

Abstract
Chronic use of levodopa, the most effective treatment for Parkinson’s disease, causes abnormal involuntary movements named dyskinesias, which are linked to maladaptive changes in plasticity and disturbances of dopamine and glutamate neurotransmission in the basal ganglia. Dyskinesias can be modeled in rats with unilateral 6-hydroxydopamine lesions by repeated administration of low doses of levodopa (6 mg/kg, s.c.). Previous studies from our lab showed that sub-chronic treatment with the cannabinoid agonist WIN55,212-2 attenuates levodopa-induced dyskinesias at doses that do not interfere with physiological motor function. To investigate the neurochemical changes underlying WIN55,212-2 anti-dyskinetic effects, we used in vivo microdialysis to monitor extracellular dopamine and glutamate in the dorsal striatum of the both hemisphere of freely-moving 6-hydroxydopamine-treated, SHAM-operated and intact rats receiving levodopa acutely or chronically (11 days), and studied how sub-chronic WIN55,212-2 (1 injection × 3 days, 20 min before levodopa) affected these neurochemical outputs.
Our data indicate that: 1) the 6-hydroxydopamine lesion decreases dopamine turnover in the denervated striatum; 2) levodopa injection reduces extracellular glutamate in the side ipsilateral to the lesion of dyskinetic rats; 3) sub-chronic WIN55,212-2 prevents levodopa-induced glutamate volume transmission unbalances across the two hemispheres; 4) levodopa-induced dyskinesias are inversely correlated with glutamate levels in the denervated striatum. These data indicate that the anti-dyskinetic properties of WIN55,212-2 are accompanied by changes of dopamine and glutamate outputs in the two brain hemispheres of 6-hydroxydopamine-treated rats.
Parkinson’s disease (PD) is a progressive neurodegenerative disorder characterized by the loss of nigrostriatal neurons. Although the dopamine (DA) precursor levodopa represents the golden standard for PD therapy, its long-term use causes disabling abnormal involuntary movements (AIMs) known as dyskinesias. In rodents, levodopa-induced dyskinesias can be mimicked by chronic administration of low doses of levodopa following nigrostriatal denervation by unilateral injection of the neurotoxin 6-hydroxydopamine (6-OHDA). This model has been pharmacologically validated (Lundblad et al. 2002) and displays behavioral phenotypes and cellular responses similar to those observed in dyskinetic non-human primates and PD patients (Di Monte et al. 2000; Lundblad et al. 2004).
Although the pathophysiology of dyskinesias is not completely understood, experimental evidence indicates that non-physiological stimulation of DA receptors, as well as alterations in the plasticity and neurochemistry of the basal ganglia (Carta et al. 2006; Cenci and Lindgren 2007), may contribute to the expression of these motor disturbances.
Numerous studies point to the endocannabinoid system as an important neuromodulator of basal ganglia function (Giuffrida et al. 1999; Beltramo et al. 2000; Gerdeman et al. 2002; Sidlo et al. 2008; Kofalvi et al. 2005), and stimulation of CB1 cannabinoid receptors has been shown to alleviate levodopa-induced dyskinesias in animal models of PD and PD patients (Sieradzan et al. 2001; Ferrer et al. 2003; Morgese et al. 2007). CB1 receptors are highly expressed in brain areas regulating motor behaviors, including the basal ganglia (Tsou et al. 1998; Mackie 2005), and co-expressed with D1 and D2 dopamine receptors in the striatum. This co-localization has functional implications as (1) CB1 and D2 receptors share common pools of G-proteins (Glass and Felder 1997) and may form heterodimers and change their original signal transduction pathways (Kearn et al. 2005); (2) CB1 receptor stimulation inhibits D1-mediated cellular (Meschler and Howlett 2001) and behavioral (Martin et al. 2008) responses. CB1 receptors also exert a modulatory control on striatal glutamate (GLU) release, GABAergic activity and synaptic plasticity (Gubellini et al. 2002; Kofalvi et al. 2005; Kreitzer and Malenka 2007).
Previous studies showed that sub-chronic administration of the cannabinoid agonist WIN55,212-2 (WIN) reduces levodopa-induced AIMs in 6-OHDA-treated rats in a CB1 receptor-dependent fashion (Ferrer et al. 2003; Morgese et al. 2007), and causes changes of GLU and DA transmission in intact animals. Specifically, WIN inhibits GLU (Domenici et al. 2006; Takahashi and Castillo 2006) and DA (Cadogan et al. 1997; Sidlo et al. 2008) release in the striatum ex vivo, whereas it induces GLU release in rat cerebral cortex (Ferraro et al. 2001) and increases DA output in the dorsal striatum in vivo (Price et al. 2007). To date, there is no information on the effects of systemic administration of WIN on striatal GLU and DA release in animal models of dykinesia. In this study we used in vivo microdialysis to monitor striatal DA and GLU extracellular levels in both hemispheres of dyskinetic rats, before and after sub-chronic treatment with WIN. We also correlated changes in dopaminergic and glutamatergic transmission with the severity of levodopa-induced AIMs to identify possible neurochemical correlates accompanying the anti-dyskinetic activity of WIN.
1. Experimental procedures
1.1. Materials
Desipramine hydrochloride, levodopa methyl ester, 6-OHDA hydrochloride, benserazide and amphetamine were purchased from Sigma Chemicals Co. (St. Louis, MO); WIN55,212-2 mesylate was from Tocris Bioscience (Ellisville, MI). All HPLC chemicals were from Sigma-Aldrich (Milan, Italy).
1.2. Animals and 6-OHDA Lesion
Animal care and all experiments were conducted in accordance with the guidelines of the National Institutes of Health (“Guide for the Care and Use of Laboratory Animals”), the European Communities Council Directive of 24 November 1986 (86/609/EEC) and the Italian Department of Health (D.L. 116/92), and approved by the Institutional Animal Care and Use Committees of the University of Texas Health Science Center at San Antonio and the University of Foggia, Italy. All efforts were made to minimize animal suffering and to reduce the number of animals used in the study.
Male Wistar rats (225–250 g; Charles River Laboratories, Wilmington, MA; Harlan, San Pietro al Natisone, Udine, Italy) were housed on 12-h dark-light cycle, at 22±1° C with food and water available ad libitum and habituated to housing conditions for 1 week before the experiments. DA-denervating lesions were performed by unilateral injection of 6-OHDA into the left medial forebrain bundle (MFB) as previously reported (Morgese et al. 2007). Briefly, on the day of the surgery (see Fig. 1), rats received an intraperitoneal (i.p.) injection of the noradrenaline transporter inhibitor, desipramine (25 mg/kg, 30 min before surgery) and were anesthetized with equithesin (3 ml/kg, i.p.) [Na-pentobarbital (0.972 g), chloral hydrate (4.251 g), magnesium sulfate (2.125 g), ethanol (12.5 ml), and propylene glycol (42.6 ml) in distilled water (total volume, 100 ml)] and positioned on a stereotaxic frame (Kopf Instruments, Tujunga, CA). 6-OHDA (4 μg/μl) was dissolved in 0.1% ascorbate saline. 2 μl of 6-OHDA solution, or a corresponding volume of saline (SHAM-operated rats), were injected into the left MFB [anteroposterior (AP) −4.3, mediolateral (ML) +1.6, dorsoventral (DV) −8.3, tooth bar −2.4 (relative to bregma and midline, in mm) [according to (Paxinos and Watson 1998)] at a flow rate of 0.5 μl/min using a 10 μl Hamilton microsyringe with a 32-gauge needle. A third group of intact rats was housed and pharmacologically treated as the 6-OHDA- and SHAM-operated rats. Two weeks after surgery (day -7, see Fig. 1), 6-OHDA-treated rats received an acute injection of amphetamine (2.5 mg/kg, i.p.) and were immediately screened for amphetamine-induced rotational behavior to assess the efficacy of the lesion. Net ipsi-lateral turns were calculated by subtracting the number of contra-lateral from ipsi-lateral rotations. Only rats displaying more than 5 full ipsi-lateral rotations per min (Winkler et al. 2002) were included in the study.
Fig. 1.

Experimental design of the study. ACT 6-OHDA, SHAM-operated and intact animals received saline chronically and levodopa or vehicle acutely during the microdialysis experiment (day 11). CHR 6-OHDA, SHAM and intact animals were treated chronically with levodopa (11 days, light gray shade) and WIN+levodopa (3 days, dark grey shade). On day 11, rats received the last injection of vehicle/WIN and/or levodopa during the microdialysis experiment. Numbers represents days after the 6-OHDA/SHAM lesion.
