

Abstract
Voltage-gated potassium channel subunit Kv3.3 is prominently expressed in cerebellar Purkinje cells and is known to be important for cerebellar function, as human and mouse movement disorders result from mutations in Kv3.3. To understand these behavioral deficits, it is necessary to know the role of Kv3.3 channels on the physiological responses of Purkinje cells. We studied the function of Kv3.3 channels in regulating the synaptically evoked Purkinje cell complex spike, the massive postsynaptic response to the activation of climbing fiber afferents, believed to be fundamental to cerebellar physiology. Acute slice recordings revealed that Kv3.3 channels are required for generation of the repetitive spikelets of the complex spike. We found that spikelet expression is regulated by somatic, and not by dendritic, Kv3 activity, which is consistent with dual somatic–dendritic recordings that demonstrate spikelet generation at axosomatic membranes. Simulations of Purkinje cell Na+ currents show that the unique electrical properties of Kv3 and resurgent Na+ channels are coordinated to limit accumulation of Na+ channel inactivation and enable rapid, repetitive firing. We additionally show that Kv3.3 knock-out mice produce altered complex spikes in vitro and in vivo, which is likely a cellular substrate of the cerebellar phenotypes observed in these mice. This characterization presents new tools to study complex spike function, cerebellar signaling, and Kv3.3-dependent human and mouse phenotypes.
Keywords: Kv3, potassium channels, Purkinje cell, complex spike, spinocerebellar ataxia, excitability
Introduction
Cerebellar Purkinje cells (PCs) transmit the output of the cerebellar cortex. The complex spike is the PC response to the massive all-or-none EPSP generated after activation of the single climbing fiber it receives from the inferior olive (Eccles et al., 1966) and is thought to have critical functions in cerebellar activity. The stereotyped complex spike consists of a fast, prominent first spike followed by two to six smaller, high-frequency spikelets. PCs possess multiple intrinsic somatic and dendritic conductances that may interact with synaptic inputs to sculpt membrane voltage signals (Llinas and Sugimori, 1980a,b; Raman and Bean, 1997; Womack and Khodakhah, 2002; Swensen and Bean, 2003; McKay and Turner, 2004; Khavandgar et al., 2005). However, discrepancies remain regarding the ionic mechanisms responsible for the generation of the complex spike, including whether spikelets are generated at somatic or dendritic membranes and, more generally, the importance of somatic versus dendritic excitability to the somatic complex spike waveform (Callaway et al., 1995; Callaway and Ross, 1997; Hansel and Linden, 2000; Schmolesky et al., 2002; Weber et al., 2003; Davie and Hausser, 2004).
Recent observations have demonstrated that potassium channels containing Kv3.3 subunits are essential components of cerebellar function. Waters et al. (2006) reported that mutations in the coding sequence of Kv3.3 are the cause of human spinocerebellar ataxia (SCA13). Furthermore, mice deficient in Kv3.3 protein expression [Kv3.3 knock-out (KO) mice] display cerebellar deficits, including gait and motor abnormalities (Matsukawa et al., 2003; Joho et al., 2006) and resistance to harmaline-induced tremor (McMahon et al., 2004). It is not yet known, however, how altered activity of Kv3.3 channels results in these human and mouse cerebellar phenotypes.
Potassium channels of the Kv3 subfamily are remarkable for their fast kinetics of activation and deactivation and depolarized voltage of activation (Rudy et al., 1999; Rudy and McBain, 2001). Accordingly, in multiple neuronal populations, Kv3 channel expression has been found to enable sustained high-frequency tonic firing (Martina et al., 1998; Wang et al., 1998; Erisir et al., 1999; Lau et al., 2000; Lien and Jonas, 2003).
Kv3.3 expression throughout rodent brain is most prominent in cerebellar PCs (Goldman-Wohl et al., 1994; Weiser et al., 1994). Within PCs, Kv3.3 subunits are expressed prominently in somas, axons, and distal dendrites (Martina et al., 2003; McMahon et al., 2004; Chang et al., 2007). In vitro studies of Kv3 function in PCs have revealed important roles in spike repolarization for both Na+ and Ca2+ spikes (Martina et al., 2003, 2007; McKay and Turner, 2004; McMahon et al., 2004) and supporting high-frequency spontaneous activity (Raman and Bean, 1999; Akemann and Knopfel, 2006). Furthermore, various preparations have been used to study intrinsic bursting in PCs, and Kv3-like currents are important for generating these burst outputs (Raman and Bean, 1999; Swensen and Bean, 2003; McKay and Turner, 2004). The contribution of Kv3 channels to the synaptically evoked complex spike has not been studied. Moreover, the relative contributions of somatic versus dendritic Kv3 conductances to burst outputs have not been investigated.
Materials and Methods
Acute slice preparation.
Postnatal Postnatal day 15–21 mice were anesthetized with pentobarbital and decapitated after loss of pain reflexes. Kv3.3 KO mice were kindly provided by Dr. Nathaniel Heintz (Rockefeller University, New York, NY). Brains were rapidly removed from the skull and bathed in ice-cold artificial CSF (ACSF) (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 1 MgSO4 2 CaCl2, and 20 glucose, pH 7.4 (saturated with 95% O2, 5% CO2). Sagittal slices, 350 μm thick, of cerebellar vermis were prepared using a Vibratome 3000 and were allowed to recover in a submerged holding chamber with continuous bubbling of ACSF for at least 1 h before transfer to the recording chamber. The submersion-type chamber was perfused at a rate of 2–3 ml/min with ACSF. Recordings were conducted at room temperature or 34–36°C, controlled by an in-line solution heater (Warner Instruments, Hamden, CT).
Whole-cell recordings.
Cerebellar PCs were visually targeted using an upright BX-51 Olympus Optical (Tokyo, Japan) microscope equipped with infrared differential interference contrast optics. Recording pipettes of 2–5 MΩ (for somatic recordings) or 6–10 MΩ (for dual somatic–dendritic recordings) were made from borosilicate glass using a horizontal pipette puller (model P-97; Sutter Instruments, Novato, CA) and filled with a K+-gluconate-based internal solution (in mm): 144 K+-gluconate, 0.2 EGTA, 3 MgCl2, 10 HEPES, 4 MgATP, and 0.5 NaGTP, pH adjusted to 7.4 with KOH. Recordings in whole-cell configuration were obtained from PC somas or simultaneously from the soma and dendrite of the same cell. Dual recordings were verified by the electrotonic spread of negative current injections from the soma into the dendrite. Transmembrane voltage was recorded in bridge mode of Axoclamp 2B or fast current-clamp mode of Axopatch 200B (Molecular Devices, Sunnyvale, CA), sampled at 20 kHz, and filtered at 10 kHz. Access resistance was monitored during break-in and throughout the experiment, and only recordings with access resistance below 20 MΩ were used for analysis; bridge was balanced. pClamp9.2 software (Molecular Devices) was used for data acquisition, and analysis was performed with Clampfit9.2 software (Molecular Devices) and OriginPro7.0 software (Microcal Software, Northampton, MA).
All PCs were spontaneously active in acute slice recordings, and negative current was applied through the somatic recording electrode to maintain a resting membrane potential near −65 mV. A bipolar tungsten stimulating electrode (125 μm in diameter; FHC, Bowdoin, ME) was placed in the granule cell layer directly beneath the targeted PC, and single stimulations delivered by ISO-Flex stimulus isolator (A.M.P.I., Jerusalem, Israel) were used to activate climbing fibers (0.2 ms, 20–120 μA). Repetitive stimulations were conducted at 0.1 Hz to minimize potential synaptic plasticity (Hansel and Linden, 2000). Complex spike waveforms were extremely stable throughout the duration of the recording session (up to 1.5 h).
Drug application.
