

Abstract
Cyclin-dependent kinase 4 (CDK4)/cyclin D complexes are expressed early in the G1 phase of the cell cycle and stimulate the expression of genes required for G1 progression by phosphorylation of the product of the retinoblastoma gene, pRb. To elaborate the molecular pathway of CDK4 activation and substrate selection we have determined the structure of nonphosphorylated CDK4/cyclin D3. This structure of an authentic CDK/cyclin complex shows that cyclin binding may not be sufficient to drive the CDK active site toward an active conformation. Phosphorylated CDK4/cyclin D3 is active as a pRb kinase and is susceptible to inhibition by p27Kip1. Unlike CDK2/cyclin A, CDK4/cyclin D3 can be inactivated by treatment with λ-phosphatase, implying that phosphorylated T172 is accessible to a generic phosphatase while bound to a cyclin. Taken together, these results suggest that the structural mechanism of CDK4/cyclin D3 activation differs markedly from that of previously studied CDK/cyclin complexes.
Keywords: cell cycle, kinase, phosphorylation, X-ray crystallography, G1 phase
The transition of cells through the early G1 stage of the cell cycle is coordinated by the activities of cyclin-dependent kinase 4 (CDK4) and CDK6 complexes that are formed after the mitogen-dependent expression of cyclin D. CDK4/cyclin D and CDK6/cyclin D stimulate the expression of genes required for G1 progression by phosphorylation of the product of the retinoblastoma gene, pRb (reviewed in ref. 1). In wild-type cells it is considered that CDK2, which is found associated first with cyclin E and then with cyclin A, subsequently coordinates events as cells progress from G1 through S phase (reviewed in ref. 2). This model of sequential CDK activation controlling cell cycle progression has been elaborated by studies of CDK and cyclin knockout mice that showed that various mouse cell types derived from knockout animals are able to proliferate in the absence of several CDKs. This work further revealed that, although the CDK and cyclin families exhibit considerable functional redundancy, certain cell cycle CDKs have essential cell-type specific roles through their ability to phosphorylate specific substrates (3) or bind to specific cyclins (2).
The principal CDK substrate during early G1 is pRb. Sequential CDK-dependent pRb phosphorylation results in dissociation of pRb-transcription regulator complexes and concomitant enhancement of expression of genes whose products are required for G1 progression (4, 5). pRb is initially phosphorylated on multiple sites by CDKs 4 and 6 and then additionally phosphorylated by CDK2 bound to cyclins E and A (6–8). Few authentic CDK4 substrates have been identified: CDK4 phosphorylates members of the pRb family (9) and the transcription factor Smad3 (10) but does not phosphorylate the canonical CDK substrate histone H1 (9, 11, 12) or the cyclin-dependent kinase inhibitor (CKI) p27Kip1 (13).
A model for CDK regulation by cyclin and CKI binding and by phosphorylation has been developed from a series of structures of CDK2/cyclin A complexes (14–18). These structures have also revealed the molecular interactions that dictate substrate sequence requirements within the CDK substrate-peptide binding sites around the site of phosphotransfer and at the cyclin recruitment site (16, 19). Monomeric CDK2 is inactive as a result of the disposition of the C-helix and the activation segment (20). Cyclin A binding and T160 phosphorylation result in rearrangements within the CDK2 active site that correctly orientate key ATP binding and catalytic residues and create the peptide substrate binding site. For those CDKs that bind to cyclins A, B, E and D, additional interactions at the cyclin “recruitment site” (21) contribute to substrate selectivity. This hydrophobic patch binds the motif R/KXLX[0–1]Φ (where X denotes any residue and Φ denotes an hydrophobic residue) that is present in certain recruited substrates and in the Cip/Kip class of CKIs (16, 19). However, additional factors play a role in dictating CDK cellular activity and these must be particularly significant for CDK4 given its apparently strict substrate preference.
Subsequent determination of the structures of CDK5/p25 (22), CDK6/V-cyclin (23), and CDK9/cyclin T (24) have revealed significant structural differences among the CDKs, suggesting that their mechanisms of activation and regulation have diverged with expansion of the family. This might be particularly the case for CDK4 where early in vitro and cellular studies revealed that, unlike CDK1/ and CDK2/cyclin complexes, ectopically expressed CDK4 and cyclin D do not readily associate in quiescent cells, and a mitogen-dependent step is required to assemble them into active complexes (25–27). Notably, CDK4 is one of a number of protein kinases for which association with the Hsp90 chaperone system is an essential step in activation (reviewed in ref. 28). It has also been proposed that CDK4–cyclin D association is promoted by binding to members of the p21/p27/p57 Cip/Kip family, suggesting an additional role for this CKI family as assembly factors (29, 30). In one study, p21 and p27 binding were found to increase the Ka for CDK4–cyclin D association by 35- and 80-fold, respectively (29).
