

Abstract
The chemical environment of the extracellular matrix may influence the tissue-selective deposition observed there in gelsolin amyloid disease. Previously, we have identified the proteases that generate the amyloidogenic fragments from the full length gelsolin variants, and demonstrated that heparin is capable of accelerating gelsolin amyloidogenesis. Herein, we identify the structural features of heparin that promote the 8 kDa disease-associated gelsolin fragments (residues 173-243) generated at the cell surface to form amyloid. In conjunction with electron microscopy analyses, our kinetic studies demonstrate that heparin efficiently accelerates the formation of gelsolin amyloid by enabling intermolecular β-sheet formation. The use of heparin analogues reveal that sulfation is important in accelerating amyloidogenesis and that the extent of acceleration is proportional to the molecular weight of heparin. In addition, heparin accelerated aggregation at both early and late stages of amyloidogenesis. Dynamic light scattering coupled to size exclusion chromatography showed that heparin promotes the formation of soluble aggregates. Collectively, these data reveal that heparin templates fibril formation and affords solubility to the aggregating peptides through its sulfated structure. By extension, the biochemical results herein suggest that tissue-selective deposition characteristic of the gelsolin amyloidoses are likely influenced by the extracellular localization of distinct glycosaminoglycans.
Amyloid diseases are characterized by the extracellular deposition of a given protein into a variety of structures, including cross β-sheet-rich fibrillar aggregates called amyloid. More than twenty non-homologous human proteins become amyloidogenic later in life. The clinically most important example is the amyloid β-peptide (Aβ)1, the deposits of which are associated with Alzheimer's disease. Many amyloid diseases exhibit distinct tissue-selective deposition of a given amyloidenic protein–the exact tissue targeted being dependent on the biological and chemical characteristics of the amyloidogenic protein (1). Familial gelsolin amyloidosis is an autosomal dominantly inherited neurodegenerative disease. Heterozygotes usually have a normal lifespan with clinical manifestations presenting in the third to fifth decade. In contrast, homozygotes generally do not survive past their thirties. Extensive deposition of mutant gelsolin fragments as amyloid in the basement membrane of the skin, blood vessel walls, central nervous system, and the ocular system causes cutis laxa, cranial neuropathy, and corneal lattice dystrophy (2-4).
The endoproteolytic cascade generating the amyloidogenic gelsolin peptides is distinct from that affording Aβ, in that the mutations (D187N/Y) causing gelsolin amyloidosis enable aberrant proteolysis to occur by disrupting Ca2+-binding within domain 2. The conformational alterations within domain 2 of D187N/Y gelsolin render it susceptible to aberrant cleavage by furin in the slightly acidic trans-Golgi compartment yielding a secreted 68 kDa fragment referred to as C68 (5, 6). C68 is subsequently cleaved by a membrane associated type I matrix metalloprotease located proximal to the extracellular matrix (ECM) where amyloid deposition is observed in patients (7). The C68 fragment is cleaved in at least two positions by metalloprotease(s) producing 8 (residues A173-M243) and 5 kDa (residues A173-R225) gelsolin fragments which compose the amyloid deposits in patients (8, 9). The deposition of these peptides is an early pathological event. The distinctive connective tissue distribution of the gelsolin amyloid deposits (i.e. heavy deposition in the skin) strongly suggests that amyloidogenesis may be enabled by the local chemical environment of the ECM.
Glycosaminoglycans (GAGs) are a unique class of linear polysaccharides that also compose the polysaccharide-protein conjugates called proteoglycans, which are among the major macromolecules of the ECM. The GAG polysaccharides consist of disaccharide repeating units that are arranged in an alternating unbranched sequence. In proteoglycans, the GAGs are coupled to the protein core through a trisaccharide linker by an O-glycosidic bond to a serine residue in the protein (GAG-Gal-Gal-Xyl-O-CH2-Cα of Ser-protein). Each GAG differs in its disaccharide repeating unit(s) and in the linkages between the monosaccharide unit. The structures of the repeating disaccharide units of the GAGs used in this study (heparan sulfate (HS)/heparin, chondroitin sulfate (Chon) A, C, D, E, dermatan sulfate (DS) also known as ChonB, keratan sulfate and hyaluronic acid) are depicted in Figure 1. Distinct biological distributions and functions result from the structural variations among these GAGs.
Figure 1.

Heparan sulfate (HS) and heparin have the same repeating disaccharide subunits of glucuronic acid (GlcA) or iduronic acid (IdoA) and glucosamine (GlcN) or N-acetylated glucosamine (GlcNAc) linked by an α-1,4 glycosidic linkage with various sulfation patterns (box). IdoA is formed by epimerization of GlcA at the C5 position. In both heparin and HS, these subunits can have variable N-acetylation and N- or O- sulfation at C2 or C6 positions. HS has fewer sulfates, is less negatively charged, and has a more heterogeneous structure than heparin. Chondroitin sulfate A (ChonA) contains 4-O-sulfo GalNAc; dermatan sulfate (DS) also known as chondroitin sulfate B (ChonB) contains IdoA, 4-O-sulfo GalNAc; chondroitin sulfate C (ChonC) contains 6-O-sulfo GalNAc; chondroitin sulfate D (ChonD) contains 2-O-sulfo IdoA and 6-O-sulfo GalNAc; chondroitin sulfate E (ChonE) contains 4, 6-O-sulfo GalNAc. Hyaluronic acid is a copolymer of GlcA and GlcNAc and is the only GAG that is not sulfated and is not linked to a core protein Keratan sulfate contains galactose units (Gal) instead of hexuronic acid units. R1 indicates that the substituent can be either a hydrogen (-H) or a sulfo group (-SO3-). R2 indicates that the the substituent can be either a hydrogen (-H), a sulfo group (-SO3-) or an acetyl group (-COCH3). Negatively charged groups are shown in red (-COO-, SO3-).
It has been suggested that the GAG substructure of proteoglycans associates with amyloid-forming polypeptides and plays an important role in initiating and sustaining amyloidogenesis (10-16). In particular, the GAGs HS and heparin have been extensively associated with the process of amyloidogenesis. Heparin and HS cannot be distinguished based on their core disaccharide substructures, however their level of sulfation is different. The disaccharide from heparin contains 3 sulfate groups on average, compared to 1 sulfate per disaccharide from HS (17). Heparin also contains a higher content of iduronate (IdoA) residues (epimerization of glucuronate (GlcA) at C5 position forms IdoA) than HS and less N-acetylation at its glucosamine (GlcN) residues than HS. HS is ubiquitously expressed in mammalian cells and is found in the extracellular membrane and ECM proteoglycans. In contrast, heparin is generally considered a product of mast cells, stored in intracellular granules and secreted as a free soluble GAG (18, 19). The HS component of proteoglycans are known to promote the aggregation of peptides including Aβ (Alzheimer's disease) (15, 20, 21), the prion protein (spongiform encephalopathies) (11) and serum amyloid A protein (inflammation-associated amyloid) (14). Heparin and HS induce the formation of β-structure within the amyloidogenic Aβ peptide and dramatically accelerate the growth of α-synuclein fibrils associated with Parkinson's disease (22, 23). HS has also been implicated as an amyloid stabilizer, protecting amyloid against proteolytic degradation (24, 25) and against microglial phagocytosis (26). Furthermore, a recent mouse model has demonstrated direct association of HS with amyloid deposits (27). Despite the apparent roles of heparin and HS in amyloidogenesis, the mechanism of GAG-mediated amyloidogenesis remains unclear.