Drugs were applied by either superfusion of the recording chamber (bath) or selective perfusion using glass micropipette pressure pulses controlled by Picospritzer III (Parker Hannifin, Cleveland, OH). In all selective perfusion experiments, the micropipette was directed in the same direction as the outflow of the recording chamber and perpendicular to the PC soma–dendrite axis. For somatic targeting, either 5 mm tetraethylammonium (TEA) for 200 ms or 1 mm TEA for 2 s was applied close to the soma within the PC layer immediately before climbing fiber stimulation. Dendritic conductances were selectively targeted by applying 1 mm TEA onto the PC dendrites at a depth one-third into the molecular layer closer to the pial surface, for 2 s before climbing fiber stimulation. In initial experiments, 300 μm Alexa Fluor 594 was added to the perfusate to determine that, for short pulses (up to 10 s), the perfusate was well confined to targeted regions (data not shown). Furthermore, bath application of 5 μm CNQX [IC50 < 1 μm for climbing fiber synaptic inputs (Llano et al., 1991)] is sufficient to fully block the complex spike by blocking AMPA receptors at climbing fiber synapses, which are widely distributed throughout the large-caliber dendrites (Larramendi and Victor, 1967). We selectively perfused 50 μm CNQX at the PC soma, and this did not alter the complex spike (data not shown), further suggesting that effective drug concentration was limited to the intended target. Additional controls are presented in supplemental Figure 1 (available at www.jneurosci.org as supplemental material).
Iberiotoxin (IbTx), dendrotoxin-K (DTX-K), and dendrotoxin-I (DTX-I) were purchased from Alomone Labs (Jerusalem, Israel), linopirdine and CNQX were from Tocris Bioscience (Ellisville, MO), and all other salts and reagents were from Sigma (St. Louis, MO). Effects of bath application of TEA were reversible after 20 min washout, whereas other bath-applied drugs showed partial or no reversibility after 1 h of washout.
In vivo preparation.
Three-month-old wild-type and Kv3.3 KO mice were anesthetized with an initial intraperitoneal injection of ketamine (100 mg/kg) and xylazine (8 mg/kg). Supplemental anesthesia at the same dosage was provided after surgery and at ∼2 h intervals throughout the recording session. Temperature was monitored rectally and maintained at ∼37°C by use of an electric heating pad.
After loss of pain reflexes, the mouse was placed in a stereotactic apparatus, and the skin, muscle, and skull overlying the cerebellum were resected. Gelfoam soaked in Ringer's solution bordered the exposed dura, and warmed agar was applied to the region and allowed to solidify for enhanced stability and temperature control.
In vivo recordings.
Single glass microelectrodes filled with 1 m NaCl with ∼1 MΩ resistance were inserted into the cerebellar vermis using a joystick-controlled three-axis micromanipulator (Marzhauser, Wetzlar, Germany). The recording electrode was oriented either vertically to target posterior cerebellar lobules or angled ∼30° relative to the dorsal surface of the brain to target anterior cerebellar lobules. Extracellular voltage signals were recorded using an alternating current amplifier (gain 1000×) at a sampling rate of 25 kHz (Multichannel Systems). Activity was monitored on an oscilloscope, and a virtual oscilloscope was built using the MCRack software (MultiChannel Systems). Raw data and 200 Hz high-pass-filtered data were simultaneously acquired. Data analysis was performed with programs written in IgorPro (WaveMetrics, Lake Oswego, OR).
The recording electrode was slowly lowered through the cerebellar cortex until activity from a single PC was well isolated. Specific criteria were established to determine that we were recording PC activity from sites close to, or at, the level of the PC soma. Climbing fiber responses can be recorded by extracellular electrodes throughout most of the molecular layer depth; however, only at the deepest portion of the molecular layer and in the PC layer are both complex and simple spikes present (Thach, 1967). Therefore, only recordings containing both complex and simple firing patterns at similar amplitudes were further analyzed. Moreover, simple spikes had to have an initial positive-going phase, indicating close proximity to the cell body (Thach, 1967). The PC recordings meeting these criteria (n = 8 wild type, n = 9 Kv3.3 KO) were analyzed for specific properties of their complex spike waveforms.
A spikelet was defined as a sharp negative-going impulse after the first spike, of at least two times the amplitude of the noise.
Simulations of Na+ currents.
Simulations were performed using the Neuron5.9 simulation environment (Hines and Carnevale, 1997). State-dependent Na+ channel models are based on the models of Raman and Bean (2001) and Khaliq et al. (2003) who performed recordings in wild-type and Nav1.6 mutant PCs, using the implementation in the Neuron platform by Akemann and Knopfel (2006) (http://senselab.med.yale.edu/modeldb/ShowModel.asp?model=80769). Voltage-clamp simulations were conducted using resurgent and nonresurgent Na+ channel models to predict Na+ current availability during the complex spike. Voltage waveforms were adapted from experimental recordings of the complex spike in control conditions and after bath application of 1 mm TEA. For data presented in Figure 6C showing the proportion of channels in each state, we combined all closed and all inactivated states explicit in the model (Raman and Bean, 2001) into single “closed” and “inactivated” conditions.
Figure 6.
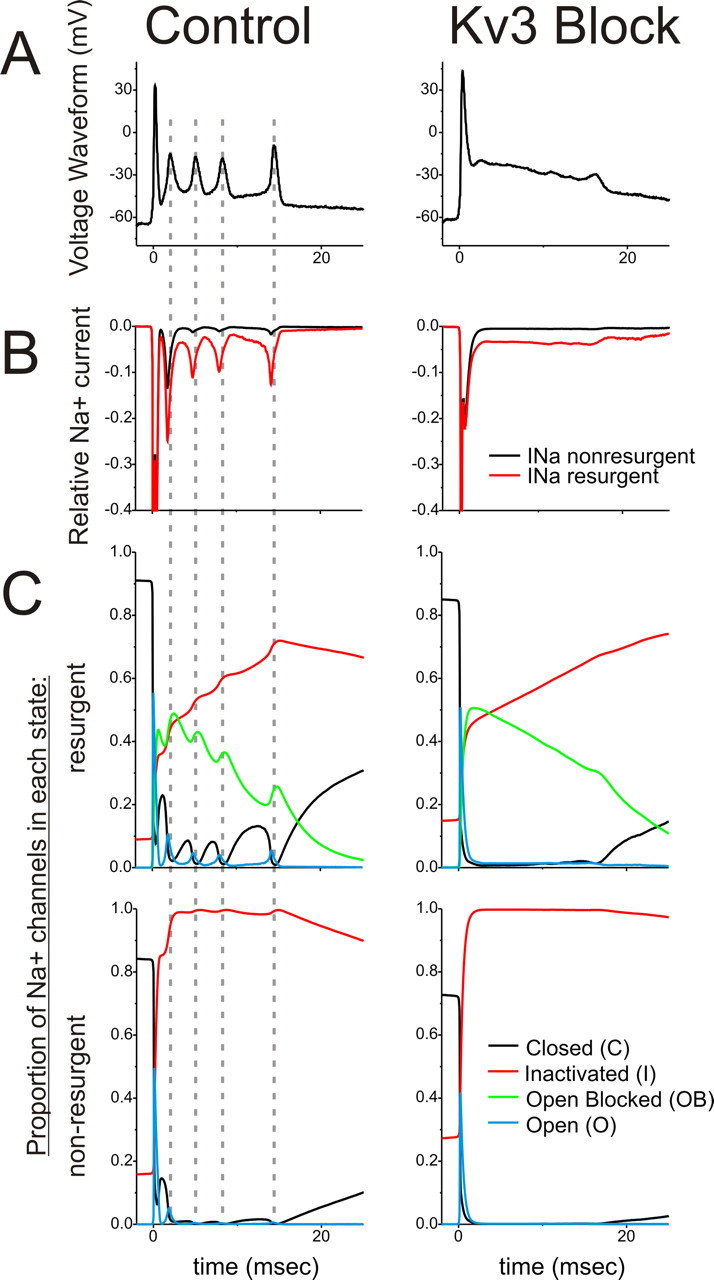
Kv3-mediated spike repolarization and resurgent Na+ channel properties cooperatively promote Na+ current availability for repetitive activity. A, Complex spike recordings from acute slices in control and 1 mm TEA (Kv3 Block) conditions were used as voltage waveforms in simulations with state models of Na+ channel gating. B, Na+ currents from resurgent (red) and nonresurgent (black) channels during the simulated complex spike, normalized to the peak amplitude of Na+ currents during the first spike (−1.0) for each simulation. C, Proportion of Na+ channels in each state during the simulations. Dashed lines are aligned to the peak of each spikelet. Rapid recovery only occurred for Na+ channels in the open blocked state, as opposed to inactivated states, and only for Kv3-intact control complex spike simulations. Also, transition out of the open blocked state for control complex spike simulations was concomitant with increases in the proportion of Na+ channels in closed states.