1.3. Microdialysis and Treatment Schedule
After anaesthesia (as reported above), animals were placed on a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA). Two custom-made microdialysis probes of concentric design (AN69 Hospal S.p.A; 20 kDa cut-off; membrane length, 3-mm) were implanted into the right and the left striatum according to the following stereotaxic coordinates: AP +1.0, ML ±2.8 from bregma and DV −6.8 from dura (Paxinos and Watson, 1998). The microdialysis probes were fixed to the skull with stainless steel screws and methylacrylic cement. Twenty-four hours after surgery (day 11, see Fig. 1), the microdialysis probes were perfused with Krebs Ringer solution (NaCl 145 mmol/L, KCl 2.7 mmol/L, CaCl2 2H2O 1.2 mmol/L, MgCl2 6H2O 1 mmol/L, Na2HPO4 2 mmol/L, pH 7.4) at a constant flow rate of 2 μl/min. Perfusates were collected every 20 min into mini-vials containing 3 μl of 10% acetic acid. After a wash-out period of 2 hours, 4 samples were collected to determine the baseline levels of the neurotransmitters studied (no more than 10% difference among 4 consecutive samples). Rats were then treated according to their assigned experimental protocol (Fig. 1) and consecutive microdialysate samples were collected every 20 min. DA and GLU concentrations, obtained from the same samples, were detected and quantified by HPLC.
Intact, 6-OHDA-treated and SHAM-operated rats were assigned to 2 experimental groups (Fig. 1). The first group (ACT) received a daily injection of saline (1 ml/kg) from day 1 to 10 (Fig. 1). 24 hours after the insertion of the probes into the left and right striatum (day 10), and once a stable neurotransmitter baseline was obtained rats received a single injection of levodopa+ the inhibitor of aromatic aminoacid decarboxilase, benserazide (6 mg/kg and 12.5 mg/kg s.c, respectively.) or vehicle (saline), and dialysates were collected over a 2-hour period.
The second group (CHR) received a daily injection of levodopa+benserazide (6 mg/kg and 12.5 mg/kg, respectively s.c.) from day 1 to day 11 (Fig. 1). Chronic administration of this dose of levodopa induces a gradual development of dyskinesia (AIMs) in 6-OHDA-treated rats (Lundblad et al. 2002). On day 8 of levodopa, AIMs were scored using a severity scale as previously described (Morgese et al. 2007). For the following three consecutive days (days 9 to 11), all rats received an injection of WIN (1 mg/kg, i.p.) or vehicle (5/5/90% of Tween80/PEG/saline) 20 minutes before levodopa. On day 11 (Fig. 1), after receiving the last injection of WIN or vehicle followed by levodopa+benserazide (20 min later), microdialysates were collected over a 2 hour period, while dyskinetic behaviors were monitored simultaneously. At the end of each experiment, the correct placement of dialysis probes was verified histologically.
1.4. HPLC Analyses
The levels of DA and its metabolite, homovanillic acid (HVA), were determined by microbore HPLC using a SphereClone 150-mm × 2-mm column (3-μm packing) with a Unijet cell (BAS, Bioanalytical Systems, Kenilworth Warwickshire, United Kingdom) equipped with a 6-mm-diameter glassy carbon electrode (set at + 650 mV) and connected to an electrochemical amperometric detector (INTRO, Antec Leyden, The Netherlands). The flow rate of the mobile phase [85 mM sodium acetate, 0.34 mM EDTA, 15 mM sodium chloride, 0.81 mM octanesulphonic acid sodium salt, 5% methanol (v/v), pH 4.85] was 220 μl/min and the total runtime 15 min.
GLU and Aspartate (ASP) levels were quantified using a HPLC/fluorimetric detection system, including pre-column derivatization with o-phtaldialdehyde reagent and a Chromsep 5 (C18) column as previously described (Ferraro et al. 2001). The flow rate of the mobile phase (0.1 M sodium acetate, 10% methanol and 2.5% tetrahydrofurane, pH 6.5) was 1 ml/min and the total runtime 6 min.
1.5. AIMs recordings
Axial, limb, locomotive and oro-facial AIMs were monitored between 10:00 a.m. and 4:00 p.m. as described (Lundblad et al. 2002). Briefly, animals were placed individually in a Plexiglas box and observations were carried out by a trained researcher blind to the treatment schedule. AIMs were scored on a severity scale ranging from 0 to 4 (Lundblad et al. 2002; Morgese et al. 2007). Only animals displaying AIMs were included in the study.
In the case of the animals undergoing microdialysis, AIMs were scored for 1 min every 20 min after levodopa injection throughout the microdialysis experiment (2 hours).
1.6. Statistical Analysis
Microdialysis data were analyzed by one- or two-way analysis of variance (ANOVA) for repeated measures with “treatment” (6-OHDA injection, or SHAM operation, or no-surgical procedure) or “time” as the between variables and “hemisphere” as the within variable (basal neurotransmitter comparisons or post-challenge comparisons). Dunnett’s and Tukey’s post hoc comparisons were used where appropriate. Violations of the sphericity assumption were corrected using the Greenhouse-Geisser epsilon correction to adjust the degrees of freedom for each test. Basal extracellular values of each neurotransmitter were defined as the average of 4 consecutive samples before drug administration. Comparisons of basal neurotransmitter levels between levodopa-treated (CHR) and saline-treated (ACT) groups were analyzed by two-way ANOVA, followed by the Tukey’s multiple comparison test. The overall effect of drug treatments on neurotransmitter outputs in each brain hemisphere was estimated by two-way ANOVA and by comparing the areas under the curve (AUC) by the paired Student’s t-test. The AUC was calculated using the standard trapezoid method (Gibaldi and Perrier 1975) using neurotransmitter levels over a time window of 20–120 min for the ACT group, and 40–140 min for the CHR group.
DA turnover was expressed as HVA/DA ratio. Comparisons of DA turnover between the ipsi-and contra-lateral hemispheres in each experimental group were analyzed using the Student paired t-test. Neurochemical data were expressed as mean ± SEM. Dyskinesias were expressed as median of total AIMs score and analyzed using the Kruskal-Wallis test followed by the Dunn’s multiple comparison test. Pearson’s linear correlation analyses were performed between GLU levels in the ipsi-lateral striatum of 6-OHDA-treated rats (CHR) and the severity of AIMs. The threshold for statistical significance was set at p<0.05.
2. Results
2.1. Effects of 6-OHDA lesion on striatal DA and GLU outputs
Table 1 shows the basal levels of DA, HVA and GLU in the dorsal striatum of each hemisphere. ASP levels were omitted for clarity. These values reflect the mean neurotransmitter concentrations of 4 consecutive microdialysates collected on day 11, 2 hours before the acute injection of levodopa or its vehicle (saline, ACT).
TABLE 1.
Basal extracellular neurotransmitter levels in rats treated according to ACT and CHR schedules. In ACT, basal levels were monitored 2 hours before acute levodopa (or the last saline) injection. In CHR, basal levels reflect extracellular neurotransmitter concentrations observed 2 hours before the last injection of chronic levodopa or WIN+levodopa.
| INTACT | SHAM - OPERATED | 6-OHDA-TREATED | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ACT | CHR |
ACT | CHR |
ACT | CHR |
|||||
| Levodopa |
Levodopa |
Levodopa |
||||||||
| veh |
WIN |
veh |
WIN |
veh |
WIN |
|||||
| DA (pg/ml) | Ipsi | 532±79 | 444±22 | 510±109 | 525±45 | 406±91 | 407±80 | 127±13a | 60±6a,b | 92±39a |
| Contra | 545±59 | 469±48 | 571±170 | 537±68 | 408±76 | 437±63 | 513±65 | 288±98b | 283±84 | |
|
| ||||||||||
| HVA (ng/ml) | Ipsi | 167±18 | 173±36 | 165±9 | 137±29 | 152±18 | 173±38 | 5.3±2.1a | 2±0.4a | 10±7a |
| Contra | 151±25 | 148±29 | 155±36 | 128±17 | 161±22 | 211±56 | 137±16 | 223±44b | 151±27 | |
|
| ||||||||||
| GLU (μM) | Ipsi | 0.3±0.1 | 0.3±0.1 | 0.3±0.1 | 0.3±0.1 | 0.4±0.1 | 0.7±0.2 | 0.38±0.1 | 0.57±0.2 | 0.57±0.2 |
| Contra | 0.3±0.1 | 0.3±0.1 | 0.3±0.1 | 0.3±0.1 | 0.3±0.04 | 0.4±0.2 | 0.37±0.1 | 0.79±0.1b | 0.62±0.2 | |
Two-way ANOVA followed by Tukey’s multiple comparison test:
p<0.05 vs contro-lateral hemisphere within the same experimental group;
p<0.05 vs corresponding hemisphere of the same group.