Herein, we study the means by which heparin accelerates gelsolin (173-243) amyloidogenesis. We demonstrate an increase in the rate of wild type (WT), D187N and D187Y gelsolin amyloid fibril formation upon the addition of heparin and an accelerated increase in cross β-sheet content. Using modified synthetic heparin derivatives, we show that the sulfated structure of heparin and in particular the O-sulfo groups of heparin are critical for accelerating gelsolin amyloidogenesis. Heparin also appears to promote the formation of soluble aggregates and thus we propose that heparin increases the solubility of aggregates and templates their assembly into fibrils.
Experimental Procedures
Preparation of recombinant 8 kDa WT, D187N and D187Y gelsolin amyloid peptides
The peptide corresponding to amino acids A173-M243 of WT plasma gelsolin and the D187N, D187Y variants were recombinantly expressed and purified as described previously (28). Prior to use, the peptide was pretreated to ensure an aggregate-free starting solution. The solution pH was adjusted to 10.5 with aqueous NaOH (100 mM) and the sample was sonicated (30 min, 25 °C) then centrifuged at 100,000 × g before the supernatant was transferred to a new Eppendorf tube. The concentration of the peptide was determined by UV absorption at 280 nm (ε = 1,280 M-1· cm-1) and used immediately by diluting into the aggregation buffer.
Fibril formation monitored by Thioflavin T (TfT) fluorescence (plate-reader assay)
Fibrilization assays were performed with WT, D187N and D187Y gelsolin173-243 peptides (10 μM) in 50 mM HEPES (pH 7) or sodium acetate (pH 5), 100 mM NaCl, 20 μM TfT (29), with or without GAGs (0.05-5 μM). The components were mixed together in an Eppendorf tube, and 100 μL was transferred into a well of a 96-well microplate (black plastic with clear bottom, Corning Inc, Corning, NY). The plate was sealed and loaded into a Gemini SpectraMax EM fluorescence plate reader (Molecular Devices, Sunnyvale, CA), where it was incubated at 37 °C. The fluorescence (excitation at 440 nm, emission at 485 nm) was measured from the bottom of the plate at 10 min intervals, with 5 sec of shaking before each reading. Each value obtained was the average of thirty individual scans. The data from triplicate wells (error range < 10%) were averaged, corrected for baseline, normalized and plotted as fluorescence vs. time. All experiments were repeated on at least 5-6 independent occasions. All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) and/or synthesized unless indicated otherwise. GAGs used were 17-19 kDa heparin sodium, 5 kDa heparin (Calbiochem, San Diego, CA), porcine intestine heparin, HS, ChonA, DS (ChonB) (Celsus Laboratories, Cincinnati, OH), ChonC, Chon D, ChonE (Seikagaku, Tokyo), keratan sulfate (Associates of Cape Cod Inc., East Falmouth, MA), and hyaluronic acid. Fully desulfated heparin, N-desulfated heparin and 2-O-desulfated IdoA heparin were prepared based on Yates et al. (30). Fully O-sulfated-N-acetylated heparin was prepared by O-sulfonation followed by N-sulfonation (31). Heparin oligosaccharides dp4, dp8, dp12 and dp20 of defined degree of polymerization (dp4 to dp20) were prepared from controlled partial heparin lyase 1 treatment of bovine lung heparin followed by size fractionation (32).
Morphology assessment by transmission electron microscopy (TEM)
TEM samples were taken from the aggregation assays described above and prepared as described previously, using a Phillips CM-100 transmission electron microscope (Phillips, Eindhoven, The Netherlands) at 80 kV (33).
Circular dichroism (CD) and TfT fluorescence to monitor β-sheet structure
Pre-treated D187N gelsolin173-243 (36 μM) samples were subjected to constant rocking (30 cycles/min) on a Labquake shaker (Barnstead/Thermolyne, Dubuque, IA) at 37 °C in pH 7 buffer (50 mM Tris-HCl, 100 mM NaCl) in a total volume of 1.5 mL. The aggregation process was monitored by diluting a 12 μL sample of the reaction into 108 μL of 20 μM TfT (at 0, 12, 23, 37, 49, 64 and 88 h) and measuring the fluorescence (excitation at 440 nm, emission at 485 nm; Varian Cary Eclipse Fluorometer, Walnut Creek, CA). Far-UV CD spectra of the aggregation reaction samples at these time points were recorded from 200 to 250 nm at 25 °C using an AVIV model 202SF stopped flow CD spectrometer (AVIV Associates Inc., Lakewood, NJ) equipped with Peltier temperature-controlled cell holder using a 0.1 cm path length Suprasil quartz cell (Hellma, Forest Hills, NY). The wavelength step size used was 0.5 nm with an averaging time of 5 s per scan at each wavelength step.
Size exclusion chromatography coupled to dynamic light scattering to monitor monomer disappearance and aggregate size distribution
The aggregated samples (200 μL; prepared as described for CD measurements) were transferred into an Eppendorf tube and centrifuged (13,000 rpm for 5 mins) in an Eppendorf microcentrifuge. The supernatant (100 μL) was analyzed by size exclusion chromatography on a Superdex 75 HR, NJ10/30 column (1 × 30 cm, 13 μm) (Amersham Biosciences, Piscataway, NJ) at 25 °C utilizing UV detection at 280 nm. The mobile phase comprised PBS with 0.02% sodium azide (pH 7). Samples were passed through a 0.1 μm membrane filter prior to injection onto the column and column eluates were directly injected into a Dawn EOS light scattering photometer (Wyatt Technology, Santa Barbara, CA) connected to the FPLC. The UV 280 nm absorption peak area, molar mass, and the hydrodynamic radius of the samples were calculated using the Astra software package from Wyatt Technology.
Results
The pre-treated gelsolin 173-243 fragments were analyzed by analytical ultracentrifugation sedimentation velocity experiments, by analytical size exclusion chromatography coupled to dynamic light scattering, and atomic force microscopy, which collectively revealed the absence of detectable aggregates and the presence of ∼93% of monomeric peptides along with approximately ∼7% of dimeric and tetrameric species in the starting solution (supporting information).