Statistics.
Mean values are reported with their SD. Student's t test, paired or unpaired as appropriate, was used for comparisons of population means. Spikelet amplitude was measured from the base of the plateau potential to peak of the spikelet, for both somatic and dendritic recordings. High-frequency character of the spikelets of the complex spike was calculated as SD of the first derivative of the voltage trace for 10 ms after the first spike of the complex spike, minus the same procedure for 10 ms preceding the complex spike to control for noise within the same recordings.
Results
Kv3.3 channels are required for the expression of the spikelets of the complex spike
Patch-clamp recordings in whole-cell configuration were established at PC somas in acute slices of mouse cerebellar vermis. To evoke complex spikes, the climbing fiber was stimulated within the granule cell layer (experimental configuration in Fig. 1A). The all-or-none nature of the complex spike may be appreciated in Figure 1B. Recordings with complex spikes fully isolated from parallel fiber inputs, as determined by lack of a discernable EPSP in traces with complex spike failures, were further studied.
Figure 1.

TEA blocks the high-frequency spikelets of the complex spike. A, Acute slice recordings from PC somas were established in whole-cell configuration, and complex spikes were evoked by stimulation of the climbing fiber (illustrated in red) in the granule cell layer of the cerebellar cortex. B, The highly stereotyped, all-or-none nature of the complex spike may be appreciated at near-threshold stimulations. Hash marks indicate stimulation artifact. Lack of EPSP in complex spike failures indicated isolation of climbing fiber from parallel fiber inputs. C, D, Bath application of 1 mm TEA drastically altered the complex spike, reversibly eliminating the repetitive spikelets resulting in an extended plateau after the first spike. Seen at expanded timescales (right traces), 1 mm TEA reduced the rate and depth of repolarization of the first spike of the complex spike. Effects of TEA were similar at room temperature (C) and physiological temperatures (D; 36°C). Lower calibration values in C and D refer to colored traces in right columns.
Bath application of 1 mm TEA, which blocks Kv3 channels [IC50 150–300 μm (Rudy et al., 1999)], drastically altered the complex spike waveform (Fig. 1). The most striking difference in the complex spike after application of TEA was the absence of repetitive spikelets. Whereas the overall duration of the complex spike did not appreciably change, spikelets were replaced by a slowly decrementing voltage plateau. This established that currents sensitive to 1 mm TEA are essential in supporting the stereotyped high-frequency components of the complex spike.
Apparent at expanded timescales (Fig. 1C,D, right column), 1 mm TEA significantly reduced the rate and depth of repolarization of the first spike of the complex spike (n = 9; rate, 51 ± 15% reduction; t test, p < 0.001; depth, 24 ± 7mV difference; p < 0.00001) but did not affect its rate of rise (p = 0.28). The first spike of the complex spike is known to be a Na+ spike initiated in the axon and dependent on axosomatic conductances (Stuart and Hausser, 1994). These effects on the first spike are therefore consistent with Kv3.3 expression in somata of PCs significantly contributing to the repolarization of axosomatic spikes. TEA also significantly increased the amplitude of the first spike (12 ± 8.7% increase; p = 0.003), suggesting that, in control conditions, TEA-sensitive repolarizing currents limit the influence of Na+ channel-mediated depolarization, contrary to Na+ channel inactivation alone determining action potential amplitude. Effects of TEA on the first spike and spikelets were similar at room temperature and physiological temperatures (34–36°C) (Fig. 1C,D).
TEA at a concentration of 1 mm blocks 80–90% Kv3 channel activity but is also sufficient to significantly block other K+ channel types, including homomeric Kv1.1, BK, and KCNQ2 channels (Coetzee et al., 1999; Rudy et al., 1999). To determine whether the effects of TEA described above were a consequence of Kv3 channels, we tested the effects of bath application of specific blockers of the non-Kv3 TEA-sensitive channels on the complex spike waveform, a strategy used previously to isolate the effects of Kv3 currents (Erisir et al., 1999; McKay and Turner, 2004). IbTx (Fig. 2A; n = 3) and DTX-K (Fig. 2B; n = 3), which block BK- and Kv1.1-containing channels, respectively, had no effect on the rate of repolarization of the first spike and did not abolish the high-frequency spikelet sequence. These drugs did affect total duration of the complex spike, typically increasing it and producing an additional spikelet (iberiotoxin, 3 of 3; DTX-K, 2 of 3; no effect on 1 of 3). The KCNQ blocker linopirdine had no effect on the complex spike (Fig. 2C; n = 3). These data support the conclusion that the dramatic effects of TEA on the complex spike spikelets and first spike repolarization result from block of Kv3 channels. Moreover, Kv3 channels are not expressed in the inferior olive (Weiser et al., 1994), and therefore these effects are most likely attributable to changes in PC excitability.
Figure 2.
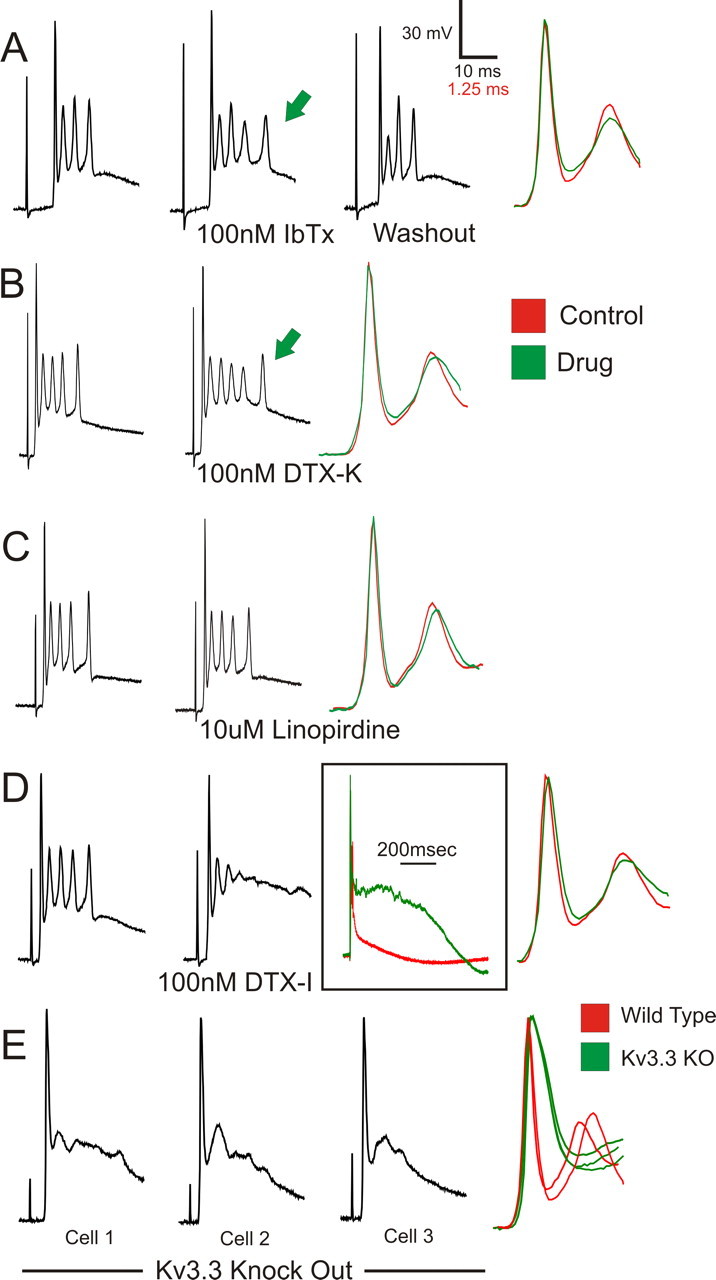
Effects of TEA on the complex spike are mediated by block of Kv3 channels. Specific blockers of the non-Kv3 TEA-sensitive ion channels were bath applied to determine contributions of these channels to the effects of TEA on the complex spike. IbTx at 100 nm (A), which blocks BK channels, and 100 nm DTX-K (B), which blocks channels containing Kv1.1 subunits, mildly affected the complex spike waveform, typically increasing duration of the complex spike and yielding an additional spikelet (green arrows). C, Linopirdine at 10 μm, blocker of KCNQ channels, had no effect on the complex spike waveform. Unlike the effects of 1 mm TEA, none of these blockers eliminated spikelet expression or considerably affected rate of repolarization of the first spike (right traces). D, DTX-I at 100 nm, which blocks Kv1.1, Kv1.2, and Kv1.6 subunits, did not affect repolarization of the first spike but compromised repetitive spikelet expression and dramatically extended the duration of the complex spike. E, Complex spike recordings from PCs in Kv3.3 KO mice show disrupted spikelet generation and affected repolarization of the first spike. Right traces are overlaid recordings from three Kv3.3 KO and two wild-type PCs at expanded timescales and normalized amplitudes.
