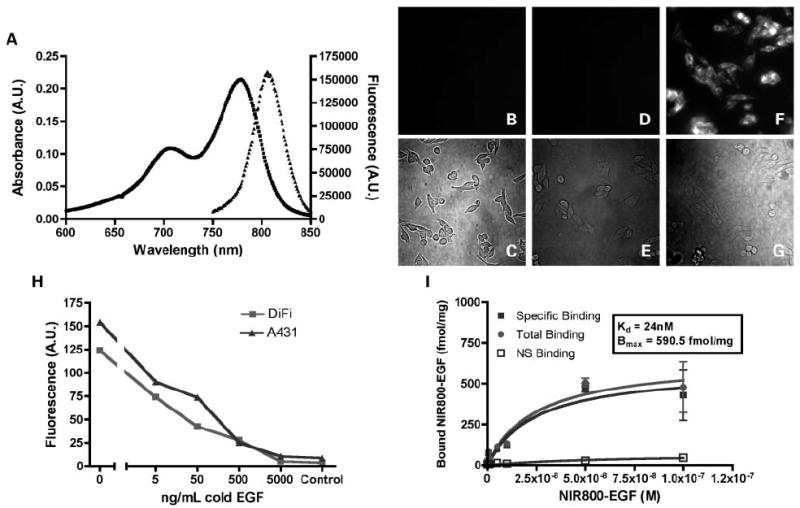
Spectroscopy, specificity, and intracellular visualization of the NIR800-EGF imaging agent. A, absorbance (■) and fluorescence (▲) emission of 1 μmol/L aqueous solution. B to G, live DiFi cells labeled with NIR800-EGF and imaged by fluorescence microscopy. B and C, no appreciable autofluorescence was observed in unlabeled DiFi cells (blank). D and E, DiFi cells pretreated with unlabeled (“cold”) EGF followed by NIR800-EGF were nonfluorescent, indicating EGFR specificity. F and G, plasma membrane of DiFi cells incubated with the agent was brightly fluorescent, suggesting localized binding at the cell surface. H, in vitro competitive displacement assay in DiFi and A431cells. I, saturation binding isotherms for NIR800-EGF in DiFi cells (Kd = 24 nmol/L, Bmax = 591fmol/mg).
To further examine the specific binding of NIR800-EGF, we examined agent uptake in EGFR-positive cell lines using a fluorescence plate reader. Live-cell competitive binding assays between NIR800-EGF and unlabeled EGF were done in A431 and DiFi cells, which express very high levels of EGFR (both 2 × 106-4 × 106 receptors/cell; refs. 30, 31). We found that both A431 and DiFi cells exhibited quantifiable uptake of NIR800-EGF that was competitively displaceable to near-background fluorescence levels with administration of excess cold EGF (Fig. 2H).
Next, the binding variables Kd and Bmax for NIR800-EGF were measured in DiFi cells (Fig. 2I). Cellular binding of NIR800-EGF in DiFi cells followed a typical parabolic binding curve, and Kd was calculated to be ~24 nmol/L for NIR800-EGF, comparable with published results (32). Bmax for NIR800-EGF in DiFi cells was calculated to be 591 fmol/mg, or ~3 × 106 binding events per DiFi cell at saturation, correlating well with the known EGFR expression profile of DiFi cells (30, 31, 33).
In vivo biodistribution of NIR800-EGF and tumor uptake were evaluated in three xenograft models with varying EGFR expression. Animals bearing HCT-116 (moderate EGFR expression; n = 16), DiFi (high EGFR expression; n = 14), and SW620 (EGFR-negative control; n = 9) xenograft tumors were administered equimolar quantities (0.5 nmol) of NIR800-EGF and imaged repetitively over a 24 h time course. As shown in Fig. 3A and B, significant accumulation of the imaging probe was observed in HCT-116 and DiFi tumors compared with SW620 tumors, which do not express EGFR (34). Minor accumulation of the imaging probe was also noted in the kidneys of all animals imaged. Representative in vivo fluorescence images shown in Fig. 3A were captured 24 h post-injection and were the basis for the quantification shown in Fig. 3B. Importantly, tumor uptake of NIR800-EGF as reflected by the average CCD pixel intensity/unit area (average fluorescence/area) was found to agree closely with relative EGFR immunoreactivity in tumor tissues (Fig. 3C). Thus, this method of quantification, which is reflective of EGFR activity and normalized to be independent of tumor burden, was used to assess treatment response in later studies. Alternately, further NIR800-EGF imaging experiments in DiFi xenografts showed close correlation between the total integrated NIR800-EGF fluorescence intensity (total photons) in tumors with direct measurement of tumor burden, including total tumor mass (Supplementary Fig. S1) and volume (data not shown), with both metrics showing a linear correlation (r2 = 0.881 for tumor mass).
Fig. 3.