As expected, the 6-OHDA lesion produced a significant DA depletion in the striatum ipsi-lateral to the lesion compared to the contra-lateral side (p<0.05, Tukey’s test), whereas no differences were found across the two hemispheres of intact and SHAM-operated rats (Table 1). Basal HVA levels followed a similar pattern (Table 1).
The 6-OHDA lesion did not affect the basal output of GLU and ASP (Table 1 and data not shown).
2.2. Effects of acute (ACT) and chronic (CHR) Levodopa on DA and GLU outputs
To investigate the effect of acute levodopa administration on DA and GLU outputs, 6-OHDA-treated, SHAM-operated and intact rats received a single injection of levodopa+benserazide or vehicle (saline) (ACT rats) on day 11, immediately after the collection of 4 basal microdialysates (baseline). Sample collection was continued for the following 120 min. Two-way ANOVA showed that DA levels were significantly lower in the denervated striatum, independently from acute levodopa (Fig. 2a; F(time)5,70=0.164, n.s., F(time × hemi)5,70=0.092, n.s., F(hemi)1,14=10.267, p<0.01) or vehicle (Fig. 2b; F(time)5,55=1.507, n.s., F(time × hemi)5,55=0.860, n.s., F(hemi)1,11=25.544, p<0.001) administration. Moreover, acute levodopa did not modify DA efflux in SHAM-operated or intact rats (data not shown).
Fig. 2.

Effects of levodopa (a) or its vehicle (saline) (b) on DA extracellular concentrations in the ipsi-lateral (open squares) and contra-lateral (filled triangles) striatum of 6-OHDA-treated rats treated according to ACT schedule (see Table 1 legend). Inserts represent AUCs values (ipsi-lateral striatum, open bars; contra-lateral striatum, filled bars). The bottom panels show the effects of acute levodopa (c) or saline (d) on DA turnover (HVA/DA ratios) in 6-OHDA-treated rats. Data are expressed as mean±SEM of n=8 experiments. Panels a and b, two-way ANOVA repeated measures **p<0.01 and ***p<0.001 ipsi vs contra. Inserts and panel c and d, Student’s paired t test *p<0.05, **p<0.01 and ***p<0.001 ipsi vs contra. Arrows indicate the time of levodopa or saline administration.
As the difference in DA levels across the two hemispheres of 6-OHDA-treated rats was time-independent, we estimated and compared the AUCs from the ipsi- and contra-lateral striata by paired Student’s t-test (Fig. 2a, b inserts) and confirmed that the difference in DA output between the two hemispheres (p<0.01 and p<0.001, respectively) was unaffected by levodopa or vehicle.
DA turnover was decreased in the ipsi-lateral striatum of 6-OHDA-treated rats (p<0.05, Student’s paired t test) following acute levodopa or vehicle (Fig 2c, d), whereas no changes were observed in SHAM-operated or intact animals (data not shown). Similarly, ASP or GLU efflux were unchanged in all experimental groups (data not shown).
6-OHDA-treated rats receiving chronic levodopa (CHR rats) showed a reduction of DA basal levels (day 11, before the last injection of levodopa) in both hemispheres and a decrease of basal HVA in the hemisphere ipsi-lateral to the lesion, (p<0.05, compared to 6-OHDA-treated animals receiving saline only) (Table 1). Chronic levodopa had no effect on basal DA output of intact and SHAM-operated rats (Table 1), while it significantly increased basal DA turnover (SHAM, CHR: 513±130, 541±130, vs ACT: 240±37, 267±50; intact: 468±59, 404±73 vs ACT: 254±32, 230±48, left and right, respectively) in both hemispheres, suggesting that this effect is independent of the 6-OHDA lesion. In CHR 6-OHDA-treated rats, the last injection of levodopa did not modify the difference in DA output observed between the two brain hemispheres (40–140 min sampling period), (Fig. 3a) (two-way ANOVA: F(hemi)1,12=9.152, p<0.05; F(time)5,60=0.726, n.s., F(time × hemi)5,60=1,059, n.s.). The analysis of the corresponding AUCs further confirmed this result (p<0.05, Student’s paired t-test, Fig. 3a, insert). Similarly, the last injection of levodopa did not affect DA output in CHR SHAM-operated and intact rats (data not shown).
Fig. 3.

Effects of chronic levodopa on DA (a) or GLU (c) release in the ipsi-lateral (open squares) and contra-lateral (filled triangles) striatum of 6-OHDA-treated rats treated according to CHR schedule (see Table 1 legend). Panel b shows the effects of chronic levodopa on DA turnover (HVA/DA ratios) in the ipsi-lateral (open bar) and contra-lateral (filled bar) striatum in the same rats. Data are expressed as mean±SEM (n=9). Panels a and c, two-way ANOVA repeated measures *p<0.05 ipsi vs contra. Inserts and panel b, Student’s paired t test, *p<0.05 ipsi vs contra. Panel c, one-way ANOVA repeated measures followed by Dunnett’s multiple comparison test, #p<0.05 and ##p<0.01 post challenge values vs baseline. Arrows indicate the time of vehicle (PEG/tween 80/saline 5/5/90%) and levodopa administrations.
Chronic levodopa further increased the disparity in DA turnover across the two hemispheres of 6-OHDA-treated rats (Fig. 3b) (p<0.05, Student’s paired t test) (see Fig. 2c for comparison with ACT rats), and elevated DA turnover by ~80% in CHR SHAM-operated rats (data not shown).
Concerning the effects of chronic levodopa on basal aminoacid neurotransmission (day 11, before last levodopa injection), two-way ANOVA revealed a significant effect of treatment on GLU output in CHR 6-OHDA-treated rats (Table 1) (F(treatment)1,48=7.136, p<0.05, F(hemi)1,48=0.926, n.s., F(treatment × hemi)1,48=1.058, n.s.). Although the GLU increase occurred in both hemispheres, statistical significance was reached only in the intact striatum (Table 1) (p<0.05, Tukey’s test). Chronic levodopa had no effect on basal ASP output in all CHR experimental groups (data not shown).
In CHR 6-OHDA-treated rats, the last injection of levodopa produced a disparity in GLU efflux between the two hemispheres (Fig. 3c; two-way ANOVA: F(time)5,95=0.761, n.s., F(time × hemi)5,95=0.162, n.s., F(hemi)1,19=6.984, p<0.05), as revealed by the smaller AUC of the ipsi-lateral vs the contra-lateral striatum (p<0.01 Student’s paired t test, Fig. 3c, insert). Comparison of the post-challenge vs baseline GLU values revealed that such difference (p<0.05, Dunnett’s test) resulted from GLU decrease in the denervated side beginning at 40 min after levodopa administration (Fig. 3c). No changes were observed in CHR SHAM-operated and intact rats (data not shown).
2.3. Sub-chronic WIN reduces neurotransmitter unbalances in dyskinetic rats
As previously reported (Morgese et al. 2007), rats with unilateral 6-OHDA lesion chronically treated with levodopa develop severe AIMs, whereas no dyskinetic behaviors are observed in either SHAM-operated or intact animals. Fig. 4 shows the time course of levodopa-induced AIMs (total score) in CHR 6-OHDA-treated rats measured during microdialysis (day 11). In agreement with our previous study, sub-chronic administration of WIN had a significant anti-dyskinetic effect (Fig. 4) at a dose that is most effective and does not produce motor suppression (1 mg/kg, i.p., 1 injection per day, for 3 days) (Morgese et al. 2007).
Fig. 4.

Time course of levodopa-induced abnormal involuntary movements (AIMs) in 6-OHDA-treated rats receiving chronic levodopa (CHR). Dyskinesias were scored during the microdialysis experiments. Rats received WIN (open diamonds) or vehicle (filled squares) 20 min before levodopa. Arrows indicate the time of vehicle/WIN and levodopa administrations. Data are expressed as median score (interquartile ranges have been omitted for clarity) of n=9 experiments. Kruskal-Wallis followed by Dunn’s multiple comparison test, *p<0.05 and ***p<0.001, vs dyskinetic rats treated with vehicle.
To investigate the neurochemical changes underlying the behavioral effects of WIN, we measured DA and aminoacid outputs in the same group of 6-OHDA-treated animals, as well as in CHR SHAM-operated and intact rats. All experimental groups received an injection of WIN (1 mg/kg, i.p.) or vehicle (5/5/90% of Tween80/PEG/saline) on days 9–11, 20 minutes before levodopa. On day 11, WIN was injected after the collection of 4 microdialysates (baseline), and the measurements of neurochemical outputs were carried out for a total of 2 hours.