Interestingly, DTX-I, which blocks Kv1 channels containing Kv1.1, Kv1.2, or Kv1.6 subunits, massively affected the complex spike but unlike the effects of 1 mm TEA. DTX-I did not affect repolarization of the first spike but limited the expression of robust repetitive spikelets and increased the plateau duration ∼14-fold (Fig. 2D; n = 2). This result highlights the importance of Kv1 channels in shaping the complex spike, although block of Kv1 channels cannot account for the effects of TEA because DTX-K caused only minor changes in complex spike properties. Kv1 channels selectively regulate dendritic excitability of PCs (Khavandgar et al., 2005), and therefore this result highlights the importance of certain dendritic conductances in shaping the somatic complex spike waveform.
Acute slice recordings were also obtained from Kv3.3 KO mice. Climbing fiber-evoked complex spikes in these PCs displayed altered repolarization of the first spike and disrupted spikelets compared with wild-type recordings, changes similar to those caused by TEA on wild-type PCs (Fig. 2E; n = 5). Altered complex spike generation in Kv3.3 KO PCs was also observed at physiological temperatures (data not shown; n = 5). These data implicate Kv3.3 subunits as essential components of the Kv3 channels that are necessary for the generation of the synaptically evoked complex spike. The results also show that disrupted complex spike expression is not compensated in the Kv3.3 KO mouse, which may reflect the paucity of expression of other Kv3 subunits in PCs (Weiser et al., 1994) and the functional importance of unique properties of Kv3 channels.
Somatic but not dendritic Kv3.3 channels are necessary for spikelet generation
Because PCs prominently express Kv3.3-containing channels in both somatic and dendritic membranes (Martina et al., 2003; McMahon et al., 2004; Chang et al., 2007), we next investigated the relative contributions of Kv3 activity in these compartments to the complex spike waveform. To do this, we took advantage of the highly laminar organization of the cerebellar cortex in which the planar arborization of the PC dendritic tree is primarily segregated from the cell body. We attempted to block somatic or dendritic conductances selectively by applying short-duration perfusions of TEA at the soma within the PC layer or to the dendrites in the molecular layer, immediately before climbing fiber stimulation (configuration in Fig. 3A). Precautions were taken to ensure that drug targeting was specific yet effective within the intended compartment (see Materials and Methods) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Figure 3.

Somatic Kv3 channel activity is required for spikelet expression. A, A series of local TEA perfusion experiments were conducted to determine the contributions of somatic and dendritic Kv3 channels to complex spike properties. Pressure injection from a micropipette was applied immediately before climbing fiber stimulation in the PC layer or molecular layer to target the PC soma or dendrites, respectively. B, TEA at 5 mm applied to the PC soma for the 200 ms before stimulation (B2) severely disrupted spikelet formation and compromised repolarization of the first spike, similar to bath application of 1 mm TEA in the same cell (B4). C, Selective perfusion of 1 mm TEA for 2 s at PC dendrites (C2) mildly affected the complex spike waveform, whereas the same perfusion of the soma (C3) eliminated repetitive spikelet activity and substantially reduced repolarization of the first spike. Bottom time calibration refers to overlaid traces in B5.
Somatic targeting of TEA reduced the rate and depth of repolarization of the first spike of the complex spike and disrupted spikelet expression, resulting in a voltage plateau that slowly repolarized to resting membrane potentials (Fig. 3; n = 4). These effects were similar to bath application of TEA in the same cells (Fig. 3B). In additional experiments (n = 3), CNQX was added at a higher effective concentration than TEA to the perfusate (50 μm CNQX, ∼50 times the IC50; 5 mm TEA, ∼25 times the IC50) as an internal control for restricted perfusion of the soma. CNQX did not block the complex spike, indicating that the perfusion was primarily limited to somatic and perisomatic regions (see Materials and Methods), yet the complex spike waveform was affected by TEA similar to experiments without CNQX in the perfusate (data not shown). The absence of repetitive spikelets in somatic perfusion experiments suggested that Kv3 conductances at the soma are essential for the generation of spikelets. Furthermore, local dendritic TEA application only mildly affected the complex spike, slightly limiting repolarization of the first spike and spikelets (Fig. 3C; n = 3). This is unlikely attributable to lack of TEA effects in the dendritic tree, because control experiments demonstrated the ability of local dendritic TEA application to significantly alter dendritic excitability (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Moreover, because climbing fiber synapses are distributed along the large-caliber dendrites, the minimal effect on the complex spike of TEA application to the molecular layer further suggests that effects of bath-applied TEA on the complex spike are attributable to disturbances of postsynaptic excitability and not to effects on synaptic release from climbing fiber terminals.
Whereas the PC soma is clearly endowed with the ability to generate burst outputs (Llinas and Sugimori, 1980a,b; Raman and Bean, 1997; Swensen and Bean, 2003), discrepancies exist in the field as to whether spikelets of the synaptically evoked complex spike are generated at somatic or dendritic compartments (for review, see Schmolesky et al., 2002) (Callaway et al., 1995; Callaway and Ross, 1997; Hansel and Linden, 2000; Schmolesky et al., 2002; Weber et al., 2003). To test this, we recorded in whole-cell configuration simultaneously from the soma and dendrite of a PC while stimulating its associated climbing fiber afferent (Fig. 4). Dendritic recordings were obtained from 45–75 μm from the origin of the dendrite at the soma, from primary to tertiary dendrites and bifurcation points. We compared spikelet properties from the soma and dendrites for all but the first spikelet in each recording, which in some dendritic recordings was obscured by the peak of the EPSP (n = 4 dual recorded cells). Spikelets were always larger in amplitude at the soma (soma, 16.3 ± 4.0 mV; dendrites, 5.3 ± 1.7 mV; p = 0.000016) (Fig. 4D). Moreover, the spikelets rose faster and peaked first in the soma (Fig. 4B,C). By plotting the lag of spikelet peak in the dendrite relative to the soma versus distance of the dendritic recording site from the soma (Fig. 4E), a well-fit linear relationship emerged with the slope of the fitted line giving a propagation speed of 0.18 m/s (R2 = 0.88). These data strongly argue that spikelets are generated at axosomatic membranes and spread passively into the dendritic tree.
Figure 4.