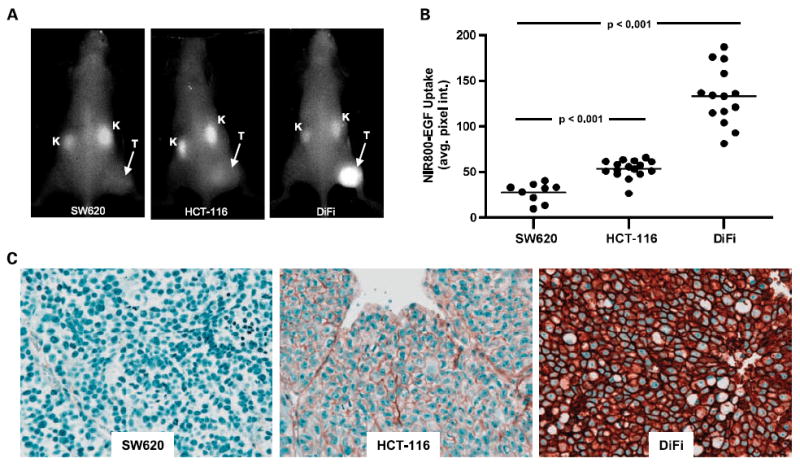
Quantitative in vivo imaging of NIR800-EGF uptake reflects the relative EGFR immunoreactivity in colorectal cancer xenografts. A, representative in vivo fluorescence images of SW620 (EGFR-negative), HCT-116 (EGFR-moderate), and DiFi (EGFR-high) tumor-bearing mice 24 h post-administration of NIR800-EGF. T, tumor; K, kidney. B, relative in vivo uptake of NIR800-EGF in SW620, HCT-116, and DiFi tumors. C, total EGFR immunoreactivity in tumor specimens from SW620, HCT-116, and DiFi xenografts shown in A and B.
Imaging apoptosis
An apoptosis-signaling optical imaging agent was prepared by conjugating Annexin V with a NIR dye. Figure 4A illustrates the aqueous-phase spectroscopic properties of the resulting imaging probe, NIR700-Annexin V. As shown in Fig. 4B, NIR700-Annexin V uptake in DiFi cells treated with cetuximab was concentration dependent and correlated with caspase-3/7 activity, thus indicating that NIR700-Annexin V functions as an accurate reporter of apoptosis in vitro.
Fig. 4.
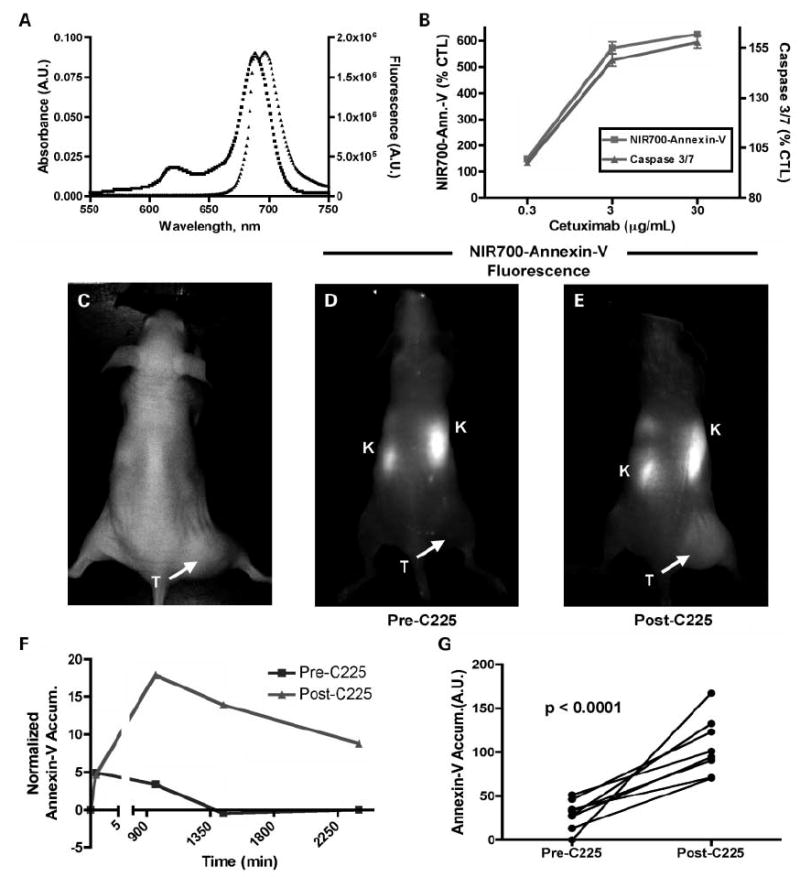
Characterization and validation of NIR700-Annexin V. A, absorbance (■) and fluorescence (▲) emission of 1 μmol/L aqueous solution NIR700-Annexin V. B, cetuximab-dependent apoptosis in DiFi cells measured by NIR-Annexin V and validated by caspase-3/7 activity. C, white-light and (D and E) fluorescence images of a mouse bearing a DiFi xenograft tumor and administered NIR700-Annexin V either before (D) or after (E) a cetuximab regimen. Fluorescence images were recorded 24 h post-administration of the imaging probe. F, quantitative clearance profile for NIR-Annexin V pre-cetuximab (squares) and post-cetuximab (triangles) treatment illustrating the enhanced NIR-Annexin V accumulation and longer clearance profile following treatment with cetuximab. G, in vivo treatment response indicated by elevated NIR700-Annexin V accumulation in DiFi tumors following cetuximab. Before treatment, minimal accumulation of the imaging agent was observed in tumors. However, following treatment with cetuximab, significantly increased tumor accumulation of NIR-Annexin V was observed (n = 8; P < 0.0001). C225 synonymous with cetuximab.