Table 1 shows the basal DA, GLU and ASP levels collected on day 11. The last injection of WIN had no effect per se on DA output in both striata of CHR 6-OHDA-treated rats (Fig. 5a), but completely abolished the difference in DA levels between the two sides (Fig. 5a). No effect in either CHR SHAM-operated or intact rats were observed (data not shown). WIN also reduced the disparity in DA turnover between left and right hemispheres induced by the last injection of levodopa in CHR 6-OHDA-treated rats (Fig. 5b), whereas it did not change DA turnover in the two hemispheres of either CHR SHAM-operated or intact rats (data not shown).
Fig. 5.
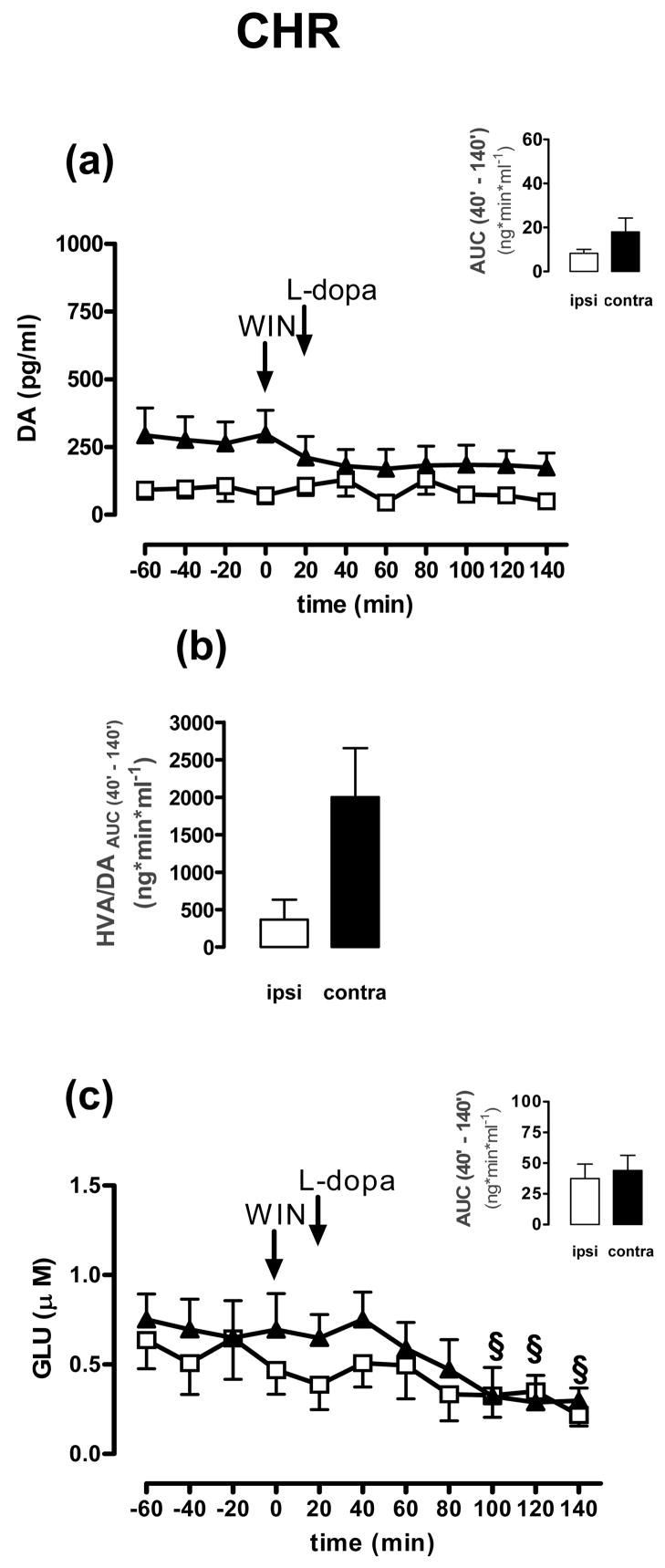
Effects of WIN+levodopa on DA (a) or GLU (c) release in the ipsi-lateral (open squares) and contra-lateral (filled triangles) striatum of 6-OHDA-treated rats treated according to CHR schedule (see Table 1 legend). Panel b shows the effects of vehicle+levodopa on DA turnover (HVA/DA ratios) in the ipsi-lateral (open bar) and contra-lateral (filled bar) striatum of the same rats. Data are expressed as mean±SEM (n=9). Panel c, one-way ANOVA repeated measures followed by Dunnett’s multiple comparison test, §p<0.05 post challenge values vs baseline. Arrows indicate the time of WIN and levodopa administrations.
Finally, WIN prevented levodopa-induced GLU reduction in the denervated side of CHR 6-OHDA-treated rats (Fig. 5c and insert; see Fig. 3c for comparison) (F(time)5,100=5.048, p<0.01, F(time × hemi)5,100=0.361, n.s., F(hemi)1,20=0.479, n.s., two-way ANOVA). We also found that the GLU levels in this side were inversely correlated with levodopa-induced AIMs (Fig. 6) (p<0.05, rs=0.0586, Pearson’s correlation), whereas no significant correlation was observed contralaterally (data not shown). In this side, sub-chronic WIN significantly reduced GLU levels beginning at 80 min after the last injection of levodopa (Fig 5c) (p<0.05 vs baseline, Dunnett’s test). No differences in GLU and ASP outputs were found in CHR SHAM-operated and intact animals following WIN+levodopa treatment (data not shown).
Fig. 6.

Spearman’s correlation between AIMs total score and GLU extracellular levels observed in the ipsi-lateral striatum of 6-OHDA-treated rats (CHR) receiving vehicle+levodopa. p<0.05, rs=0.05860.
3. Discussion
This study shows that systemic administration of levodopa and/or levodopa+WIN change DA and GLU outputs in the striatum of 6-OHDA-treated rats. In particular, we found that chronic levodopa significantly decreases GLU levels in the denervated striatum of dyskinetic rats and that this effect is correlated to the severity of levodopa-induced dyskinesias. We also showed that sub-chronic WIN attenuates (1) the differences in DA-turnover across the two hemispheres and (2) levodopa-induced GLU decrease in the striatum ipsilateral to the lesion.
As expected, basal DA levels were significantly decreased in the denervated striatum of 6-OHDA-treated rats. In these animals we found no increase in DA efflux immediately after acute levodopa injection as well as after chronic levodopa treatment, which is accompanied by overt dyskinetic behavior. Although some reports have shown increased extracellular DA after chronic levodopa in dyskinetic rats (Meissner et al. 2006; Buck and Ferger 2008), the discrepancy with our study may depend on the dose and/or route or schedule of levodopa administration, which were different from those used in our experiments. For example, Meissner et al. used a dose that was 4-fold higher than ours (25 vs 6 mg/kg) and delivered intraperitoneally. All our experiments were carried out using 6 mg/kg (s.c.) of levodopa, since this dose is clinically relevant and produces dyskinesias which gradually develop over time (Cenci and Lundblad 2005; Cenci and Lundblad 2007; Lindgren et al. 2007). In addition, in the study of Buck and Ferger, levodopa was administered per os and for a longer period of time (21 days), whereas we used subcutaneous injections for 11 days. As different routes of administration are known to affect levodopa absorption and produce fluctuations in the severity of dyskinesia and wearing-off phenomena (Lindgren et al. 2007), it is likely that variations in levodopa pharmacokinetics may account for the differences in striatal DA output reported in these studies. Nevertheless, our results are in line with a recent study showing that high doses of levodopa (33 and 66 mg/kg, i.p.) do not increase DA levels in 6-OHDA-treated rats (Rodriguez et al. 2007). Interestingly, we found that the 6-OHDA lesion decreased basal DA turnover in the denervated striatum, suggesting an ongoing compensatory mechanism for the lower DA production. A similar result is obtained when calculating the HVA/DA ratio using the DA and HVA basal levels reported by Buck and Ferger. By contrast, chronic levodopa enhanced DA metabolism in all experimental groups, without affecting the difference in DA turnover across the two hemispheres of 6-OHDA-treated rats. These data indicate that the conversion of levodopa into DA is accompanied by a higher DA metabolism, which may explain the gradual loss of levodopa efficacy over time (Marsden and Parkes 1976; Carey 1991). Furthermore, the increased DA metabolism, as well as the reduction in DA transporter (DAT) expression in both hemispheres of dyskinetic rats (unpublished observations), is in line with the observation that DAT function and DA turnover are inversely correlated in PD patients (Sossi et al. 2007).