Heparin most effectively accelerates fibril formation
The rate of D187N gelsolin amyloidogenesis as a function of GAG concentration was probed using an aggregation assay in a plate-reader described above, as well as other methods described herein. The influence of various GAGs (Figure 2) on amyloidogenesis was monitored. Aggregation was quantified by the binding of TfT, which fluoresces upon selective binding to cross-β-sheet structures (29). GAG-mediated acceleration of aggregation was observed irrespective of whether aggregation was carried out under quiescent conditions or with agitation (i.e., shaking for a duration of 5 s every 10 min or constant rocking at 30 cycles/min) and irrespective of buffer conditions employed (i.e. 50-300 mM NaCl, pH 4-7). D187N 173-243 amyloidogenesis was observed over a peptide and GAG concentration range of 5-50 μM and 0.05-5 μM, respectively. In all cases, the presence of fibrils was confirmed upon the completion of aggregation by TEM (data for heparin shown below). Even though WT gelsolin is not cleaved by furin and/or MT1-MMP and is therefore not involved in amyloid disease, aggregation assays with WT gelsolin 173-243 peptides demonstrated that this peptide is as amyloidogenic as the D187N fragment (supporting information), implying that the D187N mutation enables aberrant cleavage but does not confer additional amyloidogenecity above and beyond the WT sequence. Analogous experiments with the D187Y gelsolin 173-243 peptide revealed that unlike the WT and D187N sequences, which aggregate under both neutral and acidic conditions, D187Y does not efficiently aggregate at neutral pH on a convenient laboratory time scale (34), suggesting that this disease associated variant is actually slightly less amyloidogenic than the WT sequence. However, heparin does accelerate D187Y gelsolin173-243 amyloidogenesis under acidic conditions (supporting information).
Figure 2.
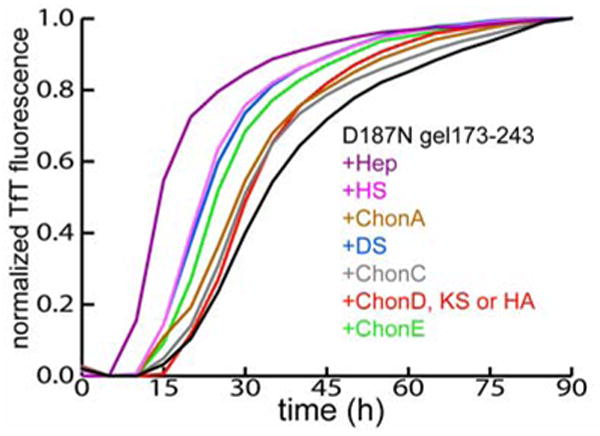
TfT monitoring of D187N gelsolin173-243 (10 μM) amyloidogenesis (37 °C, pH 7, 5 s of shaking every 10 min) with (colored lines) or without (black line) GAGs. Heparin (Hep), heparin sulfate (HS), chondroitin A (ChonA), dermatan sulfate (DS), chondroitin C (ChonC), chondroitin D (ChonD), chondroitin E (ChonE), keratan sulfate (KS) and hyaluronic acid (HA) were added at the initiation of aggregation. Increasing the GAG concentrations (0.05 - 5 μM) increased the rates of the reactions (5 μM data shown). The data is representative of 5 to 6 independent experiments, each in triplicates. The GAGs alone did not affect the TfT fluorescence.
Heparin is the most effective gelsolin amyloidogenesis accelerator, shortening the time to reach 50% completion (t50) by ∼2-fold. The mean t50 value for D187N gelsolin 173-243 fragment aggregation in the absence of GAGs is 31.8 ± 7 h, while in the presence of heparin the mean t50 is 15.4 ± 2.9 h (Figure 2) (two-sample t-test: P < 0.001, n = 6). The GAGs HS and DS are also effective (mean t50 value = 23.2 ± 3.3 h and 22.9 ± 2.6 h, respectively, P < 0.001). ChonE is a slightly less effective aggregation promoter (mean t50 value = 26.4 ± 1 h, P < 0.005). All other GAGs are less effective at accelerating D187N gelsolin173-243 amyloidogenesis (Figure 2). Heparin, HS and DS consist of disaccharide subunits containing IdoA residues, suggesting that this monosaccharide residue may be important for accelerating amyloidogenesis (Figure 1). IdoA residues have more conformational flexibility than GlcA residues and are believed to be important in many GAG-protein interactions (35). Supporting this notion, ChonA, which is less effective than ChonB at accelerating D187N gelsolin173-243 amyloidogenesis, has GlcA in place of IdoA residues (Figure 1). The O-sulfo groups of the hexosamine residue also appear to be important for accelerating amyloidogenesis. ChonE, with both a 4-O- and 6-O-sulfo group substituted on the GalNAc residue is more effective than ChonA and ChonC, which each contain only a single 4-O- or 6-O-sulfo GalNAc, respectively (Figure 1, 2). Heparin and HS also contain O-sulfo groups at the C2 position of the IdoA (or GlcA) residue and C6 position of the GlcNAc residue, consistent with their effect on amyloidogenesis. To understand the mechanistic basis for the GAG-mediated acceleration of D187N gelsolin173-243 amyloidogenesis, we have chosen to utilize heparin, the most effective GAG.
Heparin enhances fibril formation through cross β-sheet formation
The time dependence of D187N gelsolin173-243 cross β-sheet quaternary structure formation (a hallmark of amyloidogenesis) was followed by far-UV CD spectroscopy in the presence and absence of heparin. All samples exhibited a predominantly monomeric random coil structure at the start of the aggregation reaction (Figure 3A, B). The percentage of the cross β-sheet structural transition completed at each time point was calculated (Figure 3C). After 23 h, the transition from random coil to a cross β-sheet structure was 74% complete in the presence of heparin, whereas it was only 55% complete in the absence of heparin. After 37 h, transition towards cross β-sheet was 92% complete in the presence of heparin, whereas it was only 77% complete in the absence of heparin. Moreover, the 220 nm ellipticity value at 88 h in the absence of heparin (θ220nm = -22.0) did not reach the intensity observed in the presence of heparin (θ220nm = -24.3). Therefore, aggregating samples in the presence of heparin appear to have higher cross β-sheet content than those without heparin. Qualitatively, the increase in TfT fluorescence measured in these samples was consistent with the increase in cross β-sheet content revealed by far-UV CD (Figure 3D). Thus, heparin accelerates the structural transition of D187N gelsolin173-243 from random coil to what appears to be a cross β-sheet structure.
Figure 3.
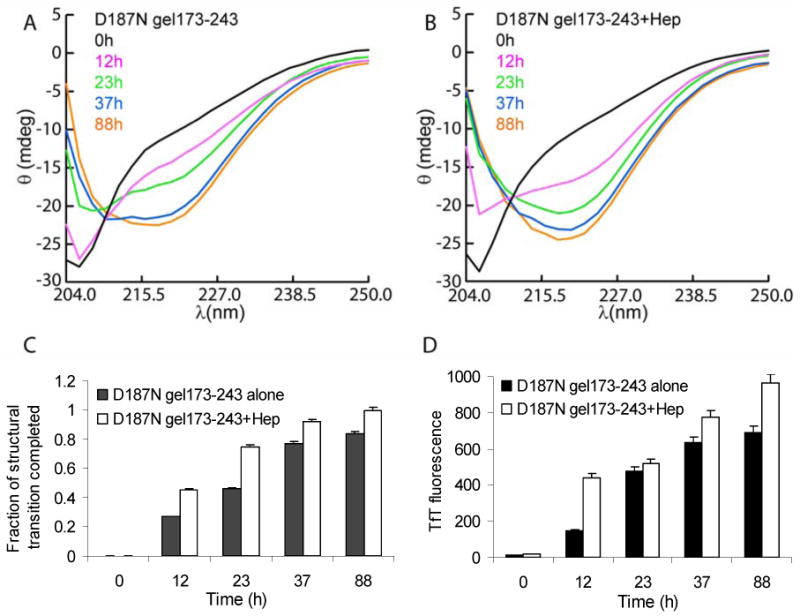
CD-spectra of D187N gelsolin173-243 (36 μM) samples aggregated alone (A) or with 5 μM heparin (Hep) (B) (37 °C, pH 7, constant rocking at 30 cycles/min) were taken at the indicated time points. The CD spectra minima are observed at 215-220 nm for a cross β-sheet structure. The transition of the structure towards a β-sheet rich structure was more pronounced in samples with heparin. (C) The fraction of structural transition completed for each time point was calculated as the ratio of ellipticity at 220 nm to its maximum value observed and normalized. (D) TfT fluorescence of samples were taken from the CD samples and diluted (see experimental procedures). Samples in the presence of heparin exhibited increased TfT fluorescence compared to D187N gelsolin173-243 samples aggregated alone. The data is representative of at least 5 to 6 independent experiments. Analysis of heparin alone (∼5 μM) did not lead to a CD signal at 220 nm.