Building on these experiments, we evaluated NIR700-Annexin V as an in vivo marker of treatment response by monitoring apoptosis. As shown in Fig. 4C to G, a group of DiFi xenograft-bearing animals (n = 8) was followed independently through a multidose, 1-week cetuximab regimen (3 treatments, 40 mg/kg, every 3 days). Pretreatment uptake of the imaging probe served as a baseline measure of tumor apoptosis and was compared with post-treatment uptake. Following administration of the probe, fluorescence images were recorded repetitively over a 40 h time course to characterize uptake and clearance properties of the agent. Figure 4 shows representative white-light (Fig. 4C) and fluorescence (Fig. 4D and E) images from these investigations (images shown were collected 24 h post-administration of NIR700-Annexin V). As shown in Fig. 4D, pretreatment tumor uptake of NIR700-Annexin V was minimal with some accumulation noted in the kidneys. In contrast, following cetuximab treatment, significant accumulation of NIR700-Annexin V was observed in the tumor (Fig. 4E). Clearance profile analysis (Fig. 4F) showed enhanced uptake and retention of the probe in DiFi tumors following treatment and illustrated that imaging data collected 24 h post-injection (1440 min) maximize the difference between treatment-induced probe accumulation and minor probe retention unrelated to therapy. A statistically significant difference was observed in NIR700-Annexin V uptake pretreatment and post-treatment across multiple animals (n = 8; P < 0.0001, paired t test; Fig. 4G). Histologic analysis of caspase-3 activity was used to validate tumor-related apoptosis (data not shown).
Concomitant imaging assessment of EGFR blockade: assessment of EGF uptake, apoptosis, and proliferation
Three complementary noninvasive molecular imaging readouts (EGF uptake, apoptosis, and proliferation) were profiled as potential biomarkers of response to pharmacologic blockade of EGFR signaling with cetuximab in DiFi (n = 26) and HCT-116 (n = 16) xenografts. As shown in Fig. 5A and B, cetuximab treatment of DiFi xenografts resulted in significantly decreased tumor uptake of NIR800-EGF (P < 0.0001) and correspondingly increased retention of NIR700-Annexin V compared with untreated controls (P = 0.0006). In contrast to what we observed in vitro, [18F] FLT uptake was not significantly different between treated and untreated mice (P = 0.425; Fig. 5C). Representative NIR800-EGF, NIR700-Annexin V, and [18F] FLT PET images collected from an individual control (Fig. 5D, F, and H) and treated (Fig. 5E, G, and I) mouse are shown. Importantly, each of the imaging-based physiologic readouts was validated by analysis of tissue histology. Following the cetuximab regimen, no discernable difference in tumor EGFR was noted between control (Fig. 5J) and treated (Fig. 5K) animals, suggesting that decreased NIR800-EGF uptake in treated animals was due primarily to EGFR occupancy by cetuximab. Additionally, tumors collected from cetuximab-treated animals exhibited significantly increased caspase-3 staining compared with untreated controls (Fig. 5L and M), confirming our in vivo observations with NIR700-Annexin V that the efficacy of cetuximab on DiFi cells was primarily through induction of apoptosis. In agreement with [18F] FLT PET imaging, we found no discernible difference between Ki-67 staining of tumors collected from control or treated mice (Fig. 5N and O), indicating that cetuximab was ineffective at reducing cellular proliferation in this model.
Fig. 5.
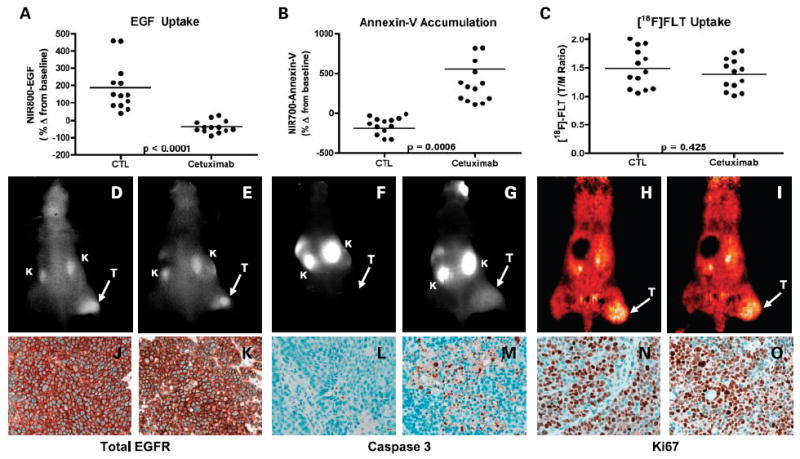
Noninvasive imaging assessment of response to EGFR blockade with cetuximab in DiFi xenograft-bearing mice. Treated and untreated cohorts bearing DiFi xenograft tumors were simultaneously imaged with NIR800-EGF, NIR700-Annexin V, and [18F] FLT PET. Following cetuximab treatment, DiFi tumors exhibited significantly reduced NIR800-EGF uptake (A) and increased NIR700-Annexin V uptake (B) compared with untreated controls. No statistical difference in [18F] FLTuptake was observed between treated and untreated mice. Units are defined as tumor (T)/muscle (M) Ratio (C). Representative NIR800-EGF, NIR700-Annexin V, and [18F] FLT PET images collected from an individual control (D, F, and H) and treated (E, G, and I) mouse. Strong agreement between the imaging metrics of response and standard immunohistochemistry was observed. Tumors from control (J) and treated (K) animals exhibited similar levels of total EGFR. Treated animals (M) exhibited elevated caspase-3 staining compared with untreated cohorts (L). No discernible difference in Ki-67 staining was observed between tumors from control (N) and treated cohorts (O).