DA denervation did not change GLU basal levels in 6-OHDA-treated rats, whereas chronic levodopa increased basal GLU output in both hemispheres. This elevation, however, reached statistical significance only in the intact striatum. No changes in GLU output were observed in SHAM-operated or intact rats, suggesting that the GLU increase results from the combination of DA denervation and chronic levodopa administration, two conditions that promote the development of dyskinesias (Lundblad 2004; Carta et al. 2006). The GLU elevation may also underlie and/or participate to the dyskinesia-priming process attributed to chronic levodopa (Cenci et al. 1998; Blanchet et al. 2004). Interestingly, the last levodopa injection in chronically-treated animals produced a significant reduction in GLU output in the denervated side. This finding suggests that levodopa, at the dose used in our experimental setting, has an immediate depressing effect over GLU efflux and is followed by a long-lasting rebound, as indicated by GLU elevation upon chronic levodopa administration (Table 1). Our data differ from those reported by Robelet and colleagues (Robelet et al. 2004), which showed a significant increase in GLU levels in 6-OHDA-treated rats after the last injection of chronic levodopa. In this study, however, the dose of levodopa was ~ 17 fold higher than ours, and the length of treatment almost double (21 vs 11 days). The decrease of GLU observed by us in the ipsi-lateral striatum may result from the stimulation of dopamine D2 receptors, which are highly expressed in rat striatum (Graybiel et al. 1994; Surmeier et al. 2007), become supersensitive after DA denervation (Pierot et al. 1988; Lisovoski et al. 1992; Cai et al. 2000; Ishida et al. 2004) and negatively control GLU transmission. Indeed, (1) ex vivo studies show that stimulation of D2 receptors reduces GLU release presynaptically (Bamford et al. 2004); (2) dopaminergic and glutamatergic systems work antagonistically in the striatum to control cognitive and motor functions (Carlsson and Carlsson 1990; Riederer et al. 1992; Amalric et al. 1994). This hypothesis, however, remains controversial (Morari et al. 1998). Although in our study levodopa administration did not increase DA output, we cannot rule out that levodopa-induced increase of DA turnover might have masked DA elevation, thus making the tonic stimulation of D2 receptors still possible. Alternatively, levodopa may directly bind to D2 receptors as postulated by Silva and colleagues in PD patients (Silva et al. 2006), and confirmed by PET studies in 6-OHDA-treated rats (Ishida et al. 2004).
Our study shows that the unilateral 6-OHDA lesion causes neurochemical changes also in the intact hemisphere. Indeed, chronic levodopa elevated basal GLU output in the intact side, thus indicating that the use of the contra-lateral hemisphere as control in experimental protocols implicating unilateral lesions is generally biased. In keeping with our observations, molecular alterations have been reported in the intact hemisphere of 6-OHDA-treated rats, such as changes in striatal NMDA receptor and [3H]-Mazindol binding (O’Dell and Marshall 1996) and hypertrophy of dendritic cells in the neocortex (Miklyaeva et al. 2007). Similarly, other models utilizing unilateral lesions, such as intra-hippocampal kainic acid injections to mimic chronic seizures, produce neurochemical alterations contra-laterally (Arabadzisz et al. 2005).
In previous reports, we showed that sub-chronic administration of WIN at a dose unable to cause general motor suppression, significantly reduced levodopa-induced AIMs via a CB1-mediated mechanism (Morgese et al. 2007). To assess whether WIN also induced neurochemical changes across the brain hemispheres of 6-OHDA-treated rats, we measured DA, HVA and aminoacid outputs in dyskinetic rats receiving sub-chronic WIN from day 9 to day 11 of levodopa. Our data indicate that WIN completely reversed the disparity in DA and GLU efflux across the two hemispheres of these animals. In particular, the analysis of the AUCs in each striatum showed that WIN increased DA output by 49% in the denervated striatum and decreased it by 21% in the contra-lateral side. On the other hand, sub-chronic WIN prevented levodopa-induced GLU decrease ipsi-laterally and significantly decreased GLU output contra-laterally.
The WIN-induced increase of DA output in the denervated striatum may result from a CB1-dependent inhibition of GABA release from striatonigral terminals, producing a disinhibition of the SNc (Wallmichrath and Szabo 2002). In support of this hypothesis, CB1 receptors are expressed on striatonigral terminals (Herkenham et al. 1991) and their activation has been shown to excite dopaminergic neurons in the SNc (French et al. 1997). This phenomenon may be particularly relevant in dyskinetic animals, as chronic levodopa increases CB1 receptor mRNA levels in the denervated hemisphere (Zeng et al. 1999), thus explaining the opposite effect on DA output in the intact side. In particular, the DA decrease observed in this hemisphere cannot be attributed to either: (1) inhibition of DAT activity by WIN (Price et al. 2007), which would prevent levodopa uptake, as this effect occurs in vivo only when WIN is administered at higher doses (4 mg/kg) (Price et al. 2007); or (2) changes in DA turnover, since WIN did not affect DA metabolism in the contra-lateral hemisphere. Therefore, as previously postulated, WIN-induced DA decrease may be an indirect consequence of a cannabinoid-mediated reduction of GLU release, possibly from glutamatergic terminals projecting to the SN from the subthalamicus nucleus, which in turn may decrease the firing of nigrostriatal neurons (Freiman and Szabo 2005). Alternatively, WIN may decrease striatal DA release via multiple non-synaptic mechanisms, including GABA release inhibition (Sidlo et al. 2008).
CB1 receptor stimulation has been associated with a decrease of GLU release in rat striatum (Gerdeman and Lovinger 2001; Kofalvi et al. 2005; Kreitzer and Malenka 2005; Adermark and Lovinger 2007). In agreement with these studies, we found that WIN significantly decreased GLU output in the intact striatum of 6-OHDA-treated rats receiving chronic levodopa. By contrast, WIN attenuated levodopa-induced GLU decrease in the denervated side. This unexpected result suggests that, following DA denervation and chronic levodopa administration, cannabinoid agonists may evoke different responses due to changes in CB1 receptor function. CB1 receptors are coupled to Gi/o proteins and they are co-expressed with D1 and D2 receptors in the striatum. This co-expression has functional consequences as CB1 and D2 receptors share common pools of G-proteins (Meschler and Howlett 2001; Brotchie 2003). In particular, in vitro studies have shown that concomitant application of cannabinoid and dopaminergic agonists promotes the formation of CB1 and D2 heterodimers (Kearn et al. 2005) and switches the CB1 receptor coupling from Gi (Howlett and Fleming 1984; Childers and Deadwyler 1996) to stimulatory Gs proteins (Glass and Felder 1997), which in turn may affect cannabinoid-mediated responses. This hypothesis fits with the pharmacological treatment received by dyskinetic rats; indeed, the concomitant stimulation of DA and CB1 receptors by levodopa and WIN may change the coupling of CB1 receptors and prevent in turn a further decrease of GLU efflux in the denervated striatum. This phenomenon, however, appears to be relevant only in this side, where D2 receptors are supersensitive (Pierot et al. 1988; Ishida et al. 2004), have altered coupling (Hossain and Weiner 1993) and CB1 receptors are over-expressed following chronic levodopa treatment (Zeng et al. 1999). Nevertheless, we cannot rule out that other receptor systems in the striatum, i.e. GABA receptors, might affect CB1 receptor signaling as already shown in the hippocampus (Cinar et al. 2008). Further studies aiming at identifying changes in CB1 receptor coupling under our experimental conditions are necessary to test this hypothesis.