Heparin sulfation is important for accelerating D187N gelsolin173-243 aggregation
The specific sulfate substituents composing GAGs are known to be critical for the acceleration of Aβ amyloidogenesis. However, in gelsolin amyloid formation it seems clear that it is not simply the polyanionic nature of heparin that hastens amyloidogenesis, since other highly charged macromolecules such as polyglutamic acid (polyE) and dextran sulfate did not accelerate D187N gelsolin173-243 fibril formation (supporting information). To scrutinize the hypothesis that it is the appropriate spatial presentation of the sulfate groups in heparin that are important for accelerating gelsolin amyloidogeneis, heparin molecules with various sulfate substitutions were prepared (Figure 4A). Complete desulfation of heparin nearly eliminates acceleration of D187N gelsolin173-243 amyloidogenesis, whereas 2-O-desulfated heparin composed exclusively of iduronic acid and N-desulfated heparin exhibit modest levels of acceleration (Figure 4B). Consistent with the hypothesis above, a fully O-sulfated and N-acetylated heparin is the most potent accelerator, decreasing the time to reach 50% completion (t50) by 4 fold (Figure 4B, mean t50 value= 7.8 ± 0.7 h, P < 0.001, n = 6). The importance of O-sulfation is also reflected in the influence of the various O-sulfated GAGs previously described (Figure 2). The influence of N-acetylation of the hexosamine residue remains unclear; in heparin less than 5% of these residues contain N-acetyl groups while heparan sulfate contains ∼95% N-acetyl groups and dermatan sulfate is 100% N-acetylated.
Figure 4.
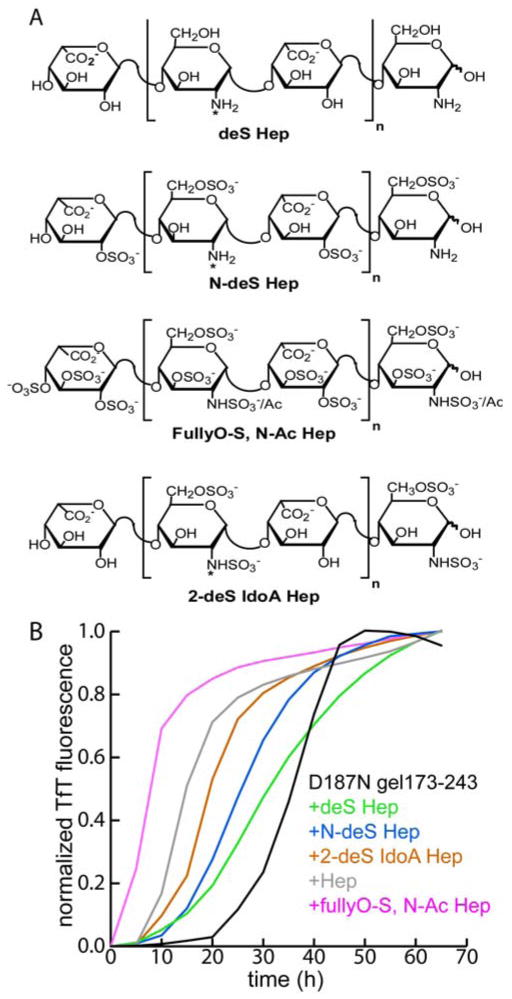
(A) Heparin derivatives synthesized with indicated sulfate substitutions; desulfated heparin (deS Hep), N-desulfated heparin (N-deS Hep), fully O-sulfated-N-acetylated heparin (fullyO-S, N-Ac Hep), 2-O-desulfated iduronic acid heparin (2-deS IdoA Hep). ‘*’ indicates positions where variable N-acetylation occurs. (B) TfT monitoring of D187N gelsolin173-243 aggregation (10 μM) (37 °C, pH 7, 5 s of shaking every 10 min) in the presence or absence of the synthetic heparin derivatives (5 μM).
Heparin molecular weight influences its ability to accelerate amyloidogenesis
In addition to the chemical heterogeneity of heparin observed in vivo, there are substantial variations in the molecular weight distribution (36). Therefore, we compared the influence of heparin molecules of different sizes with regard to their ability to accelerate D187N gelsolin173-243 amyloidogenesis. Heparin molecules of 12-19 kDa, 5 kDa (low molecular weight (LMW) heparin) and heparin oligosaccharides of varying lengths (4, 8, 12, 20 saccharides; dp4, dp8, dp12, dp20, respectively) were tested (Figure 5A). The results demonstrate that heparin oligomers as small as 4 saccharide units (dp4) can accelerate gelsolin amyloidogenesis. However, amyloidogenesis was generally enhanced in proportion to chain length up to 20 saccharide units (mean t50 value in the presence of dp4 = 19.1 ± 1.86 h; mean t50 value in the presence of dp20 = 13.1 ± 0.3, n = 6) consistent with the expected polyvalent interactions between gelsolin assemblies and the sulfated GAGs (Figure 5B). The morphologies of the fibrils formed in the presence of the different sized heparin molecules were examined by TEM. Fibrils of 8-10 nm diameter were observed and a statistical analysis (Student's t-test; data not shown) confirmed that no significant differences between the sizes of the fibrils formed were discernable employing heparin molecules of different molecular weights (data shown for dp4 and 17-19 kDa heparin, Figure 5C, D).
Figure 5.
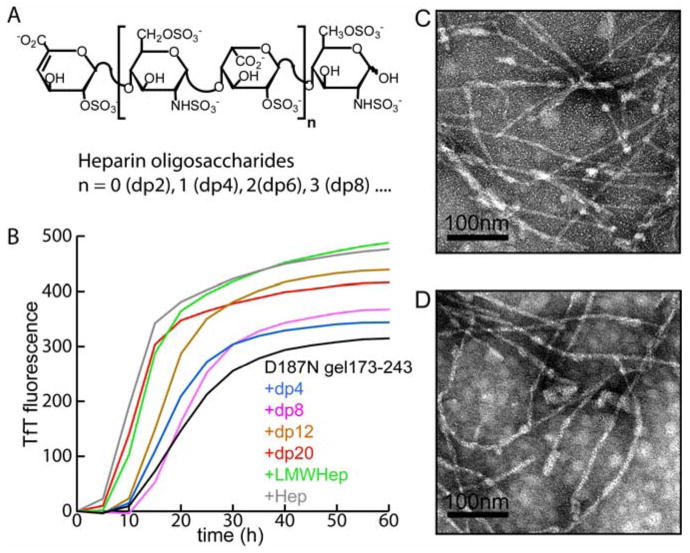
Synthesized heparin molecules (A) (0.1 mg/mL) having 4 saccharide units (dp4), 8 saccharide units (dp8), 12 saccharide units (dp12), 20 saccharide units (dp20), as well as a 5 kDa low molecular weight heparin (LMW Heparin), and 12-19 kDa heparin (heparin) were added to the D187N gelsolin173-243 (10 μM) aggregation reaction to discern their influence. (B) TfT assays were performed (37 °C, pH 7, constant rocking) to monitor the role of aggregation. TEM image of D187N gelsolin173-243 fibrils formed in the presence of dp4 (C) and 12-19 kDa heparin (D). Contrasts of the images shown were optimized using the auto-level function of Adobe Photoshop. Original images are presented in the supporting information.