Similarly, we evaluated the ability of the three reported imaging modalities to assess treatment response in a cetuximab-resistant colorectal cancer model, HCT-116 xenografts. HCT-116 cells express mutant KRAS, a negative predictor of response to cetuximab in colorectal cancer (20, 21, 35, 36). As shown in Fig. 6A, cetuximab treatment of HCT-116 xenografts resulted in somewhat diminished tumor uptake of NIR800-EGF, although the downward trend in treated mice did not reach significance. Furthermore, NIR700-Annexin V uptake was similar in control and treated mice (Fig. 6B), suggesting minimal tumor apoptosis following cetuximab treatment. Like NIR800-EGF uptake, [18F] FLT uptake was also somewhat reduced in treated mice compared with untreated controls, although the trend was not significant (P = 0.110; Fig. 6C). Representative NIR800-EGF, NIR700-Annexin V, and [18F] FLT PET images collected from the HCT-116 animals are shown in Fig. 6D to I. As with the DiFi studies, each of the imaging-based physiologic readouts was validated by analysis of tissue histology (Fig. 6J-O), where we noted similar tumor EGFR, caspase-3, and Ki-67 staining of treated and untreated HCT-116 tumor tissues. Over the course of these studies, high-resolution ultrasound imaging was used to document changes in tumor size. Following cetuximab treatment, we observed measurable tumor regression in DiFi xenografts but only modest growth arrest in HCT-116 xenografts (Supplementary Figs. S2 and S3). Further tissue-level confirmation of treatment response was afforded by p-EGFR immunoreactivity, where we noted significantly reduced p-EGFR in treated DiFi tumors compared with untreated controls but only modestly reduced p-EGFR staining in treated HCT-116 tumors compared with untreated controls. Both treated DiFi and HCT-116 tumors, in contrast to untreated controls, showed evidence of increased EGFR internalization as noted by the presence of cytosolic p-EGFR (Supplementary Fig. S4).
Fig. 6.
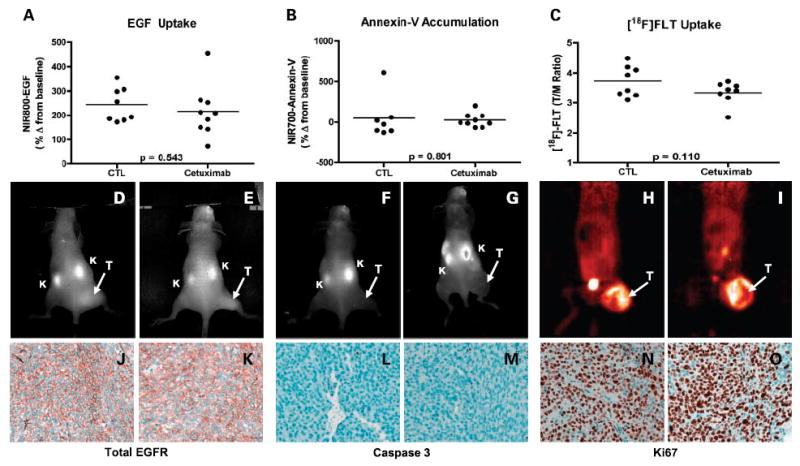
Noninvasive imaging assessment of response to EGFR blockade with cetuximab in HCT-116 xenograft-bearing mice. Treated and untreated cohorts bearing HCT-116 xenograft tumors were simultaneously imaged with NIR800-EGF, NIR700-Annexin V, and [18F] FLT PET. Following cetuximab treatment, NIR800-EGF uptake in HCT-116 tumors appeared modestly reduced, although the difference was not significant (A). NIR700-Annexin V uptake (B) was similar in both treated and untreated mice. C, cetuximab-treated HCT-116 mice displayed modestly reduced [18F] FLTuptake compared with untreated controls that approached statistical significance. Representative NIR800-EGF, NIR700-Annexin V, and [18F] FLT PET images were collected from an individual control (D, F, and H) and treated (E, G, and I) mouse. Strong agreement between the imaging metrics of response and standard immunohistochemistry was observed. Tumors from control (J) and treated (K) animals exhibited similar levels of total EGFR. Caspase-3 staining was similar in both control (L) and treated (M) tumors. No discernible difference in Ki-67 staining was observed between tumors from control (N) and treated (O) cohorts.