To understand the pathway of CDK4/cyclin D3 activation we have determined the structure of CDK4/cyclin D3. This structure is of the nonphosphorylated form of the complex, which has no measurable catalytic activity. The structure explains this lack of activity, by revealing that cyclin D3 binding does not bring about rearrangement of the CDK active site. However, phosphorylated CDK4/cyclin D3, as purified from insect cells, is active as a pRb kinase and we show that it is a substrate for λ phosphatase. Taken together, these results confirm that structural peculiarities of the CDK4/cyclin D3 complex compared with other CDK/cyclin complexes are reflected in different substrate selectivity and different phosphatase-accessibility of the activation-segment threonine.
Results
CDK4/Cyclin D3 Can Be Expressed in Insect Cells as an Active Complex That Highly Depends on Cyclin-Mediated Recruitment for Substrate Selection.
Full-length CDK4 and cyclin D3 (Fig. S1) were coexpressed in insect cells as N-terminally tagged GST and flag-tagged fusion proteins, respectively. After purification of the complex by affinity chromatography, the GST tag was removed by treatment with protease (Fig. S2A). Initial analysis by nonequilibrium pH-gradient electrophoresis (NEpHGE) revealed that the CDK4/cyclin D3 was a mixture of phosphorylated and nonphosphorylated CDK4/cyclin D3 complexes (Fig. S3A). Quantitative analysis of the corresponding gel indicated that the preparation used in crystallization contained ≈62% phosphorylated and 38% unphosphorylated CDK4. Liquid chromatography (LC)/MS/MS analysis of CDK4/cyclin D3 (Fig. S3B), which yielded a high degree of sequence coverage (89.1%), indicated that the only CDK4 site to be phosphorylated is T172 (Fig. S4). As expected, CDK4/cyclin D3 phosphorylates a C-terminal fragment of pRb and is inhibited by the CKI p27Kip1 (Fig. S2 B and C). Unlike fully activated CDK2/cyclin A phosphorylated on T160 (T160pCDK2/cyclin A), CDK4/cyclin D3 does not phosphorylate histone H1 (Fig. S2B).
The activity of CDK1 or CDK2 toward recruited substrates has been shown to depend on the identity of the associated cyclin subunit, the local epitope around the site of phospho-transfer (respectively, either a canonical S/TPXR/K or noncanonical S/TPXX motif) (31, 32), and the distance between the RXLΦ and S/TPXK/R motifs (33). In particular, the RXL motif has been found to make a poor CDK substrate a better one, an effect that is enhanced if the substrate lacks a basic residue at P+3 (refs. 31 and 34 and references therein and ref. 32).
To assess the role of the RXL motif in directing substrate specificity in the context of either a noncanonical or canonical substrate sequence, we used as CDK substrates a set of GST fusions of fragments of p107 in which the native noncanonical phosphorylation site at S640 (S640PIS) and the native KRRLF recruitment motif were present or had been changed to SPIR (canonical) or AAAAF (nonrecruited) motifs, respectively (Fig. 1B). Unlike CDK2/cyclin A and CDK2/cyclin B (32), the activity of CDK4/cyclin D3 heavily depends on the presence of a substrate recruitment motif and, unlike the case for CDK2/cyclin A, even the introduction of a canonical sequence around the site of phosphotransfer does not remove this dependence. These results are consistent with the characteristics of previously identified authentic substrates of CDK4, although in vivo substrate selection might also depend on additional CDK4/cyclin D3–substrate interactions that remain to be identified.
Fig. 1.

Structure and substrate selection in CDK4/cyclin D3. (A) The fold of CDK4/cyclin D3 (left side) is compared with that of CDK2/cyclin A in complex with a substrate peptide (PDB ID code 1QMZ; right side). Important structural and regulatory elements referred to in the text are indicated on each structure and correspond to the CDK N-lobe (light gray), the C-helix (cyan), the CDK C-lobe (yellow), the activation segment (red), and the cyclin subunit (green). (B) The activity of CDK4/cyclin D3 toward pRb family members highly depends on substrate recruitment. See Results for details.
CDK4/Cyclin D3 Crystallizes as an Intimate Dimer of Binary Complexes.