Heparin influences both early and late stages of gelsolin amyloidogenesis
Heparin was also introduced at later stages of the TfT-based aggregation assay and changes in the kinetics were monitored to evaluate whether heparin affects amyloid formation in the presence of pre-aggregated species. Addition of heparin at 43 h increased the amplitude of the TfT fluorescence when compared to samples with no heparin added (Figure 6A). In the absence of heparin, the TfT fluorescence trends towards a gradual decrease late in the time course, which is likely due to the formation of visibly large aggregates interfering with the fluorescence. White insoluble pellets were seen by eye in centrifuged (13,800 × g, 5 min) D187N gelsolin173-243 samples aggregated for 90 h in the absence of heparin. However, when samples aggregated with heparin from the start were centrifuged at 90 h, such insoluble aggregates were not obvious (data not shown). The TfT intensity eventually reached a plateau for samples with heparin introduced at 43 h, but was further increased if heparin was added again at 90 h (data not shown). To further test whether heparin imparts solubility to the fibrils, these samples were characterized by TEM. At 43 h into the aggregation time course, the morphology of aggregates without heparin appears as a mesh of laterally associated fibrillar aggregates (Figure 6B) and by 90 h progressed into larger aggregated clumps with few distinguishable fibrils (Figure 6C). In contrast, the presence of heparin added at 0 h produced evenly distributed 8-10 nm diameter fibrils with rope-like morphology (Figure 6D). Heparin introduced at 43 h into an aggregating sample induced changes in the aggregate morphology and well-dispersed aggregates containing fibrils were observed at 90 h (Figure 6E) (P < 0.001, n = 12). Thus, heparin also has an effect in the late stages of D187N gelsolin173-243 fibril formation.
Figure 6.
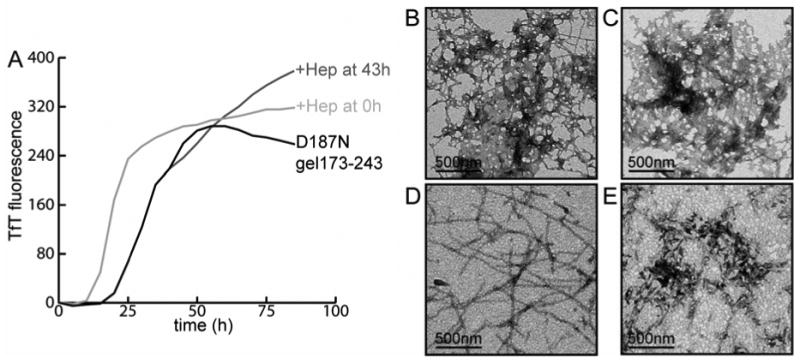
(A) TfT analysis of D187N gelsolin173-243 (10 μM) aggregation was performed (37 °C, pH 7, 5 s of shaking every 10 min) and the averages of triplicate samples are shown (n = 4). The aggregation process was accelerated when heparin was added at the initiation of aggregation. When heparin was added at 43 h, a continuous increase in the TfT fluorescence was detected in contrast to D187N gelsolin173-243 aggregated alone. TEM images of D187N gelsolin173-243 aggregated at 43 h (B), and at 90 h (C). (D) D187N gelsolin173-243 aggregated with heparin present from the start; image taken at 43 h. (E) Aggregated sample at 90 h after heparin is introduced at 43 h. Contrasts of the images shown were optimized using the auto-level function of Adobe Photoshop. Original images are presented in the supporting information.
Heparin induces the formation of gelsolin assembly intermediates
We monitored the aggregation process over time by analytical size exclusion chromatography coupled to dynamic light-scattering (DLS). In the absence of heparin or HS, the monomer peak (7500 ± 1300 g/mol by DLS) eluting at ∼13 mL decreased without the appearance of a peak corresponding to aggregates (Figure 7A), which is almost certainly the case because the aggregates formed quickly and were centrifuged/filtered away. In the samples with heparin or HS, larger aggregate species were observable in addition to the monomeric peak, and eluted in the void volume (∼7 mL) (Figure 7B, C). The calculated molar mass of the aggregated D187N gelsolin173-243 void volume peak was 3.252 (± 0.155) × 107 g/mol with HS (hydrodynamic radius (Rh) = 91 ± 1 nm) and 1.9 (± 0.104) × 107 g/mol with heparin (Rh = 47 ± 0.2 nm). Importantly, the LS data suggests a distribution of heterogeneous species in both the “monomer” and “aggregate” peaks in samples with GAGs implying that these GAGs may be able to mediate the association of monomeric gelsolin (supporting information). The rate of monomer disappearance quantified by UV absorbance was similar in all cases (Figure 7D). In contrast, the rate of large aggregate formation as discerned by the void volume peak was ∼2-fold higher in the presence of heparin/HS in comparison to gelsolin alone (Figure 7E). The conservation of mass was noted in the presence of heparin suggesting that the monomeric species aggregate into the soluble aggregate species. The explanation given above for the absence of mass balance in samples aggregated without heparin/HS is consistent with the previous observations of insoluble pellets upon centrifugation of samples, whereas pellets were not observed in samples with heparin. HS was not as effective as heparin at producing all soluble aggregates, thus the mass balance of the injected and eluted protein was not entirely conserved with HS in this experiment (Figure 7D, E).
Figure 7.
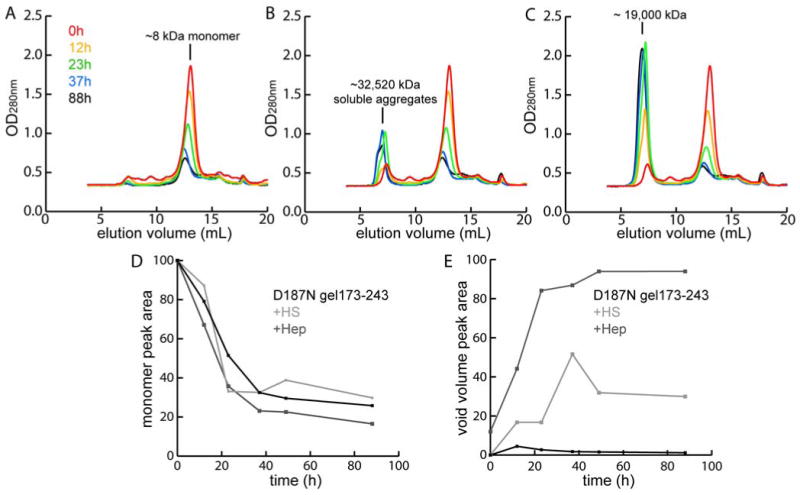
Overlaid gel filtration UV absorption traces (280 nm) of D187N gelsolin173-243 (36 μM) aggregation time course (37 °C, pH 7, constant rocking) in the absence of added GAGs (A), with HS (B) and with heparin (C) obtained at 0, 12, 23, 37 and 88 h. The monomer peak and the soluble aggregate peak (molar mass derived from light-scattering) are indicated. Fractions of monomers (D) and soluble aggregates (E) were calculated from UV absorption. The mass balance is poor in A (no aggregate peak growing in) because the aggregates are removed by the centrifugation/filtration step prior to FPLC injection.