Discussion
Currently, imaging criteria for clinical evaluation of therapeutic response in cancer are based on Response Evaluation Criteria in Solid Tumors guidelines (37). Designed predominantly to characterize changes in tumor size, Response Evaluation Criteria in Solid Tumors assessments rely on anatomic imaging modalities such as computed tomography, planar X-ray, or magnetic resonance imaging, which do not provide substantial information regarding specific cellular and molecular events relevant to treatment response (38). Biological changes that may occur in the tumor within hours of treatment may not manifest as detectable changes in tumor size until much later as assessed by anatomic imaging techniques. However, these changes can be followed noninvasively and longitudinally with molecular imaging metrics, including glucose metabolism (fluorodeoxyglucose PET; refs. 39-43), cellular proliferation ([18F] FLT PET; refs. 26, 27), and apoptosis (Annexin V; refs. 44, 45). Application of these methodologies can significantly improve physiologic profiling of treatment response compared with conventional Response Evaluation Criteria in Solid Tumors, with one of the most dramatic examples being the assessment of response of gastrointestinal stromal tumors to inhibitors of c-kit. Gastrointestinal stromal tumors rarely shrink in size but undergo rapid metabolic response to c-kit inhibition as reflected by a reduction in fluorodeoxyglucose uptake after treatment (46, 47).
In this preclinical study, we have shown the utility of concomitant use of three molecular imaging metrics as potential biomarkers of treatment response to a molecularly targeted therapy employed in metastatic colorectal cancer. Each of the employed imaging metrics was selected to assess a unique and important aspect of antitumor therapeutic response and was evaluated within the context of both cetuximab-sensitive (DiFi) and cetuximab-resistant (HCT-116) human colorectal cancer cell line xenografts. Specifically, we synthesized and validated an optical imaging probe to assess the molecular targeting and tumor cell EGFR occupancy by cetuximab in vivo (NIR800-EGF) as well as a spectroscopically distinct optical imaging probe to assess the ensuing treatment-induced apoptosis (NIR700-Annexin V). Additionally, changes in tumor proliferation occurring in response to cetuximab were assessed in vivo by [18F] FLT PET imaging. Importantly, the combined noninvasive imaging data illustrate the potential to collect multiple relevant physiologic readouts simultaneously in individual animals. This analysis paradigm revealed that cetuximab, in contrast to its effect on both apoptosis and proliferation in cetuximab-sensitive colorectal cancer cells in vitro, induced significant levels of tumor cell apoptosis but was surprisingly ineffective at reducing tumor cell proliferation in vivo (48). Our in vivo assessment of both cetuximab-sensitive (DiFi) and cetuximab-resistant (HCT-116) cell lines showed that complementary molecular imaging techniques can provide important and accurate information on the biological effect of a therapy on the tumor. The tight correlation of these imaging measures with direct measurement of EGF binding, proliferation, and apoptosis in the tumor tissue, the ability to detect these changes soon after and throughout dosing, and the ability to obtain this information on a longitudinal, noninvasive basis represent very attractive features of this technology.
We envision that the molecular imaging strategy presented here, which emphasizes simultaneous evaluation of multiple physiologic readouts, could be highly useful in the preclinical setting to optimize dosing and administration of EGFR-directed agents as part of innovative molecularly targeted therapeutic regimens in colorectal cancer, including rational selection of complementary therapeutic agents that may affect tumor cell proliferation as well as apoptosis. Furthermore, substitution of the optical probe by a radiolabel for PET or single-photon emission computed tomography imaging will enable each of these molecular imaging metrics to be considered for future advancement to the clinical setting for dose optimization and patient management. For example, we recently reported a randomized phase II trial comparing the clinical and biological effects of two dose levels of gefitinib, a small-molecule EGFR tyrosine kinase inhibitor, in patients with previously treated metastatic colorectal cancer (9). Surprisingly, post-treatment tumor biopsies revealed that gefitinib did not inhibit activation of its proximal target, EGFR. Additionally, expression of total or activated EGFR, activated Akt, activated mitogen-activated protein kinase, or Ki-67 did not decrease following 1 week of gefitinib. These findings suggest that the clinical failure of gefitinib in this setting was of a pharmacologic nature in that the agent proved insufficiently potent to inhibit phosphorylation of EGFR at these doses. Importantly, however, in the subset of patients with longer-than-average progression-free survival, there was a trend toward greater reduction of tumor Ki-67 staining and thus decreased proliferation. In another trial, Townsley et al. found that erlotinib, a more potent EGFR tyrosine kinase inhibitor, effectively inhibited both EGFR autophosphorylation and phosphorylation of ERK but did not reduce proliferation, as measured by p27 and Ki-67/MIB-1, in colorectal cancer metastases (49); however, no clinical responses were observed in 31 evaluable patients. These observations raise the possibility that an EGFR tyrosine kinase inhibitor (that may affect proliferation) combined with an EGFR mAb (that may affect apoptosis) could be a promising approach for treatment of advanced colorectal cancer. We have initiated a phase I clinical trial to test this hypothesis, evaluating the combination of cetuximab and erlotinib in patients with advanced solid tumors (phase I) and with progressive colorectal cancer (phase II). We recently reported that in vitro pharmacologic blockade of the EGFR axis in HCA-7 cells, a human polarizing colorectal cancer cell line, with EGFR mAb, EGFR tyrosine kinase inhibitor and a selective TACE inhibitor (which inhibits cell surface cleavage of EGFR ligands), resulted in a concentration-dependent reduction of cell proliferation and active, phosphorylated mitogen-activated protein kinase. Combining suboptimal concentrations of these agents achieved cooperative growth inhibition, increased apoptosis, and greater reduction in mitogen-activated protein kinase pathway activation (50).