Our findings show that the levodopa-dependent decrease of GLU efflux in the denervated striatum of dyskinetic rats is positively correlated with the severity of levodopa-induced AIMs. By contrast, no correlation between these two variables was observed in the intact side. By preventing levodopa-induced GLU reduction in the denervated side, WIN may positively affect striatal synaptic plasticity, which is dysfunctional in 6-OHDA-treated rats (Picconi et al. 2003; Cenci 2007). In particular, these animals show decreased concentrations of the endocannabinoid anandamide (Ferrer et al. 2003) and lack of endocannabinoid-mediated long-term depression (LTD) (Kreitzer and Malenka 2007). Since activation of metabotropic GLU receptors is required to trigger endocannabinoid-mediated LTD, the GLU decrease induced by chronic levodopa may explain its pro-dyskinetic effect. On the other end, WIN-induced reduction of GLU efflux in the intact side may contribute to its anti-dyskinetic properties. Indeed, in intact animals, the motor suppressive effects of cannabinoid agonists have been linked to stimulation of CB1 receptors located on glutamatergic cortico-striatal terminals (Monory et al. 2007), which in turn inhibits GLU release (Huang et al. 2001). Finally, we cannot exclude that WIN might modulate other neurotransmitter/neuromodulator systems regulating basal ganglia function, such as acetylcholine, serotonin, nitric oxide and GABA (Malone and Taylor 1999; Kofalvi et al. 2005; Bambico et al. 2007; Narushima et al. 2007; Sergeeva et al. 2007; Balazsa et al. 2008). In particular, physical manifestations of AIMs have been linked with enhanced GABA overflow in the output nuclei of the basal ganglia (Mela et al. 2007) and stimulation of CB1 receptors may affect the excitability of striatal medium spiny neurons by acting at GABAergic interneurons in the striatum (Narushima et al. 2007) where they are highly expressed (Uchigashima et al. 2007).
In conclusion, the ability of WIN to attenuate neurochemical disturbances in the denervated side of 6-OHDA-treated rats may contribute to the anti-dyskinetic properties of this drug.
Acknowledgments
This study was supported by a grant from the Italian Department of Research (FIRB) to V.C., by the National Institute of Health NS 050401-04 (to A.G.) and by the National Parkinson Foundation (to A.G.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adermark L, Lovinger DM. Retrograde endocannabinoid signaling at striatal synapses requires a regulated postsynaptic release step. Proc Natl Acad Sci U S A. 2007;104:20564–20569. doi: 10.1073/pnas.0706873104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amalric M, Ouagazzal A, Baunez C, Nieoullon A. Functional interactions between glutamate and dopamine in the rat striatum. Neurochemistry international. 1994;25:123–131. doi: 10.1016/0197-0186(94)90031-0. [DOI] [PubMed] [Google Scholar]
- Arabadzisz D, Antal K, Parpan F, Emri Z, Fritschy JM. Epileptogenesis and chronic seizures in a mouse model of temporal lobe epilepsy are associated with distinct EEG patterns and selective neurochemical alterations in the contralateral hippocampus. Exp Neurol. 2005;194:76–90. doi: 10.1016/j.expneurol.2005.01.029. [DOI] [PubMed] [Google Scholar]
- Balazsa T, Biro J, Gullai N, Ledent C, Sperlagh B. CB1-cannabinoid receptors are involved in the modulation of non-synaptic [3H]serotonin release from the rat hippocampus. Neurochemistry international. 2008;52:95–102. doi: 10.1016/j.neuint.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Bambico FR, Katz N, Debonnel G, Gobbi G. Cannabinoids elicit antidepressant-like behavior and activate serotonergic neurons through the medial prefrontal cortex. J Neurosci. 2007;27:11700–11711. doi: 10.1523/JNEUROSCI.1636-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford NS, Zhang H, Schmitz Y, Wu NP, Cepeda C, Levine MS, Schmauss C, Zakharenko SS, Zablow L, Sulzer D. Heterosynaptic dopamine neurotransmission selects sets of corticostriatal terminals. Neuron. 2004;42:653–663. doi: 10.1016/s0896-6273(04)00265-x. [DOI] [PubMed] [Google Scholar]
- Beltramo M, de Fonseca FR, Navarro M, Calignano A, Gorriti MA, Grammatikopoulos G, Sadile AG, Giuffrida A, Piomelli D. Reversal of dopamine D(2) receptor responses by an anandamide transport inhibitor. J Neurosci. 2000;20:3401–3407. doi: 10.1523/JNEUROSCI.20-09-03401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchet PJ, Calon F, Morissette M, Hadj Tahar A, Belanger N, Samadi P, Grondin R, Gregoire L, Meltzer L, Di Paolo T, Bedard PJ. Relevance of the MPTP primate model in the study of dyskinesia priming mechanisms. Parkinsonism Relat Disord. 2004;10:297–304. doi: 10.1016/j.parkreldis.2004.02.011. [DOI] [PubMed] [Google Scholar]
- Brotchie JM. CB1 cannabinoid receptor signalling in Parkinson’s disease. Current Opinion in Pharmacology. 2003;3:54–61. doi: 10.1016/s1471-4892(02)00011-5. [DOI] [PubMed] [Google Scholar]
- Buck K, Ferger B. Intrastriatal inhibition of aromatic amino acid decarboxylase prevents l-DOPA-induced dyskinesia: a bilateral reverse in vivo microdialysis study in 6-hydroxydopamine lesioned rats. Neurobiol Dis. 2008;29:210–220. doi: 10.1016/j.nbd.2007.08.010. [DOI] [PubMed] [Google Scholar]
- Cadogan AK, Alexander SP, Boyd EA, Kendall DA. Influence of cannabinoids on electrically evoked dopamine release and cyclic AMP generation in the rat striatum. J Neurochem. 1997;69:1131–1137. doi: 10.1046/j.1471-4159.1997.69031131.x. [DOI] [PubMed] [Google Scholar]
- Cai G, Zhen X, Uryu K, Friedman E. Activation of extracellular signal-regulated protein kinases is associated with a sensitized locomotor response to D(2) dopamine receptor stimulation in unilateral 6-hydroxydopamine-lesioned rats. J Neurosci. 2000;20:1849–1857. doi: 10.1523/JNEUROSCI.20-05-01849.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey RJ. Chronic L-dopa treatment in the unilateral 6-OHDA rat: evidence for behavioral sensitization and biochemical tolerance. Brain Res. 1991;568:205–214. doi: 10.1016/0006-8993(91)91399-l. [DOI] [PubMed] [Google Scholar]
- Carlsson M, Carlsson A. Interactions between glutamatergic and monoaminergic systems within the basal ganglia--implications for schizophrenia and Parkinson’s disease. Trends Neurosci. 1990;13:272–276. doi: 10.1016/0166-2236(90)90108-m. [DOI] [PubMed] [Google Scholar]
- Carta M, Lindgren HS, Lundblad M, Stancampiano R, Fadda F, Cenci MA. Role of striatal L-DOPA in the production of dyskinesia in 6-hydroxydopamine lesioned rats. J Neurochem. 2006;96:1718–1727. doi: 10.1111/j.1471-4159.2006.03696.x. [DOI] [PubMed] [Google Scholar]
- Cenci M, Lindgren H. Advances in understanding l-DOPA-induced dyskinesia. Curr Opin Neurobiol. 2007;17:665–671. doi: 10.1016/j.conb.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Cenci MA. Dopamine dysregulation of movement control in L-DOPA-induced dyskinesia. Trends Neurosci. 2007;30:236–243. doi: 10.1016/j.tins.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Cenci MA, Lundblad M. Utility of 6-Hydroxydopamine lesioned rats in the preclinical screening of novel treatment for Parkinson Disease. In: LeDoux M, editor. Animal Models of Movements Disorders. Elselvier Academic Press; Burlington: 2005. pp. 193–208. [Google Scholar]
- Cenci MA, Lundblad M. Ratings of L-DOPA-induced dyskinesia in the unilateral 6-OHDA lesion model of Parkinson’s disease in rats and mice. Curr Protoc Neurosci. 2007 doi: 10.1002/0471142301.ns0925s41. Chapter 9, Unit 9 25. [DOI] [PubMed] [Google Scholar]
- Cenci MA, Lee CS, Bjorklund A. L-DOPA-induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin- and glutamic acid decarboxylase mRNA. The European journal of neuroscience. 1998;10:2694–2706. [PubMed] [Google Scholar]