Discussion
The process of amyloidogenesis depends on the concentration of aggregation prone species, the environment supporting deposition, and the presence of numerous accessory molecules which bind to amyloid fibrils. The biological distribution of some of these molecules may influence the tissue specific amyloid deposition and turnover distinguishing different human amyloid diseases. Herein, we provide evidence that GAGs, especially heparin, accelerate gelsolin amyloid formation and we provide insight into the mechanism by which GAGs could lead to the tissue specific deposition observed in gelsolin amyloidogenesis. First, we have shown that the GAGs heparin, HS and DS are the most effective accelerators of gelsolin amyloidogenesis. These are the dominant GAGs in the skin (37, 38) and therefore may explain the heavy gelsolin amyloid load there, which is distinct from other amyloidoses. While we used soluble GAGs, the majority of GAGs and the proteoglycans in tissue are immobilized and would template amyloidogenesis in the regions where they are localized.
Second, the hastening of β-sheet quaternary structure ultimately affording fibrils is consistent with the influence of GAGs on other amyloidogenic proteins (12, 22, 23). In gelsolin amyloidogenesis, this structural transition is accelerated by heparin. It is possible that heparin may directly promote structural transitions within monomeric peptides leading to accelerated aggregation and fibril formation, although at this time we have no direct evidence for this.
Third, we have identified that the sulfated structure of heparin is critical in its role as an accelerator of gelsolin amyloid formation. In particular, the O-sulfo groups of heparin appear to be important. A fully O-sulfated heparin analogue is the most potent accelerator identified heretofore, shortening the t50 by ∼4-fold. O-sulfation of heparin has known biological functions and is critical to promote the binding of basic fibroblast growth factor to its receptors and regulates its biological activity (39). Tissue-specific modifications of heparin (40-42) are likely to influence local deposition of amyloid and thus under certain circumstances can exacerbate pathology. It is known that chondroitin sulfates, which are sulfated on a single face of the polymer, dramatically enhance Aβ amyloidogenesis (22). In contrast, the chondroitin sulfates were less effective accelerators of gelsolin amyloidogenesis. The position and distribution of sulfates on a given GAG chain appear to be a crucial determinant for accelerating and enhancing certain types of amyloid formation. Such specific interactions between GAGs and different amyloidogenic peptides may explain, at least in part, the tissue selective pathology exhibited by most amyloidogenic polypeptides.
Fourth, we have shown that the effect of heparin depends on its molecular size. Larger heparin chains probably establish multiple interactions between a single heparin molecule and multiple gelsolin peptides, thereby increasing the capacity of heparin to affect amyloid formation. These results have implications for the development of therapeutic strategies since injected LMW heparin molecules are believed to antagonize the interaction between tissue-anchored GAGs and amyloid fibrils. Therefore, the LMW GAGs and small molecule mimetics thereof have been proposed to serve as potential therapeutic agents (15, 43-45). In the context of gelsolin amyloidogenesis, our results suggest that these LMW heparin molecules actually increase the rate of fibril formation implying that the injected GAGs themselves may accelerate gelsolin amyloidogenesis and thus, therapeutic uses of GAGs will need to be optimized by modulating their sizes and chemical composition.
Fifth, heparin is also effective at late stages of aggregation and induces the formation of well-defined elongated and non-laterally assembled fibrils. Our data suggests that during the earliest aggregation events, heparin and related GAGs may template the monomers or oligomers to associate, reducing the lag time for gelsolin fibrilization. Consistent with this suggestion, GAGs and proteoglycans seem to enhance the stability and persistence of amyloid by affording protection from proteolysis (24, 25). Although our studies have been focused on the mechanism of heparin as an accelerator of gelsolin amyloid formation, other GAGs and proteoglycans are likely to affect the amyloidogenesis of gelsolin and related peptides, which may explain the features of gelsolin-related amyloidosis including amyloid localization to the cranial nerves and to the cornea. Previous studies of gelsolin fibril formation under acidic conditions reported accelerated amyloid formation utilizing short peptides (7-30 residues) from the D187N and D187Y gelsolin variants relative to the WT D187 sequence (46). In the context of amyloidogenic gelsolin173-243 fragments observed in humans, the D187N fragment is only a slightly faster aggregator than the WT. Strikingly, the D187Y fragment does not form amyloid at neutral pH on a laboratory time scale (supporting information). Collectively, these results strongly suggest that it is the ability of the D187N/Y disease-associated gelsolin proteins to be aberrantly proteolyzed, not the enhanced amyloidogenecity of the fragments that result in their pathology. Moreover, the conditions employed in these in vitro studies may not accurately represent the chemical environment presented at the GAG interface where these peptides likely form amyloid in humans. The GAGs of the extracellular matrix and the cell surface, which are potent accelerators of amyloidogenesis could be an important determinant of amyloid homeostasis and may influence which tissues are adversely affected by the process of amyloidogenesis.
Supplementary Material
Acknowledgments
The authors thank Muyun Gao and Gina Dendle for help with protein purification, Ted Foss for analytical ultra centrifugation analysis, Sarah Siegel for AFM analyses, Wim D'Haeze and Evan Powers for helpful discussions, and M. R. Ghadiri for use of his AFM.
Footnotes
We thank the Skaggs Institute of Chemical Biology, the Lita Annenberg Hazen Foundation and the National Institutes of Health (grants: AG18917, HL62244, HL52622 and GM038060) for financial support.