In conclusion, the complementary noninvasive molecular imaging metrics illustrated here can provide informative readouts of efficacy for EGFR-directed therapies such as cetuximab and thus may be useful toward the development of novel treatment regimens in the preclinical setting as well as informing the design and management of future clinical trials.
Supplementary Material
Acknowledgments
We thank Jeff Clanton and Jarrod Driskill for cyclotron production of [18F]fluoride and Annie Liu for editing the manuscript.
Grant support: National Cancer Institute grant CA 46413, GI Special Program of Research Excellence grant P50 95103, and Mouse Models of Human Cancers Consortium grant U01 084239 (R.J. Coffey); National Cancer Institute Career Development Award K25 CA127349 (H.C. Manning); NIH Predoctoral and Postdoctoral Training Grants in Imaging Science T32 EB03817 and T32 EB003817 (J.C. Gore), National Cancer Institute-funded South-Eastern Center for Small-Animal Imaging grant U24 CA126588 (J.C. Gore), a shared instrumentation award (microPET) 1S10 RR17858, and Mid-Career Award in Patient-Oriented Research K24 CA082301 (M.L. Rothenberg); Burroughs Wellcome Fund Career Award at the Scientific Interface (T.E. Peterson); and Vanderbilt University Department of Radiology nuclear medicine research fellowship (C. Shah).
Footnotes
Disclosure of Potential Conflicts of Interest No potential conflicts of interest were disclosed.
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
References
- 1.Wu X, Fan Z, Masui H, Rosen N, Mendelsohn J. Apoptosis induced by an anti-epidermal growth factor receptor monoclonal antibody in a human colorectal carcinoma cell line and its delay by insulin. J Clin Invest. 1995;95:1897–905. doi: 10.1172/JCI117871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sasaki T, Kitadai Y, Nakamura T, et al. Inhibition of epidermal growth factor receptor and vascular endothelial growth factor receptor phosphorylation on tumor-associated endothelial cells leads to treatment of orthotopic human colon cancer in nude mice. Neoplasia. 2007;9:1066–77. doi: 10.1593/neo.07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yokoi K, Thaker PH, Yazici S, et al. Dual inhibition of epidermal growth factor receptor and vascular endothelial growth factor receptor phosphorylation by AEE788 reduces growth and metastasis of human colon carcinoma in an orthotopic nude mouse model. Cancer Res. 2005;65:3716–25. doi: 10.1158/0008-5472.CAN-04-3700. [DOI] [PubMed] [Google Scholar]
- 4.Chen J, Smith M, Kolinsky K, et al. Antitumor activity of HER1/EGFR tyrosine kinase inhibitor erlotinib, alone and in combination with CPT-11 (irinotecan) in human colorectal cancer xenograft models. Cancer Chemother Pharmacol. 2007;59:651–9. doi: 10.1007/s00280-006-0320-8. [DOI] [PubMed] [Google Scholar]
- 5.Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–45. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 6.Cunningham MP, Thomas H, Fan Z, Modjtahedi H. Responses of human colorectal tumor cells to treatment with the anti-epidermal growth factor receptor monoclonal antibody ICR62 used alone and in combination with the EGFR tyrosine kinase inhibitor gefitinib. Cancer Res. 2006;66:7708–15. doi: 10.1158/0008-5472.CAN-06-1000. [DOI] [PubMed] [Google Scholar]
- 7.Harding J, Burtness B. Cetuximab: an epidermal growth factor receptor chimeric human-murine monoclonal antibody. Drugs Today (Barc) 2005;41:107–27. doi: 10.1358/dot.2005.41.2.882662. [DOI] [PubMed] [Google Scholar]
- 8.Penne K, Bohlin C, Schneider S, Allen D. Gefitinib (Iressa, ZD1839) and tyrosine kinase inhibitors: the wave of the future in cancer therapy. Cancer Nurs. 2005;28:481–6. doi: 10.1097/00002820-200511000-00012. [DOI] [PubMed] [Google Scholar]
- 9.Rothenberg ML, LaFleur B, Levy DE, et al. Randomized phase II trial of the clinical and biological effects of two dose levels of gefitinib in patients with recurrent colorectal adenocarcinoma. J Clin Oncol. 2005;23:9265–74. doi: 10.1200/JCO.2005.03.0536. [DOI] [PubMed] [Google Scholar]
- 10.Saltz LB, Meropol NJ, Loehrer PJ, Sr, Needle MN, Kopit J, Mayer RJ. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol. 2004;22:1201–8. doi: 10.1200/JCO.2004.10.182. [DOI] [PubMed] [Google Scholar]