- Childers SR, Deadwyler SA. Role of cyclic AMP in the actions of cannabinoid receptors. Biochem Pharmacol. 1996;52:819–827. doi: 10.1016/0006-2952(96)00419-4. [DOI] [PubMed] [Google Scholar]
- Cinar R, Freund TF, Katona I, Mackie K, Szucs M. Reciprocal inhibition of G-protein signaling is induced by CB(1) cannabinoid and GABA(B) receptor interactions in rat hippocampal membranes. Neurochemistry international. 2008;52:1402–1409. doi: 10.1016/j.neuint.2008.02.005. [DOI] [PubMed] [Google Scholar]
- Di Monte DA, McCormack A, Petzinger G, Janson AM, Quik M, Langston WJ. Relationship among nigrostriatal denervation, parkinsonism, and dyskinesias in the MPTP primate model. Mov Disord. 2000;15:459–466. doi: 10.1002/1531-8257(200005)15:3<459::AID-MDS1006>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Domenici MR, Azad SC, Marsicano G, Schierloh A, Wotjak CT, Dodt HU, Zieglgansberger W, Lutz B, Rammes G. Cannabinoid receptor type 1 located on presynaptic terminals of principal neurons in the forebrain controls glutamatergic synaptic transmission. J Neurosci. 2006;26:5794–5799. doi: 10.1523/JNEUROSCI.0372-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro L, Tomasini MC, Gessa GL, Bebe BW, Tanganelli S, Antonelli T. The cannabinoid receptor agonist WIN 55,212–2 regulates glutamate transmission in rat cerebral cortex: an in vivo and in vitro study. Cereb Cortex. 2001;11:728–733. doi: 10.1093/cercor/11.8.728. [DOI] [PubMed] [Google Scholar]
- Ferrer B, Asbrock N, Kathuria S, Piomelli D, Giuffrida A. Effects of levodopa on endocannabinoid levels in rat basal ganglia: implications for the treatment of levodopa-induced dyskinesias. The European journal of neuroscience. 2003;18:1607–1614. doi: 10.1046/j.1460-9568.2003.02896.x. [DOI] [PubMed] [Google Scholar]
- Freiman I, Szabo B. Cannabinoids depress excitatory neurotransmission between the subthalamic nucleus and the globus pallidus. Neuroscience. 2005;133:305–313. doi: 10.1016/j.neuroscience.2005.01.058. [DOI] [PubMed] [Google Scholar]
- French ED, Dillon K, Wu X. Cannabinoids excite dopamine neurons in the ventral tegmentum and substantia nigra. Neuroreport. 1997;8:649–652. doi: 10.1097/00001756-199702100-00014. [DOI] [PubMed] [Google Scholar]
- Gerdeman G, Lovinger DM. CB1 cannabinoid receptor inhibits synaptic release of glutamate in rat dorsolateral striatum. J Neurophysiol. 2001;85:468–471. doi: 10.1152/jn.2001.85.1.468. [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- Gibaldi M, Perrier D. Pharmacokinetics. 1. Vol. 1. Marcel Dekker; New York: 1975. pp. 37–40. [Google Scholar]
- Giuffrida A, Parsons LH, Kerr TM, Rodriguez de Fonseca F, Navarro M, Piomelli D. Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat Neurosci. 1999;2:358–363. doi: 10.1038/7268. [DOI] [PubMed] [Google Scholar]
- Glass M, Felder CC. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J Neurosci. 1997;17:5327–5333. doi: 10.1523/JNEUROSCI.17-14-05327.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM, Aosaki T, Flaherty AW, Kimura M. The basal ganglia and adaptive motor control. Science. 1994;265:1826–1831. doi: 10.1126/science.8091209. [DOI] [PubMed] [Google Scholar]
- Gubellini P, Picconi B, Bari M, Battista N, Calabresi P, Centonze D, Bernardi G, Finazzi-Agro A, Maccarrone M. Experimental parkinsonism alters endocannabinoid degradation: implications for striatal glutamatergic transmission. J Neurosci. 2002;22:6900–6907. doi: 10.1523/JNEUROSCI.22-16-06900.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, de Costa BR, Richfield EK. Neuronal localization of cannabinoid receptors in the basal ganglia of the rat. Brain Res. 1991;547:267–274. doi: 10.1016/0006-8993(91)90970-7. [DOI] [PubMed] [Google Scholar]
- Hossain MA, Weiner N. Dopaminergic functional supersensitivity: effects of chronic L-dopa and carbidopa treatment in an animal model of Parkinson’s disease. J Pharmacol Exp Ther. 1993;267:1105–1111. [PubMed] [Google Scholar]
- Howlett AC, Fleming RM. Cannabinoid inhibition of adenylate cyclase. Pharmacology of the response in neuroblastoma cell membranes. Mol Pharmacol. 1984;26:532–538. [PubMed] [Google Scholar]
- Huang CC, Lo SW, Hsu KS. Presynaptic mechanisms underlying cannabinoid inhibition of excitatory synaptic transmission in rat striatal neurons. The Journal of physiology. 2001;532:731–748. doi: 10.1111/j.1469-7793.2001.0731e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida Y, Kawai K, Magata Y, Takeda R, Hashiguchi H, Abe H, Mukai T, Saji H. Changes in dopamine D2 receptors and 6-[18F]fluoro-L-3,4-dihydroxyphenylalanine uptake in the brain of 6-hydroxydopamine-lesioned rats. Neurodegener Dis. 2004;1:109–112. doi: 10.1159/000080051. [DOI] [PubMed] [Google Scholar]
- Kearn CS, Blake-Palmer K, Daniel E, Mackie K, Glass M. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors enhances heterodimer formation: a mechanism for receptor cross-talk? Mol Pharmacol. 2005;67:1697–1704. doi: 10.1124/mol.104.006882. [DOI] [PubMed] [Google Scholar]
- Kofalvi A, Rodrigues RJ, Ledent C, Mackie K, Vizi ES, Cunha RA, Sperlagh B. Involvement of cannabinoid receptors in the regulation of neurotransmitter release in the rodent striatum: a combined immunochemical and pharmacological analysis. J Neurosci. 2005;25:2874–2884. doi: 10.1523/JNEUROSCI.4232-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci. 2005;25:10537–10545. doi: 10.1523/JNEUROSCI.2959-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- Lindgren HS, Rylander D, Ohlin KE, Lundblad M, Cenci MA. The “motor complication syndrome” in rats with 6-OHDA lesions treated chronically with L-DOPA: relation to dose and route of administration. Behav Brain Res. 2007;177:150–159. doi: 10.1016/j.bbr.2006.09.019. [DOI] [PubMed] [Google Scholar]
- Lisovoski F, Haby C, Borrelli E, Schleef C, Revel MO, Hindelang C, Zwiller J. Induction of D2 dopamine receptor mRNA synthesis in a 6-hydroxydopamine parkinsonian rat model. Brain Res Bull. 1992;28:697–701. doi: 10.1016/0361-9230(92)90248-v. [DOI] [PubMed] [Google Scholar]
- Lundblad M. A model of L-DOPA-induced dyskinesia in 6-hydroxydopamine lesioned mice: relation to motor and cellular parameters of nigrostriatal function. Neurobiology of Disease. 2004;16:110–123. doi: 10.1016/j.nbd.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Lundblad M, Picconi B, Lindgren H, Cenci MA. A model of L-DOPA-induced dyskinesia in 6-hydroxydopamine lesioned mice: relation to motor and cellular parameters of nigrostriatal function. Neurobiol Dis. 2004;16:110–123. doi: 10.1016/j.nbd.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Lundblad M, Andersson M, Winkler C, Kirik D, Wierup N, Cenci MA. Pharmacological validation of behavioural measures of akinesia and dyskinesia in a rat model of Parkinson’s disease. Eur J Neurosci. 2002;15:120–132. doi: 10.1046/j.0953-816x.2001.01843.x. [DOI] [PubMed] [Google Scholar]
- Mackie K. Distribution of cannabinoid receptors in the central and peripheral nervous system. Handb Exp Pharmacol. 2005:299–325. doi: 10.1007/3-540-26573-2_10. [DOI] [PubMed] [Google Scholar]
- Malone DT, Taylor DA. Modulation by fluoxetine of striatal dopamine release following Delta9-tetrahydrocannabinol: a microdialysis study in conscious rats. Br J Pharmacol. 1999;128:21–26. doi: 10.1038/sj.bjp.0702753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden CD, Parkes JD. “On-off” effects in patients with Parkinson’s disease on chronic levodopa therapy. Lancet. 1976;1:292–296. doi: 10.1016/s0140-6736(76)91416-1. [DOI] [PubMed] [Google Scholar]
- Martin AB, Fernandez-Espejo E, Ferrer B, Gorriti MA, Bilbao A, Navarro M, Rodriguez de Fonseca F, Moratalla R. Expression and Function of CB(1) Receptor in the Rat Striatum: Localization and Effects on D(1) and D(2) Dopamine Receptor-Mediated Motor Behaviors. Neuropsychopharmacology. 2008;33:1667–1679. doi: 10.1038/sj.npp.1301558. [DOI] [PubMed] [Google Scholar]