Abbreviations:
Aβ Amyloid β peptide
ECM Extracellular Matrix
GAG Glycosaminoglycan
HS Heparan Sulfate
Chon Chondroitin Sulfate
DS Dermatan Sulfate
WT Wild type
DLS Dynamic Light Scattering
TEM Transmission Electron Microscopy
Supporting Information Available: (1) Pre-treated D187N gelsolin173-243 fragments were analyzed to confirm a monomeric starting structure and the analytical ultracentrifugation, atomic force microscopy analyses of these samples are shown. (2) WT and D187Y gelsolin173-243 fragments were subjected to amyloid formation plate-reader assays as controls. The amyloid formation profiles of WT gelsolin173-243 are shown in the presence of various GAGs and heparin derivatives. The D187Y gelsolin173-243 only aggregated at acidic (pH 5) conditions. (3) Dextran sulfate and polyglutamic acid did not accelerate D187N gelsolin173-243 amyloidogenesis in the TfT fluorescence assays shown. (4) Full, unadjusted TEM images of the images presented in Figures 6 and 7 are shown. (5) The heterogeneous distribution of molecular weights calculated from light scattering coupled to analytical size exclusion chromatography in gelsolin aggregation samples are shown. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Sekijima Y, Wiseman RL, Matteson J, Hammarstrom P, Miller SR, Sawkar AR, Balch WE, Kelly JW. The biological and chemical basis for tissue-selective amyloid disease. Cell. 2005;121:73–85. doi: 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 2.Kiuru S. Gelsolin-related familial amyloidosis, Finnish type (FAF), and its variants found worldwide. Amyloid. 1998;5:55–66. doi: 10.3109/13506129809007291. [DOI] [PubMed] [Google Scholar]
- 3.Kiuru-Enari S, Somer H, Seppalainen AM, Notkola IL, Haltia M. Neuromuscular pathology in hereditary gelsolin amyloidosis. J Neuropathol Exp Neurol. 2002;61:565–71. doi: 10.1093/jnen/61.6.565. [DOI] [PubMed] [Google Scholar]
- 4.Kiuru-Enari S, Keski-Oja J, Haltia M. Cutis laxa in hereditary gelsolin amyloidosis. Br J Dermatol. 2005;152:250–7. doi: 10.1111/j.1365-2133.2004.06276.x. [DOI] [PubMed] [Google Scholar]
- 5.Chen CD, Huff ME, Matteson J, Page L, Phillips R, Kelly JW, Balch WE. Furin initiates gelsolin familial amyloidosis in the Golgi through a defect in Ca(2+) stabilization. EMBO J. 2001;20:6277–87. doi: 10.1093/emboj/20.22.6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kangas H, Seidah NG, Paunio T. Role of proprotein convertases in the pathogenic processing of the amyloidosis-associated form of secretory gelsolin. Amyloid. 2002;9:83–7. [PubMed] [Google Scholar]
- 7.Page LJ, Suk JY, Huff ME, Lim HJ, Venable J, Yates J, III, Kelly JW, Balch WE. Metalloprotease Cleavage within the Extracellular Matrix Triggers Gelsolin Amyloidogenesis. EMBO J. 2005;24:4124–4132. doi: 10.1038/sj.emboj.7600872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de la Chapelle A, Tolvanen R, Boysen G, Santavy J, Bleeker-Wagemakers L, Maury CP, Kere J. Gelsolin-derived familial amyloidosis caused by asparagine or tyrosine substitution for aspartic acid at residue 187. Nat Genet. 1992;2:157–60. doi: 10.1038/ng1092-157. [DOI] [PubMed] [Google Scholar]
- 9.Maury CP. Gelsolin-related amyloidosis. Identification of the amyloid protein in Finnish hereditary amyloidosis as a fragment of variant gelsolin. J Clin Invest. 1991;87:1195–9. doi: 10.1172/JCI115118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Horssen J, Wesseling P, van den Heuvel LP, de Waal RM, Verbeek MM. Heparan sulphate proteoglycans in Alzheimer's disease and amyloid-related disorders. Lancet Neurol. 2003;2:482–92. doi: 10.1016/s1474-4422(03)00484-8. [DOI] [PubMed] [Google Scholar]
- 11.Diaz-Nido J, Wandosell F, Avila J. Glycosaminoglycans and beta-amyloid, prion and tau peptides in neurodegenerative diseases. Peptides. 2002;23:1323–32. doi: 10.1016/s0196-9781(02)00068-2. [DOI] [PubMed] [Google Scholar]
- 12.Ancsin JB. Amyloidogenesis: historical and modern observations point to heparan sulfate proteoglycans as a major culprit. Amyloid. 2003;10:67–79. doi: 10.3109/13506120309041728. [DOI] [PubMed] [Google Scholar]
- 13.Castillo GM, Lukito W, Wight TN, Snow AD. The sulfate moieties of glycosaminoglycans are critical for the enhancement of beta-amyloid protein fibril formation. J Neurochem. 1999;72:1681–7. doi: 10.1046/j.1471-4159.1999.721681.x. [DOI] [PubMed] [Google Scholar]
- 14.Kisilevsky R. Review: amyloidogenesis-unquestioned answers and unanswered questions. J Struct Biol. 2000;130:99–108. doi: 10.1006/jsbi.2000.4222. [DOI] [PubMed] [Google Scholar]
- 15.Yang DS, Serpell LC, Yip CM, McLaurin J, Chrishti MA, Horne P, Boudreau L, Kisilevsky R, Westaway D, Fraser PE. Assembly of Alzheimer's amyloid-beta fibrils and approaches for therapeutic intervention. Amyloid. 2001;8 1:10–9. [PubMed] [Google Scholar]
- 16.Yamamoto S, Yamaguchi I, Hasegawa K, Tsutsumi S, Goto Y, Gejyo F, Naiki H. Glycosaminoglycans enhance the trifluoroethanol-induced extension of beta 2-microglobulin-related amyloid fibrils at a neutral pH. J Am Soc Nephrol. 2004;15:126–33. doi: 10.1097/01.asn.0000103228.81623.c7. [DOI] [PubMed] [Google Scholar]
- 17.Hileman RE, Fromm JR, Weiler JM, Linhardt RJ. Glycosaminoglycan-protein interactions: definition of consensus sites in glycosaminoglycan binding proteins. Bioessays. 1998;20:156–67. doi: 10.1002/(SICI)1521-1878(199802)20:2<156::AID-BIES8>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 18.Rabenstein DL. Heparin and heparan sulfate: structure and function. Nat Prod Rep. 2002;19:312–31. doi: 10.1039/b100916h. [DOI] [PubMed] [Google Scholar]
- 19.Powell AK, Yates EA, Fernig DG, Turnbull JE. Interactions of heparin/heparan sulfate with proteins: appraisal of structural factors and experimental approaches. Glycobiology. 2004;14:17R–30R. doi: 10.1093/glycob/cwh051. [DOI] [PubMed] [Google Scholar]
- 20.Castillo GM, Cummings JA, Yang W, Judge ME, Sheardown MJ, Rimvall K, Hansen JB, Snow AD. Sulfate content and specific glycosaminoglycan backbone of perlecan are critical for perlecan's enhancement of islet amyloid polypeptide (amylin) fibril formation. Diabetes. 1998;47:612–20. doi: 10.2337/diabetes.47.4.612. [DOI] [PubMed] [Google Scholar]