- 11.Zhu Z. Targeted cancer therapies based on antibodies directed against epidermal growth factor receptor: status and perspectives. Acta Pharmacol Sin. 2007;28:1476–93. doi: 10.1111/j.1745-7254.2007.00681.x. [DOI] [PubMed] [Google Scholar]
- 12.Cohenuram M, Saif MW. Panitumumab the first fully human monoclonal antibody: from the bench to the clinic. Anticancer Drugs. 2007;18:7–15. doi: 10.1097/CAD.0b013e32800feecb. [DOI] [PubMed] [Google Scholar]
- 13.Hecht JR, Patnaik A, Berlin J, et al. Panitumumab monotherapy in patients with previously treated metastatic colorectal cancer. Cancer. 2007;110:980–8. doi: 10.1002/cncr.22915. [DOI] [PubMed] [Google Scholar]
- 14.Williams KJ, Telfer BA, Stratford IJ, Wedge SR. ZD1839 (“Iressa”), a specific oral epidermal growth factor receptor-tyrosine kinase inhibitor, potentiates radiotherapy in a human colorectal cancer xenograft model. Br J Cancer. 2002;86:1157–61. doi: 10.1038/sj.bjc.6600182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hightower M. Erlotinib (OSI-774,Tarceva), a selective epidermal growth factor receptor tyrosine kinase inhibitor, in combination with chemotherapy for advanced non-small-cell lung cancer. Clinical Lung Cancer. 2003;4:336–8. doi: 10.1016/s1525-7304(11)70302-3. [DOI] [PubMed] [Google Scholar]
- 16.Jonker DJ, O’Callaghan CJ, Karapetis CS, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357:2040–8. doi: 10.1056/NEJMoa071834. [DOI] [PubMed] [Google Scholar]
- 17.Baselga J. The EGFR as a target for anticancer therapy—focus on cetuximab. Eur J Cancer. 2001;37(Suppl 4):S16–22. doi: 10.1016/s0959-8049(01)00233-7. [DOI] [PubMed] [Google Scholar]
- 18.Costa AF, Sander GB, Picon PD. Cetuximab in colon cancer. N Engl J Med. 2004;351:1575–6. author reply-6. [PubMed] [Google Scholar]
- 19.Lenz HJ, Van Cutsem E, Khambata-Ford S, et al. Multicenter phase II and translational study of cetuximab in metastatic colorectal carcinoma refractory to irinotecan, oxaliplatin, and fluoropyrimidines. J Clin Oncol. 2006;24:4914–21. doi: 10.1200/JCO.2006.06.7595. [DOI] [PubMed] [Google Scholar]
- 20.Khambata-Ford S, Garrett CR, Meropol NJ, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol. 2007;25:3230–7. doi: 10.1200/JCO.2006.10.5437. [DOI] [PubMed] [Google Scholar]
- 21.Lievre A, Bachet JB, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–5. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 22.Perez-Soler R, Saltz L. Cutaneous adverse effects with HER1/EGFR-targeted agents: is there a silver lining? J Clin Oncol. 2005;23:5235–46. doi: 10.1200/JCO.2005.00.6916. [DOI] [PubMed] [Google Scholar]
- 23.Czernin J, Weber WA, Herschman HR. Molecular imaging in the development of cancer therapeutics. Annu Rev Med. 2006;57:99–118. doi: 10.1146/annurev.med.57.080904.190431. [DOI] [PubMed] [Google Scholar]
- 24.DeNardo SJ. Combined molecular targeting for cancer therapy: a new paradigm in need of molecular imaging. J Nucl Med. 2006;47:4–5. [PubMed] [Google Scholar]
- 25.Rudin M, Weissleder R. Molecular imaging in drug discovery and development. Nat Rev Drug Discov. 2003;2:123–31. doi: 10.1038/nrd1007. [DOI] [PubMed] [Google Scholar]
- 26.Chen W, Cloughesy T, Kamdar N, et al. Imaging proliferation in brain tumors with 18F-FLT PET: comparison with 18F-FDG. J Nucl Med. 2005;46:945–52. [PubMed] [Google Scholar]
- 27.Choi SJ, Kim JS, Kim JH, et al. [F-18]3′-deoxy-3′-fluorothymidine PET for the diagnosis and grading of brain tumors. Eur J Nucl Med Mol Imaging. 2005;32:653–9. doi: 10.1007/s00259-004-1742-3. [DOI] [PubMed] [Google Scholar]
- 28.Dittmann H, Dohmen BM, Paulsen F, et al. [18F] FLT PET for diagnosis and staging of thoracic tumours. Eur J Nucl Med Mol Imaging. 2003;30:1407–12. doi: 10.1007/s00259-003-1257-3. [DOI] [PubMed] [Google Scholar]
- 29.Grierson JR, Schwartz JL, Muzi M, Jordan R, Krohn KA. Metabolism of 3′-deoxy-3′-[F-18]fluorothymidine in proliferating A549 cells: validations for positron emission tomography. Nucl Med Biol. 2004;31:829–37. doi: 10.1016/j.nucmedbio.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 30.Olive M, Untawale S, Coffey RJ, et al. Characterization of the DiFi rectal carcinoma cell line derived from a familial adenomatous polyposis patient. In Vitro Cell Dev Biol. 1993;29A:239–48. doi: 10.1007/BF02634191. [DOI] [PubMed] [Google Scholar]