Spikelet generation occurs first at somatic membranes and passively spreads into dendrites. A, Simultaneous whole-cell recordings were established from the soma and dendrite of PCs while stimulating the climbing fiber afferent. B, C, Shown are climbing fiber responses at the soma and dendrite of two cells, with targeted dendritic recordings from 45 μm (B) and 65 μm (C) from the cell body. By overlaying somatic (black) and dendritic (gray) recordings (B3, C3), it is apparent that spikelets are larger in the soma. At higher temporal resolution and normalization of amplitudes (B4, C4), spikelets appear to rise first and peak first in somatic recordings. D, For all recordings, spikelet amplitude was larger in the soma than the dendrites. E, Plotting the temporal lag of spikelet peak in the dendrite versus the distance of the dendritic recording electrode from the soma yields a well-fit (R2 = 0.88) linear relationship. Equation of fit is y = 0.0055 × x − 0.076; 1/slope = 0.18 m/s. These findings are consistent with spikelet generation at axosomatic membranes and passive spreading into the dendritic tree. Bottom time calibration refers to traces in B4 and C4.
Kv3 channel-dependent spike properties and Na+ current availability
To understand why elimination of somatic Kv3 activity so severely disrupts spikelet generation, we first sought a more quantitative analysis of our complex spike recordings (Fig. 5). Specifically, to quantify the high-frequency character of the spikelets, we obtained the first derivative of the voltage recording, which depicts the rate of change of transmembrane voltage. Because higher-frequency events will show greater fluctuation in rates of change, the SD of the 10 ms period containing the spikelets was used as a metric of the high-frequency character of the complex spike (Fig. 5A,B). As expected, the SD of first derivative was significantly greater for wild-type compared with 1 mm bath TEA or Kv3.3 KO complex spike recordings (Fig. 5C) (control vs TEA, p < 0.000001; control vs Kv3.3 KO, p < 0.0001; TEA vs Kv3.3 KO, p = 0.98).
Figure 5.

Properties of the first spike of the complex spike are highly correlated with robust spikelet generation. As shown for complex spike recordings from wild-type (A; WT) and Kv3.3 KO (B) PCs, we obtained the first derivative trace of the voltage recording (A2, B2) and calculated its SD over 10 ms after the first spike (bracketed regions in A and B). The value of this statistic is shown for A2 and B2 below the traces. Hash marks indicate stimulation artifact. C, SD of first derivative was significantly larger in control recordings than in 1 mm TEA or Kv3.3 KO conditions. Error bars are SD (*p < 0.0001; **p < 0.000001). D, SD of first derivative was highly correlated with depth of repolarization of the first spike within and between experimental conditions. Also plotted is the relationship between SD of first derivative and repolarization of the first spike for a single experiment during wash-in of TEA (light gray triangles).
This quantification allowed us to compare various parameters of the complex spike within and between experimental conditions. As seen in Figure 5D, SD of first derivative was highly correlated to depth of repolarization of the first spike across all experiments. This relationship between properties of the first spike and spikelets was maintained within individual experiments during wash-in of 1 mm TEA into the bath (Fig. 5D, light gray triangles). Considering the axosomatic site of spikelet initiation, we believe that robust spikelet expression depends directly on the waveform of the first spike because this waveform will determine the extent of Na+ channel availability preceding spikelet generation.
PCs express “resurgent” sodium currents with open channel block and rapid recovery through the conducting state (Raman and Bean, 1997). This resurgent property has been suggested to be important for high-frequency tonic and burst firing in PCs by providing interspike inward currents to depolarize somatic membranes toward spike initiation (Raman and Bean, 1997, 1999). Using state models of Na+ currents with and without resurgent properties (Khaliq et al., 2003; Akemann and Knopfel, 2006), we further explored the relationship between changes in the complex spike waveform from blocking Kv3 channels and availability of Na+ currents. In a series of voltage-clamp simulations, we used as the voltage waveform the complex spike we measured from current-clamp acute slice recordings in control and 1 mm TEA bath (Kv3 block) conditions (Fig. 6A). During control complex spike waveforms, resurgent Na+ channels produced robust currents associated with each of the spikelets, which were reduced in peak amplitude 75–90% compared with the first spike (Fig. 6B). However, Na+ currents were negligible during later spikelets for simulations using nonresurgent Na+ channels.
To understand the mechanism by which Kv3 and resurgent Na+ conductances maintain Na+ current availability, we plotted the proportion of channels in each state throughout the duration of the complex spike. For both control and Kv3 blocked conditions, after the first spike, nearly an equal proportion of channels accumulated in open blocked and inactivated states (Fig. 6C, top row). However, only for control complex spikes do we find rapid cycling out of the open blocked state after the first spike and each spikelet. Importantly, these transitions out of the open blocked state are associated with steep rises in the proportion of channels in closed states, which subsequently support the transient rise in Na+ currents for the following spikelet (Fig. 6C, top left). These simulations suggest that rapid cycling from the open blocked → open → closed states, which is encouraged by Kv3-dependent spike properties, is critical in maintaining Na+ currents throughout the complex spike.
Complex spikes are highly distorted in Kv3.3 KO mice in vivo
Our acute slice recordings reveal that Kv3.3 channels are necessary for generation of the synaptically evoked complex spikes and predict that complex spikes are altered in Kv3.3 KO mice, an effect that may contribute to the pathophysiology of cerebellar phenotypes observed in these mice. To test this prediction, we recorded extracellularly from PCs in anesthetized mice. Recordings were obtained throughout mouse vermis, in anterior and posterior cerebellar lobes. PCs are known to fire both simple and complex spikes spontaneously in vivo, and the presence of these patterns was used to verify that our blind recordings were targeting the somas of PCs (Thach, 1967) (see Materials and Methods). PCs from both wild-type and Kv3.3 KO mice displayed simple and complex spikes. Whereas the frequency of simple spikes was highly variable from cell to cell, complex spikes occurred near 1 Hz in both genetic backgrounds (data not shown), demonstrating that the olivocerebellar circuit is intact in Kv3.3 KO mice. However, specific properties of the PC electrical activity were altered in Kv3.3 KO mice, consistent with in vitro recordings.
Complex spikes recorded from wild-type PCs were composed of their typical high-frequency burst of spikes (Fig. 7A). These complex spikes averaged 4.49 ± 0.86 (n = 8) spikelets, with an average first interspike interval of 1.40 ± 0.6 ms and overall average interspike interval of 2.15 ± 0.39 ms. Complex spikes recorded from Kv3.3 KO PCs, however, were highly distorted, lacking the high-frequency repetitive firing characteristic of wild-type activity (Fig. 7B). Kv3.3 KO PCs fired on average 1.61 ± 0.52 spikelets (n = 9; p = 0.000005), with an average first interspike interval of 3.70 ± 0.9 ms (p = 0.00002) and overall average interspike interval of 3.58 ± 0.75 ms (p = 0.0003). These data confirm that Kv3.3 KO mice generate altered complex spikes in vivo, which presumably results in a reduction of axonal spike output associated with each complex spike.
Figure 7.

Complex spikes are altered in Kv3.3 KO mice in vivo. Extracellular recordings were conducted from the soma of PCs in anesthetized mice. A, Shown are 200 Hz high-pass-filtered spontaneous complex spikes from three wild-type PCs, which displayed rapid (up to 1 kHz), repetitive spiking. B, Complex spikes recorded from Kv3.3 KO PCs were highly distorted, with remarkably fewer and less rapid repetitive spiking events. Recordings in A and B are normalized in amplitude to match the height of the first spike.
In addition to changes in complex spikes, simple spikes were also altered in Kv3.3 KO mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Half-amplitude duration of simple spikes was increased in Kv3.3 KO recordings compared with wild-type (258 ± 58 μs, n = 8; KO, 337 ± 32 μs, n = 9; 30% increase; p = 0.003). This increase in duration is consistent with studies in acute slice suggesting that Kv3-mediated currents are important for the repolarization of single Na+ action potentials (Martina et al., 2003; McKay and Turner, 2004; McMahon et al., 2004; Akemann and Knopfel, 2006). Moreover, the shapes of the extracellularly recorded simple spikes were altered in KO mice, having a larger negative phase and a slower return to baseline, which likely reflects a prolonged duration of K+ conductance after spike repolarization in Kv3.3 KO PCs.