- 21.van Horssen J, Wilhelmus MM, Heljasvaara R, Pihlajaniemi T, Wesseling P, de Waal RM, Verbeek MM. Collagen XVIII: a novel heparan sulfate proteoglycan associated with vascular amyloid depositions and senile plaques in Alzheimer's disease brains. Brain Pathol. 2002;12:456–62. doi: 10.1111/j.1750-3639.2002.tb00462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McLaurin J, Franklin T, Zhang X, Deng J, Fraser PE. Interactions of Alzheimer amyloid-beta peptides with glycosaminoglycans effects on fibril nucleation and growth. Eur J Biochem. 1999;266:1101–10. doi: 10.1046/j.1432-1327.1999.00957.x. [DOI] [PubMed] [Google Scholar]
- 23.Cohlberg JA, Li J, Uversky VN, Fink AL. Heparin and other glycosaminoglycans stimulate the formation of amyloid fibrils from alpha-synuclein in vitro. Biochemistry. 2002;41:1502–11. doi: 10.1021/bi011711s. [DOI] [PubMed] [Google Scholar]
- 24.Gupta-Bansal R, Frederickson RC, Brunden KR. Proteoglycan-mediated inhibition of Abeta proteolysis. A potential cause of senile plaque accumulation. J Biol Chem. 1995;270:18666–71. doi: 10.1074/jbc.270.31.18666. [DOI] [PubMed] [Google Scholar]
- 25.Cotman SL, Halfter W, Cole GJ. Agrin binds to beta-amyloid (Abeta), accelerates abeta fibril formation, and is localized to Abeta deposits in Alzheimer's disease brain. Mol Cell Neurosci. 2000;15:183–98. doi: 10.1006/mcne.1999.0816. [DOI] [PubMed] [Google Scholar]
- 26.Shaffer LM, Dority MD, Gupta-Bansal R, Frederickson RC, Younkin SG, Brunden KR. Amyloid beta protein (Abeta) removal by neuroglial cells in culture. Neurobiol Aging. 1995;16:737–45. doi: 10.1016/0197-4580(95)00055-j. [DOI] [PubMed] [Google Scholar]
- 27.Coombe DR, Kett WC. Heparan sulfate-protein interactions: therapeutic potential through structure-function insights. Cell Mol Life Sci. 2005;62:410–24. doi: 10.1007/s00018-004-4293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ratnaswamy G, Koepf E, Bekele H, Yin H, Kelly JW. The amyloidogenicity of gelsolin is controlled by proteolysis and pH. Chem Biol. 1999;6:293–304. doi: 10.1016/S1074-5521(99)80075-1. [DOI] [PubMed] [Google Scholar]
- 29.Naiki H, Higuchi K, Hosokawa M, Takeda T. Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavin T1. Anal Biochem. 1989;177:244–9. doi: 10.1016/0003-2697(89)90046-8. [DOI] [PubMed] [Google Scholar]
- 30.Yates EA, Santini F, Guerrini M, Naggi A, Torri G, Casu B. 1H and 13C NMR spectral assignments of the major sequences of twelve systematically modified heparin derivatives. Carbohydr Res. 1996;294:15–27. doi: 10.1016/s0008-6215(96)90611-4. [DOI] [PubMed] [Google Scholar]
- 31.Toida T, Maruyama T, Ogita Y, Suzuki A, Toyoda H, Imanari T, Linhardt RJ. Preparation and anticoagulant activity of fully O-sulphonated glycosaminoglycans. Int J Biol Macromol. 1999;26:233–41. doi: 10.1016/s0141-8130(99)00088-4. [DOI] [PubMed] [Google Scholar]
- 32.Pervin A, Gallo C, Jandik KA, Han XJ, Linhardt RJ. Preparation and structural characterization of large heparin-derived oligosaccharides. Glycobiology. 1995;5:83–95. doi: 10.1093/glycob/5.1.83. [DOI] [PubMed] [Google Scholar]
- 33.Hurshman AR, White JT, Powers ET, Kelly JW. Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry. 2004;43:7365–81. doi: 10.1021/bi049621l. [DOI] [PubMed] [Google Scholar]
- 34.Maury CP, Nurmiaho-Lassila EL, Boysen G, Liljestrom M. Fibrillogenesis in gelsolin-related familial amyloidosis. Amyloid. 2003;10 1:21–5. [PubMed] [Google Scholar]
- 35.Capila I, Linhardt RJ. Heparin-protein interactions. Angew Chem Int Ed Engl. 2002;41:391–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 36.Esko JD, Lindahl U. Molecular diversity of heparan sulfate. J Clin Invest. 2001;108:169–73. doi: 10.1172/JCI13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Horiguchi Y, Couchman JR, Ljubimov AV, Yamasaki H, Fine JD. Distribution, ultrastructural localization, and ontogeny of the core protein of a heparan sulfate proteoglycan in human skin and other basement membranes. J Histochem Cytochem. 1989;37:961–70. doi: 10.1177/37.7.2659664. [DOI] [PubMed] [Google Scholar]
- 38.Trowbridge JM, Gallo RL. Dermatan sulfate: new functions from an old glycosaminoglycan. Glycobiology. 2002;12:117R–25R. doi: 10.1093/glycob/cwf066. [DOI] [PubMed] [Google Scholar]
- 39.Loo BM, Kreuger J, Jalkanen M, Lindahl U, Salmivirta M. Binding of heparin/heparan sulfate to fibroblast growth factor receptor 4. J Biol Chem. 2001;276:16868–76. doi: 10.1074/jbc.M011226200. [DOI] [PubMed] [Google Scholar]
- 40.Maccarana M, Sakura Y, Tawada A, Yoshida K, Lindahl U. Domain structure of heparan sulfates from bovine organs. J Biol Chem. 1996;271:17804–10. doi: 10.1074/jbc.271.30.17804. [DOI] [PubMed] [Google Scholar]
- 41.Dennissen MA, Jenniskens GJ, Pieffers M, Versteeg EM, Petitou M, Veerkamp JH, van Kuppevelt TH. Large, tissue-regulated domain diversity of heparan sulfates demonstrated by phage display antibodies. J Biol Chem. 2002;277:10982–6. doi: 10.1074/jbc.M104852200. [DOI] [PubMed] [Google Scholar]
- 42.Netelenbos T, Drager AM, van het Hof B, Kessler FL, Delouis C, Huijgens PC, van den Born J, van Dijk W. Differences in sulfation patterns of heparan sulfate derived from human bone marrow and umbilical vein endothelial cells. Exp Hematol. 2001;29:884–93. doi: 10.1016/s0301-472x(01)00653-1. [DOI] [PubMed] [Google Scholar]
- 43.Kisilevsky R, Szarek WA, Ancsin JB, Elimova E, Marone S, Bhat S, Berkin A. Inhibition of amyloid A amyloidogenesis in vivo and in tissue culture by 4-deoxy analogues of peracetylated 2-acetamido-2-deoxy-alpha- and beta-d-glucose: implications for the treatment of various amyloidoses. Am J Pathol. 2004;164:2127–37. doi: 10.1016/s0002-9440(10)63771-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rose M, Dudas B, Cornelli U, Hanin I. Glycosaminoglycan C3 protects against AF64A-induced cholinotoxicity in a dose-dependent and time-dependent manner. Brain Res. 2004;1015:96–102. doi: 10.1016/j.brainres.2004.04.048. [DOI] [PubMed] [Google Scholar]
- 45.Zhu H, Yu J, Kindy MS. Inhibition of amyloidosis using low-molecular-weight heparins. Mol Med. 2001;7:517–22. [PMC free article] [PubMed] [Google Scholar]
- 46.Maury CP, Nurmiaho-Lassila EL, Rossi H. Amyloid fibril formation in gelsolin-derived amyloidosis. Definition of the amyloidogenic region and evidence of accelerated amyloid formation of mutant Asn-187 and Tyr-187 gelsolin peptides. Lab Invest. 1994;70:558–64. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.