- Meissner W, Ravenscroft P, Reese R, Harnack D, Morgenstern R, Kupsch A, Klitgaard H, Bioulac B, Gross CE, Bezard E, Boraud T. Increased slow oscillatory activity in substantia nigra pars reticulata triggers abnormal involuntary movements in the 6-OHDA-lesioned rat in the presence of excessive extracellular striatal dopamine. Neurobiol Dis. 2006;22:586–598. doi: 10.1016/j.nbd.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Mela F, Marti M, Dekundy A, Danysz W, Morari M, Cenci MA. Antagonism of metabotropic glutamate receptor type 5 attenuates l-DOPA-induced dyskinesia and its molecular and neurochemical correlates in a rat model of Parkinson’s disease. J Neurochem. 2007;101:483–497. doi: 10.1111/j.1471-4159.2007.04456.x. [DOI] [PubMed] [Google Scholar]
- Meschler JP, Howlett AC. Signal transduction interactions between CB1 cannabinoid and dopamine receptors in the rat and monkey striatum. Neuropharmacology. 2001;40:918–926. doi: 10.1016/s0028-3908(01)00012-0. [DOI] [PubMed] [Google Scholar]
- Miklyaeva EI, Whishaw IQ, Kolb B. A golgi analysis of cortical pyramidal cells in the unilateral parkinson rat: absence of change in the affected hemisphere vs hypertrophy in the intact hemisphere. Restor Neurol Neurosci. 2007;25:91–99. [PubMed] [Google Scholar]
- Monory K, Blaudzun H, Massa F, Kaiser N, Lemberger T, Schutz G, Wotjak CT, Lutz B, Marsicano G. Genetic dissection of behavioural and autonomic effects of Delta(9)-tetrahydrocannabinol in mice. PLoS biology. 2007;5:e269. doi: 10.1371/journal.pbio.0050269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morari M, Marti M, Sbrenna S, Fuxe K, Bianchi C, Beani L. Reciprocal dopamine-glutamate modulation of release in the basal ganglia. Neurochemistry international. 1998;33:383–397. doi: 10.1016/s0197-0186(98)00052-7. [DOI] [PubMed] [Google Scholar]
- Morgese MG, Cassano T, Cuomo V, Giuffrida A. Anti-dyskinetic effects of cannabinoids in a rat model of Parkinson’s disease: role of CB(1) and TRPV1 receptors. Exp Neurol. 2007;208:110–119. doi: 10.1016/j.expneurol.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narushima M, Uchigashima M, Fukaya M, Matsui M, Manabe T, Hashimoto K, Watanabe M, Kano M. Tonic enhancement of endocannabinoid-mediated retrograde suppression of inhibition by cholinergic interneuron activity in the striatum. J Neurosci. 2007;27:496–506. doi: 10.1523/JNEUROSCI.4644-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dell SJ, Marshall JF. Chronic L-dopa alters striatal NMDA receptors in rats with dopaminergic injury. Neuroreport. 1996;7:2457–2461. doi: 10.1097/00001756-199611040-00011. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson CRR. The Rat Brain in Stereotaxic Coordinates. 1998 doi: 10.1016/0165-0270(80)90021-7. [DOI] [PubMed] [Google Scholar]
- Picconi B, Centonze D, Hakansson K, Bernardi G, Greengard P, Fisone G, Cenci MA, Calabresi P. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat Neurosci. 2003;6:501–506. doi: 10.1038/nn1040. [DOI] [PubMed] [Google Scholar]
- Pierot L, Desnos C, Blin J, Raisman R, Scherman D, Javoy-Agid F, Ruberg M, Agid Y. D1 and D2-type dopamine receptors in patients with Parkinson’s disease and progressive supranuclear palsy. J Neurol Sci. 1988;86:291–306. doi: 10.1016/0022-510x(88)90106-2. [DOI] [PubMed] [Google Scholar]
- Price DA, Owens WA, Gould GG, Frazer A, Roberts JL, Daws LC, Giuffrida A. CB1-independent inhibition of dopamine transporter activity by cannabinoids in mouse dorsal striatum. J Neurochem. 2007;101:389–396. doi: 10.1111/j.1471-4159.2006.04383.x. [DOI] [PubMed] [Google Scholar]
- Riederer P, Lange KW, Kornhuber J, Danielczyk W. Glutamatergic-dopaminergic balance in the brain. Its importance in motor disorders and schizophrenia. Arzneimittelforschung. 1992;42:265–268. [PubMed] [Google Scholar]
- Robelet S, Melon C, Guillet B, Salin P, Kerkerian-Le Goff L. Chronic L-DOPA treatment increases extracellular glutamate levels and GLT1 expression in the basal ganglia in a rat model of Parkinson’s disease. The European journal of neuroscience. 2004;20:1255–1266. doi: 10.1111/j.1460-9568.2004.03591.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez M, Morales I, Gonzalez-Mora JL, Gomez I, Sabate M, Dopico JG, Rodriguez-Oroz MC, Obeso JA. Different levodopa actions on the extracellular dopamine pools in the rat striatum. Synapse. 2007;61:61–71. doi: 10.1002/syn.20342. [DOI] [PubMed] [Google Scholar]
- Sergeeva OA, Doreulee N, Chepkova AN, Kazmierczak T, Haas HL. Long-term depression of cortico-striatal synaptic transmission by DHPG depends on endocannabinoid release and nitric oxide synthesis. The European journal of neuroscience. 2007;26:1889–1894. doi: 10.1111/j.1460-9568.2007.05815.x. [DOI] [PubMed] [Google Scholar]
- Sidlo Z, Reggio PH, Rice ME. Inhibition of striatal dopamine release by CB1 receptor activation requires nonsynaptic communication involving GABA, H2O2, and KATP channels. Neurochemistry international. 2008;52:80–88. doi: 10.1016/j.neuint.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieradzan KA, Fox SH, Hill M, Dick JP, Crossman AR, Brotchie JM. Cannabinoids reduce levodopa-induced dyskinesia in Parkinson’s disease: a pilot study. Neurology. 2001;57:2108–2111. doi: 10.1212/wnl.57.11.2108. [DOI] [PubMed] [Google Scholar]
- Silva I, Cortes H, Escartin E, Rangel C, Floran L, Erlij D, Aceves J, Floran B. L-DOPA inhibits depolarization-induced [3H]GABA release in the dopamine-denervated globus pallidus of the rat: the effect is dopamine independent and mediated by D2-like receptors. J Neural Transm. 2006;113:1847–1853. doi: 10.1007/s00702-006-0493-7. [DOI] [PubMed] [Google Scholar]
- Sossi V, de la Fuente-Fernandez R, Schulzer M, Troiano AR, Ruth TJ, Stoessl AJ. Dopamine transporter relation to dopamine turnover in Parkinson’s disease: a positron emission tomography study. Ann Neurol. 2007;62:468–474. doi: 10.1002/ana.21204. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Ding J, Day M, Wang Z, Shen W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007;30:228–235. doi: 10.1016/j.tins.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Takahashi KA, Castillo PE. The CB1 cannabinoid receptor mediates glutamatergic synaptic suppression in the hippocampus. Neuroscience. 2006;139:795–802. doi: 10.1016/j.neuroscience.2006.01.024. [DOI] [PubMed] [Google Scholar]
- Tsou K, Brown S, Sanudo-Pena MC, Mackie K, Walker JM. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience. 1998;83:393–411. doi: 10.1016/s0306-4522(97)00436-3. [DOI] [PubMed] [Google Scholar]
- Uchigashima M, Narushima M, Fukaya M, Katona I, Kano M, Watanabe M. Subcellular arrangement of molecules for 2-arachidonoyl-glycerol-mediated retrograde signaling and its physiological contribution to synaptic modulation in the striatum. J Neurosci. 2007;27:3663–3676. doi: 10.1523/JNEUROSCI.0448-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallmichrath I, Szabo B. Cannabinoids inhibit striatonigral GABAergic neurotransmission in the mouse. Neuroscience. 2002;113:671–682. doi: 10.1016/s0306-4522(02)00109-4. [DOI] [PubMed] [Google Scholar]
- Winkler C, Kirik D, Bjorklund A, Cenci MA. L-DOPA-induced dyskinesia in the intrastriatal 6-hydroxydopamine model of parkinson’s disease: relation to motor and cellular parameters of nigrostriatal function. Neurobiol Dis. 2002;10:165–186. doi: 10.1006/nbdi.2002.0499. [DOI] [PubMed] [Google Scholar]
- Zeng BY, Dass B, Owen A, Rose S, Cannizzaro C, Tel BC, Jenner P. Chronic L-DOPA treatment increases striatal cannabinoid CB1 receptor mRNA expression in 6-hydroxydopamine-lesioned rats. Neurosci Lett. 1999;276:71–74. doi: 10.1016/s0304-3940(99)00762-4. [DOI] [PubMed] [Google Scholar]