Discussion
In this study, we found that potassium channels composed of Kv3.3 subunits are required for the generation of the PC complex spike. We found that somatic, and not dendritic, Kv3 channels are critical for complex spike generation and that the spikelets in the high-frequency burst are generated at the soma and passively spread into the proximal dendritic tree. Kv3 channels and resurgent Na+ currents act to maintain Na+ channel availability, thereby enabling generation of stereotyped, high-frequency, repetitive responses to climbing fiber inputs.
Multiple lines of evidence suggest that Kv3.3 disruption alters complex spikes via effects on PC properties rather than by affecting presynaptic climbing fibers. None of the Kv3 genes are expressed in inferior olivary neurons; in contrast, Kv3.3 expression is extremely robust in PCs (Weiser et al., 1994). Local TEA perfusion experiments also argue for postsynaptic effects. Nearly all climbing fiber synaptic contacts onto PCs occur on large-caliber dendrites within the molecular layer. However, somatic perfusion of TEA disrupted the complex spike similar to bath application (Fig. 3). Functional restriction of perfused drugs to the PC layer was verified by effects on intrinsic firing patterns (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) and by demonstrating that adding CNQX at a higher effective concentration than TEA did not block the complex spike, which would be expected if the perfusate was effective within the molecular layer. Furthermore, local perfusion of TEA in the molecular layer, with access to both presynaptic climbing fiber terminals and postsynaptic PC dendrites, only mildly affected the complex spike.
Kv3 channel contributions to PC excitability
Kv3.3 (Vega-Saenz de Miera et al., 1992) is the dominant Kv3 subunit expressed in PCs (Weiser et al., 1994) and Kv3-mediated currents appear to be the major component of outward currents from PC somas (Martina et al., 2003). In PCs, Kv3 channels have been shown to repolarize single Na+ spikes rapidly and promote high-frequency spontaneous activity (Raman and Bean, 1999; McKay and Turner, 2004; McMahon et al., 2004; Akemann and Knopfel, 2006; Martina et al., 2007). The importance of Kv3.3 channels in the repolarization of simple spikes was also evident in our in vivo recordings (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Our main finding is that somatic Kv3 channels containing Kv3.3 subunits are necessary for expression of the synaptically evoked PC complex spike. Kv3 channels at the PC soma significantly contribute to repolarization of the first spike, primarily determining its rate and depth of repolarization. Furthermore, Kv3 channel activity is critical for expression of the high-frequency spikelets. We find that these two properties, first spike repolarization and spikelet expression, are intimately related through the activity and availability of Na+ currents. For fast spiking (FS) interneurons of neocortex and hippocampus, Kv3 channels enable high-frequency firing in part by mediating a rapid, large afterhyperpolarization (AHP), thereby promoting recovery of Na+ channel inactivation (Erisir et al., 1999; Lien and Jonas, 2003). The high-frequency activity of complex spikes differs from fast tonic firing of FS cells in that the PC somatic membrane does not maintain large AHPs after the first spike and each spikelet because of the extended voltage plateau on which the repetitive activity resides. Simulations of state-dependent Na+ channel models (Fig. 6) suggest that, considering the lack of prominent AHPs, the resurgent properties of PC Na+ currents are critical in maintaining Na+ current availability throughout the complex spike.
In addition to conventional inactivation states, PCs have Na+ channels that are believed to engage an open block, which recovers through the open or conducting state, thus mediating resurgent Na+ currents (Raman and Bean, 1997, 2001). After returning to the open state, Na+ channels may either close or inactivate. Exit from open block and recovery/accumulation into closed states are both enhanced by rapid and deep spike repolarization, which in PCs is determined by Kv3 activity. Because recovery from conventional inactivated states during the complex spike is restricted as a result of the voltage plateau, open block acts as a protective state in which Na+ channels may recover directly to the closed state and thereby bypass inactivation. Thus, Kv3 and resurgent Na+ channels are coordinated to induce rapid cycling between open blocked and closed states, thereby maintaining Na+ currents during the extended complex spike.
A relationship between kinetic properties of Kv3 channels and resurgent Na+ activity has also been appreciated in the context of high-frequency spontaneous firing and intrinsic burst firing in PCs (Raman and Bean, 1999; Swensen and Bean, 2003; Akemann and Knopfel, 2006). It is unknown whether the interaction between resurgent Na+ and Kv3 currents described above also contributes to repetitive firing in other high-frequency firing neurons. Additionally, similar to high-frequency tonic firing, fast deactivation rates of Kv3 channels (Rudy and McBain, 2001) are expected to be critical in determining the refractory period after each spike and spikelet and, thus, the frequency of firing within the complex spike.
Intrinsic bursting and the complex spike
PC somas generate bursts under different conditions. Prolonged, high-amplitude current injections into PC somas evokes Na+–Ca2+ spike bursts in which repetitive somatic Na+ spikes are triggered by dendritic Ca2+ electrogenesis (Llinas and Sugimori, 1980a,b). Furthermore, acutely dissociated PC somas that lack elaborated dendritic arbors respond to short-duration current injections with multi-spike bursts (Raman and Bean, 1997). Kv3 channels have been proposed to contribute to these bursting properties (Raman and Bean, 1999; Swensen and Bean, 2003; McKay and Turner, 2004). The complex spike differs from these intrinsic responses in its higher frequency of repetitive events, precise voltage trajectory, and influences of synaptic and dendritic conductances. However, whereas Kv3 channels may not contribute to burst firing in other bursting central neurons (Rudy and McBain, 2001), they appear to be critically important in PC bursting whether evoked by synaptic or intrinsic mechanisms.
Ionic mechanisms of generation of the complex spike
There are discrepancies as to whether individual spikelets of the complex spike are initiated at axosomatic membranes or triggered from repetitive Ca2+ electrogenesis in the dendrites (for review, see Schmolesky et al., 2002). Our finding that Kv3 channel activity specifically at the PC soma (and not in the dendrite) is essential for spikelet expression suggests that somatic conductances are critical determinants of spikelet generation. Moreover, the amplitude and timing of spikelets in dual somatic–dendritic complex spike recordings strongly argue for spikelet generation at somatic rather than dendritic compartments, with possible spike initiation in the axon.
Additional questions have been raised regarding the influence of active dendritic conductances on the somatic complex spike waveform. We found that blocking DTX-sensitive Kv1 channels, which are thought to be expressed in PC dendritic membranes and absent from somas (Southan and Robertson, 2000; Khavandgar et al., 2005) (but see McKay et al., 2005), dramatically compromised robust repetitive spikelet activity without affecting repolarization of the first spike (Fig. 2). This result suggests that, for somatic electrogenesis, dendritic conductances may be specifically tuned to provide axial currents large enough to limit the potential current sink imposed by the extended dendritic membrane (Llinas and Nicholson, 1976) yet not be so large that they interfere with the ability of somatic outward currents to rapidly repolarize repetitive spikes. Although we do not believe that dendritic Kv3 channels are important regulators of the somatic complex spike waveform, they may be particularly important for dendritic signaling considering their localization in distal dendrites (Chang et al., 2007).
Signaling role of the complex spike
The role of the complex spike in cerebellar function continues to be vigorously debated (Simpson et al., 1996; Schmolesky et al., 2002; Ito, 2006). The climbing fiber-evoked dendritic Ca2+ influx has been proposed to regulate strength of parallel fiber inputs through long-term depression (Ito, 1993; Linden and Connor, 1993); others have highlighted the outputs of complex spikes as important signals for motor timing and coordination by providing strong, synchronous inhibition onto deep cerebellar nuclear (DCN) neurons (Welsh et al., 1995; Blenkinsop and Lang, 2005; Lang et al., 2006).