- 31.Gross ME, Zorbas MA, Danels YJ, et al. Cellular growth-response to epidermal growth-factor in colon-carcinoma cells with an amplified epidermal growth-factor receptor derived from a familial adenomatous polyposis patient. Cancer Res. 1991;51:1452–9. [PubMed] [Google Scholar]
- 32.Mazor O, de Boisferon MH, Lombet A, et al. Europium-labeled epidermal growth factor and neurotensin: novel probes for receptor-binding studies. Analyt Biochem. 2002;301:75–81. doi: 10.1006/abio.2001.5475. [DOI] [PubMed] [Google Scholar]
- 33.Untawale S, Zorbas MA, Hodgson CP, et al. Transforming growth factor-α production and autoinduction in a colorectal carcinoma cell line (DiFi) with an amplified epidermal growth factor receptor gene. Cancer Res. 1993;53:1630–6. [PubMed] [Google Scholar]
- 34.Coffey RJ, Shipley GD, Moses HL. Production of transforming growth-factors by human-colon cancer lines. Cancer Res. 1986;46:1164–9. [PubMed] [Google Scholar]
- 35.Keller JW, Franklin JL, Graves-Deal R, Friedman DB, Whitwell CW, Coffey RJ. Oncogenic KRAS provides a uniquely powerful and variable oncogenic contribution among RAS family members in the colonic epithelium. J Cell Physiol. 2007;210:740–9. doi: 10.1002/jcp.20898. [DOI] [PubMed] [Google Scholar]
- 36.Keller JW, Haigis KM, Franklin JL, Whitehead RH, Jacks T, Coffey RJ. Oncogenic K-RAS subverts the antiapoptotic role of N-RAS and alters modulation of the N-RAS: gelsolin complex. Oncogene. 2007;26:3051–9. doi: 10.1038/sj.onc.1210103. [DOI] [PubMed] [Google Scholar]
- 37.Therasse P, Arbuck SG, Eisenhauer EA, et al. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 38.Jaffe CC. Measures of response: RECIST,WHO, and new alternatives. J Clin Oncol. 2006;24:3245–51. doi: 10.1200/JCO.2006.06.5599. [DOI] [PubMed] [Google Scholar]
- 39.Apisarnthanarax S, Chao KS. Current imaging paradigms in radiation oncology. Radiat Res. 2005;163:1–25. doi: 10.1667/rr3279. [DOI] [PubMed] [Google Scholar]
- 40.Hoh CK. Clinical use of FDG PET. Nucl Med Biol. 2007;34:737–42. doi: 10.1016/j.nucmedbio.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 41.Otsuka H, Morita N, Yamashita K, Nishitani H. FDG-PET/CT for cancer management. J Med Invest. 2007;54:195–9. doi: 10.2152/jmi.54.195. [DOI] [PubMed] [Google Scholar]
- 42.Phelps ME, Gambhir S, Herschman H, Barrio J, Cherry S, Satyamurthy N. Molecular imaging with PET: from metabolism to gene expression. FASEB J. 2000;14:A567–A. [Google Scholar]
- 43.Su H, Bodenstein C, Dumont RA, et al. Monitoring tumor glucose utilization by positron emission tomography for the prediction of treatment response to epidermal growth factor receptor kinase inhibitors. Clin Cancer Res. 2006;12:5659–67. doi: 10.1158/1078-0432.CCR-06-0368. [DOI] [PubMed] [Google Scholar]
- 44.Belhocine TZ, Blankenberg FG. 99mTc-Annexin A5 uptake and imaging to monitor chemosensitivity. Methods Mol Med. 2005;111:363–80. doi: 10.1385/1-59259-889-7:363. [DOI] [PubMed] [Google Scholar]
- 45.Schellenberger EA, Weissleder R, Josephson L. Optimal modification of Annexin V with fluorescent dyes. Chembiochem. 2004;5:271–4. doi: 10.1002/cbic.200300741. [DOI] [PubMed] [Google Scholar]
- 46.King DM. The radiology of gastrointestinal stromal tumours (GIST) Cancer Imaging. 2005;5:150–6. doi: 10.1102/1470-7330.2005.0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trent JC, Ramdas L, Dupart J, et al. Early effects of imatinib mesylate on the expression of insulin-like growth factor binding protein-3 and positron emission tomography in patients with gastrointestinal stromal tumor. Cancer. 2006;107:1898–908. doi: 10.1002/cncr.22214. [DOI] [PubMed] [Google Scholar]
- 48.Merchant NB, Voskresenk I, Rogers CM, et al. TACE/ADAM-17: a component of the epidermal growth factor receptor axis and a promising therapuetic target in colorectal cancer. Clin Cancer Res. 2008;14:1182–91. doi: 10.1158/1078-0432.CCR-07-1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Townsley CA, Major P, Siu LL, et al. Phase II study of erlotinib (OSI-774) in patients with metastatic colorectal cancer. Br J Cancer. 2006;94:1136–43. doi: 10.1038/sj.bjc.6603055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.