We report that Kv3.3 KO mice generate spontaneous climbing fiber responses in vivo that have fewer and lower frequency repetitive spikelets than control animals (Fig. 7). This is expected to reduce inhibition per complex spike onto DCN neurons, which may be compounded by increased spike propagation failure in Kv3.3 KO PCs, as Kv3.3 subunits are also expressed in PC axons (Chang et al., 2007). Interestingly, these mutant mice show a nearly abolished sensitivity to harmaline (McMahon et al., 2004), which normally produces a widespread 10 Hz tremor by massively increasing the rhythmicity and synchrony of complex spikes (de Montigny and Lamarre, 1973; Llinas and Volkind, 1973; Sasaki et al., 1989). If reduction in harmaline sensitivity in Kv3.3 KO mice is attributable to altered complex spike activity, it would argue that, even with massive synchronization of the olivocerebellar circuit, the actual quality of the burst has a critical impact on the ability of this system to drive motor activity.
Kv3.3 KO mice have other cerebellar deficits, including altered gait and reduced motor performance (Joho et al., 2006); humans with missense mutations in the Kv3.3 coding sequence develop childhood- or adult-onset ataxia (Waters et al., 2006). Future studies need to address whether alterations in PC excitability contribute to these human and mouse phenotypes.
Footnotes
This work was supported by National Institutes of Health Grants NS30989 and NS045217 (B.R.), National Science Foundation Grant IBN-0314645 (B.R.), a Hirschl Grant (E.J.L.), and National Research Service Award F31 NS59124 (E.Z.). We thank Dr. Nathaniel Heintz (Rockefeller University) for providing the Kv3.3 knock-out mice. We thank Dr. Rodolfo Llinas, Dr. Jerry Simpson, and Dr. Chris Leonard for many insightful discussions, Tim Blenkinsop for technical assistance with in vivo recordings, and Dr. Ilya Kruglikov for helpful comments during the preparation of this manuscript. We thank Stephen Larson for help implementing Neuron simulations. We also thank the Hausser laboratory and staff of the Dendritic Patching Workshop for training on dendritic recordings.
References
- Akemann W, Knopfel T. Interaction of Kv3 potassium channels and resurgent sodium current influences the rate of spontaneous firing of Purkinje neurons. J Neurosci. 2006;26:4602–4612. doi: 10.1523/JNEUROSCI.5204-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blenkinsop TA, Lang EJ. Synchronous complex spike activity is associated with changes in deep cerebellar nuclear firing patterns. Soc Neurosci Abstr. 2005;31:179.1. [Google Scholar]
- Callaway JC, Ross WN. Spatial distribution of synaptically activated sodium concentration changes in cerebellar Purkinje neurons. J Neurophysiol. 1997;77:145–152. doi: 10.1152/jn.1997.77.1.145. [DOI] [PubMed] [Google Scholar]
- Callaway JC, Lasser-Ross N, Ross WN. IPSPs strongly inhibit climbing fiber-activated [Ca2+]i increases in the dendrites of cerebellar Purkinje neurons. J Neurosci. 1995;15:2777–2787. doi: 10.1523/JNEUROSCI.15-04-02777.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SY, Zagha E, Kwon ES, Ozaita A, Bobik M, Martone ME, Ellisman MH, Heintz N, Rudy B. Distribution of Kv3.3 potassium channel subunits in distinct neuronal populations of mouse brain. J Comp Neurol. 2007;502:953–972. doi: 10.1002/cne.21353. [DOI] [PubMed] [Google Scholar]
- Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, Moreno H, Nadal MS, Ozaita A, Pountney D, Saganich M, Vega-Saenz de Miera E, Rudy B. Molecular diversity of K+ channels. Ann NY Acad Sci. 1999;868:233–285. doi: 10.1111/j.1749-6632.1999.tb11293.x. [DOI] [PubMed] [Google Scholar]
- Davie JT, Hausser M. Determinants of the complex spike waveform in cerebellar Purkinje cells. Soc Neurosci Abstr. 2004;30:169.10. doi: 10.1523/JNEUROSCI.0559-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Montigny C, Lamarre Y. Rhythmic activity induced by harmaline in the olivo-cerebello-bulbar system of the cat. Brain Res. 1973;53:81–95. doi: 10.1016/0006-8993(73)90768-3. [DOI] [PubMed] [Google Scholar]
- Eccles JC, Llinas R, Sasaki K. The excitatory synaptic action of climbing fibres on the Purkinje cells of the cerebellum. J Physiol (Lond) 1966;182:268–296. doi: 10.1113/jphysiol.1966.sp007824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erisir A, Lau D, Rudy B, Leonard CS. Function of specific K+ channels in sustained high-frequency firing of fast-spiking neocortical interneurons. J Neurophysiol. 1999;82:2476–2489. doi: 10.1152/jn.1999.82.5.2476. [DOI] [PubMed] [Google Scholar]
- Goldman-Wohl DS, Chan E, Baird D, Heintz N. Kv3.3b: a novel Shaw type potassium channel expressed in terminally differentiated cerebellar Purkinje cells and deep cerebellar nuclei. J Neurosci. 1994;14:511–522. doi: 10.1523/JNEUROSCI.14-02-00511.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansel C, Linden DJ. Long-term depression of the cerebellar climbing fiber–Purkinje neuron synapse. Neuron. 2000;26:473–482. doi: 10.1016/s0896-6273(00)81179-4. [DOI] [PubMed] [Google Scholar]
- Hines ML, Carnevale NT. The NEURON simulation environment. Neural Comput. 1997;9:1179–1209. doi: 10.1162/neco.1997.9.6.1179. [DOI] [PubMed] [Google Scholar]
- Ito M. Synaptic plasticity in the cerebellar cortex and its role in motor learning. Can J Neurol Sci. 1993;20(Suppl 3):S70–S74. [PubMed] [Google Scholar]
- Ito M. Cerebellar circuitry as a neuronal machine. Prog Neurobiol. 2006;78:272–303. doi: 10.1016/j.pneurobio.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Joho RH, Street C, Matsushita S, Knopfel T. Behavioral motor dysfunction in Kv3-type potassium channel-deficient mice. Genes Brain Behav. 2006;5:472–482. doi: 10.1111/j.1601-183X.2005.00184.x. [DOI] [PubMed] [Google Scholar]
- Khaliq ZM, Gouwens NW, Raman IM. The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: an experimental and modeling study. J Neurosci. 2003;23:4899–4912. doi: 10.1523/JNEUROSCI.23-12-04899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khavandgar S, Walter JT, Sageser K, Khodakhah K. Kv1 channels selectively prevent dendritic hyperexcitability in rat Purkinje cells. J Physiol (Lond) 2005;569:545–557. doi: 10.1113/jphysiol.2005.098053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang EJ, Sugihara I, Llinas R. Olivocerebellar modulation of motor cortex ability to generate vibrissal movements in rat. J Physiol (Lond) 2006;571:101–120. doi: 10.1113/jphysiol.2005.102764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larramendi EM, Victor T. Synapses on the Purkinje cell spines in the mouse. An electronmicroscopic study. Brain Res. 1967;5:15–30. doi: 10.1016/0006-8993(67)90216-8. [DOI] [PubMed] [Google Scholar]
- Lau D, Vega-Saenz de Miera EC, Contreras D, Ozaita A, Harvey M, Chow A, Noebels JL, Paylor R, Morgan JI, Leonard CS, Rudy B. Impaired fast-spiking, suppressed cortical inhibition, and increased susceptibility to seizures in mice lacking Kv3.2 K+ channel proteins. J Neurosci. 2000;20:9071–9085. doi: 10.1523/JNEUROSCI.20-24-09071.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien CC, Jonas P. Kv3 potassium conductance is necessary and kinetically optimized for high-frequency action potential generation in hippocampal interneurons. J Neurosci. 2003;23:2058–2068. doi: 10.1523/JNEUROSCI.23-06-02058.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden DJ, Connor JA. Cellular mechanisms of long-term depression in the cerebellum. Curr Opin Neurobiol. 1993;3:401–406. doi: 10.1016/0959-4388(93)90133-j. [DOI] [PubMed] [Google Scholar]
- Llano I, Marty A, Armstrong CM, Konnerth A. Synaptic- and agonist-induced excitatory currents of Purkinje cells in rat cerebellar slices. J Physiol (Lond) 1991;434:183–213. doi: 10.1113/jphysiol.1991.sp018465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R, Nicholson C. Reversal properties of climbing fiber potential in cat Purkinje cells: an example of a distributed synapse. J Neurophysiol. 1976;39:311–323. doi: 10.1152/jn.1976.39.2.311. [DOI] [PubMed] [Google Scholar]
- Llinas R, Sugimori M. Electrophysiological properties of in vitro Purkinje cell somata in mammalian cerebellar slices. J Physiol (Lond) 1980a;305:171–195. doi: 10.1113/jphysiol.1980.sp013357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R, Sugimori M. Electrophysiological properties of in vitro Purkinje cell dendrites in mammalian cerebellar slices. J Physiol (Lond) 1980b;305:197–213. doi: 10.1113/jphysiol.1980.sp013358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R, Volkind RA. The olivo-cerebellar system: functional properties as revealed by harmaline-induced tremor. Exp Brain Res. 1973;18:69–87. doi: 10.1007/BF00236557. [DOI] [PubMed] [Google Scholar]
- Martina M, Schultz JH, Ehmke H, Monyer H, Jonas P. Functional and molecular differences between voltage-gated K+ channels of fast-spiking interneurons and pyramidal neurons of rat hippocampus. J Neurosci. 1998;18:8111–8125. doi: 10.1523/JNEUROSCI.18-20-08111.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina M, Yao GL, Bean BP. Properties and functional role of voltage-dependent potassium channels in dendrites of rat cerebellar Purkinje neurons. J Neurosci. 2003;23:5698–5707. doi: 10.1523/JNEUROSCI.23-13-05698.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina M, Metz AE, Bean BP. Voltage-dependent potassium currents during fast spikes of rat cerebellar Purkinje neurons: inhibition by BDS-I toxin. J Neurophysiol. 2007;97:563–571. doi: 10.1152/jn.00269.2006. [DOI] [PubMed] [Google Scholar]
- Matsukawa H, Wolf AM, Matsushita S, Joho RH, Knopfel T. Motor dysfunction and altered synaptic transmission at the parallel fiber-Purkinje cell synapse in mice lacking potassium channels Kv3.1 and Kv3.3. J Neurosci. 2003;23:7677–7684. doi: 10.1523/JNEUROSCI.23-20-07677.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay BE, Turner RW. Kv3 K+ channels enable burst output in rat cerebellar Purkinje cells. Eur J Neurosci. 2004;20:729–739. doi: 10.1111/j.1460-9568.2004.03539.x. [DOI] [PubMed] [Google Scholar]
- McKay BE, Molineux ML, Mehaffey WH, Turner RW. Kv1 K+ channels control Purkinje cell output to facilitate postsynaptic rebound discharge in deep cerebellar neurons. J Neurosci. 2005;25:1481–1492. doi: 10.1523/JNEUROSCI.3523-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon A, Fowler SC, Perney TM, Akemann W, Knopfel T, Joho RH. Allele-dependent changes of olivocerebellar circuit properties in the absence of the voltage-gated potassium channels Kv3.1 and Kv3.3. Eur J Neurosci. 2004;19:3317–3327. doi: 10.1111/j.0953-816X.2004.03385.x. [DOI] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci. 1997;17:4517–4526. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J Neurosci. 1999;19:1663–1674. doi: 10.1523/JNEUROSCI.19-05-01663.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Inactivation and recovery of sodium currents in cerebellar Purkinje neurons: evidence for two mechanisms. Biophys J. 2001;80:729–737. doi: 10.1016/S0006-3495(01)76052-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy B, McBain CJ. Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci. 2001;24:517–526. doi: 10.1016/s0166-2236(00)01892-0. [DOI] [PubMed] [Google Scholar]
- Rudy B, Chow A, Lau D, Amarillo Y, Ozaita A, Saganich M, Moreno H, Nadal MS, Hernandez-Pineda R, Hernandez-Cruz A, Erisir A, Leonard C, Vega-Saenz de Miera E. Contributions of Kv3 channels to neuronal excitability. Ann NY Acad Sci. 1999;868:304–343. doi: 10.1111/j.1749-6632.1999.tb11295.x. [DOI] [PubMed] [Google Scholar]
- Sasaki K, Bower JM, Llinas R. Multiple purkinje cell recording in rodent cerebellar cortex. Eur J Neurosci. 1989;1:572–586. doi: 10.1111/j.1460-9568.1989.tb00364.x. [DOI] [PubMed] [Google Scholar]
- Schmolesky MT, Weber JT, De Zeeuw CI, Hansel C. The making of a complex spike: ionic composition and plasticity. Ann NY Acad Sci. 2002;978:359–390. doi: 10.1111/j.1749-6632.2002.tb07581.x. [DOI] [PubMed] [Google Scholar]
- Simpson JI, Wylie DR, DeZeeuw CI. On climbing fiber signals and their consequence(s) Behav Brain Sci. 1996;19:384–398. [Google Scholar]
- Southan AP, Robertson B. Electrophysiological characterization of voltage-gated K+ currents in cerebellar basket and Purkinje cells: Kv1 and Kv3 channel subfamilies are present in basket cell nerve terminals. J Neurosci. 2000;20:114–122. doi: 10.1523/JNEUROSCI.20-01-00114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart G, Hausser M. Initiation and spread of sodium action potentials in cerebellar Purkinje cells. Neuron. 1994;13:703–712. doi: 10.1016/0896-6273(94)90037-x. [DOI] [PubMed] [Google Scholar]
- Swensen AM, Bean BP. Ionic mechanisms of burst firing in dissociated Purkinje neurons. J Neurosci. 2003;23:9650–9663. doi: 10.1523/JNEUROSCI.23-29-09650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thach WT., Jr Somatosensory receptive fields of single units in cat cerebellar cortex. J Neurophysiol. 1967;30:675–696. doi: 10.1152/jn.1967.30.4.675. [DOI] [PubMed] [Google Scholar]
- Vega-Saenz de Miera E, Moreno H, Fruhling D, Kentros C, Rudy B. Cloning of ShIII (Shaw-like) cDNAs encoding a novel high-voltage-activating, TEA-sensitive, type-A K+ channel. Proc Biol Sci. 1992;248:9–18. doi: 10.1098/rspb.1992.0036. [DOI] [PubMed] [Google Scholar]
- Wang LY, Gan L, Forsythe ID, Kaczmarek LK. Contribution of the Kv3.1 potassium channel to high-frequency firing in mouse auditory neurones. J Physiol (Lond) 1998;509:183–194. doi: 10.1111/j.1469-7793.1998.183bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters MF, Minassian NA, Stevanin G, Figueroa KP, Bannister JP, Nolte D, Mock AF, Evidente VG, Fee DB, Muller U, Durr A, Brice A, Papazian DM, Pulst SM. Mutations in voltage-gated potassium channel KCNC3 cause degenerative and developmental central nervous system phenotypes. Nat Genet. 2006;38:447–451. doi: 10.1038/ng1758. [DOI] [PubMed] [Google Scholar]
- Weber JT, De Zeeuw CI, Linden DJ, Hansel C. Long-term depression of climbing fiber-evoked calcium transients in Purkinje cell dendrites. Proc Natl Acad Sci USA. 2003;100:2878–2883. doi: 10.1073/pnas.0536420100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser M, Vega-Saenz de Miera E, Kentros C, Moreno H, Franzen L, Hillman D, Baker H, Rudy B. Differential expression of Shaw-related K+ channels in the rat central nervous system. J Neurosci. 1994;14:949–972. doi: 10.1523/JNEUROSCI.14-03-00949.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh JP, Lang EJ, Suglhara I, Llinas R. Dynamic organization of motor control within the olivocerebellar system. Nature. 1995;374:453–457. doi: 10.1038/374453a0. [DOI] [PubMed] [Google Scholar]
- Womack MD, Khodakhah K. Characterization of large conductance Ca2+-activated K+ channels in cerebellar Purkinje neurons. Eur J Neurosci. 2002;16:1214–1222. doi: 10.1046/j.1460-9568.2002.02171.x. [DOI] [PubMed] [Google Scholar]