Crystals of CDK4/cyclin D3 were grown by vapor diffusion, using a reservoir solution containing 0.1 M Hepes (pH 6.6), ≈15% PEG3350, and ≈10% Tacsimate. Although the majority of crystals diffracted to <3.5-Å resolution we were able to collect a complete dataset to 3.0-Å resolution from a single crystal by screening ≈60 samples at the synchrotron. The structure of CDK4/cyclin D3 was solved by molecular replacement using an ensemble of CDK and cyclin structures in the search model. The asymmetric unit of this crystal form contains 2 binary complexes (Fig. S5) in which the 2 independent copies of the CDK4/cyclin D complex are essentially superimposable and as such were refined with tight noncrystallographic symmetry restraints. Clear electron density defines the conformation of CDK4 residues 5–295 and cyclin D3 residues 23–254, and the structure is compared with that of CDK2/cyclin A phosphorylated on T160 (T160pCDK2/cyclin A) in Fig. 1. Although adenylyl imidodiphosphate (AMP-PNP) was present in the mother liquor, there is no electron density for bound nucleotide in the CDK4 active site.
CDK4 adopts the characteristic protein kinase fold, composed of an N-lobe that is primarily formed from β-sheet and a C-lobe that is primarily α-helical. Unexpectedly, the conformation of the C-helix and the activation segment resemble more closely the inactive monomeric form of CDK2 than either partially activated cyclin-bound CDK2 or fully activated cyclin-bound and T160 phosphorylated CDK2 (Figs. 1 and 2). Fig. 1 shows that cyclin D3 binds somewhat differently to CDK4 than does cyclin A to CDK2: cyclin D3 adopts a more elevated binding position, resulting in a significantly smaller interface between CDK4 and cyclin D3 in the binary complex than is the case in CDK2/cyclin A.
Fig. 2.

Comparison of the structures of monomeric CDK2, CDK4/cyclin D3, and T160pCDK2/cyclin A. The 3 structures are viewed looking down onto the CDK C-terminal domain from the N-terminal domain and are colored according to the scheme used in Fig. 1. (A) Monomeric CDK2. (B) CDK4/cyclin D3. (C) T160pCDK2/cyclin A. The figure highlights the displacement of the CDK4/cyclin D3 activation segment away from the active conformation present in T160pCDK2/cyclin A and its similarity to the structure of the activation segment in monomeric CDK2 (monomeric T160pCDK2 PDB ID code 1HCK).
The cyclin D3 structure contains 2 copies of a cyclin box fold (CBF) and an N-terminal helix that can be aligned closely with the corresponding secondary structural elements in cyclin A. In cyclin A an ordered C-terminal extension (residues 402–432) wraps around the surface of the C-terminal CBF. The equivalent protein sequence of cyclin D3 (residues 255–C terminus) is not apparent in the electron density, presumably because of disorder. Although this disorder may be an intrinsic property of cyclin D3, it is promoted in these crystals by the fact that the surface of the C-terminal CBF against which the C-terminal tail would be expected to pack is seen here to mediate an intimate cyclin–cyclin dimerization (discussed below).
The C-terminal helices in cyclins A, B, and E appear to have no function, but in cyclin T, the cyclin that associates with CDK9 to form the complex pTEFb that regulates the elongation phase of transcription (reviewed in ref. 35), it contributes to the recognition of regulatory proteins such as HEXIM (36) and HIV Tat (37). The structures of CDK9/cyclin T1 and monomeric cyclin T2 (24) have been solved recently, and a comparative analysis shows that the cyclin C-terminal helix is also pliable and adopts different positions in the CDK9-bound and -unbound structures. This pliability has been proposed to be important for pTEFb function. By analogy, the flexibility of the cyclin D3 C-terminal helix, evidenced by its ability to unfold from the cyclin C-terminal CBF, may reflect a protein binding or regulatory role.
The cyclin D3–cyclin D3 dimerization referred to above is mediated by an extensive interface that, according to analysis by the PISA server (38), resembles the interfaces of authentic homodimers. Indeed, whereas the CDK4/cyclin D3 interface buries an area of 2,250 Å2, the cyclin D3–cyclin D3 interface is more extensive (2,880 Å2). To test whether CDK4 cyclin D3 exists as a dimer in solution we analyzed CDK4/cyclin D3 preparations by means of multiangle laser light scattering in line with gel filtration. This method showed that the CDK4/cyclin D3 complex has a solution molecular mass of 69.5 kDa and therefore is not a dimer of dimers in solution (Fig. S5C). Hence, the apparent dimerization observed in the crystals is probably a consequence of the high concentrations used in crystallization. We note, however, that such high concentrations can recapitulate the consequences of elevated protein concentrations in cellular compartments [as is the case for the kinase domain of the EGF receptor (39)], and so we do not exclude the possibility that cellular CDK4/cyclin D3 might exist in part as a dimer.
T172-Unphosphorylated CDK4/Cyclin D3 Lacks Key Structural Features Required for Catalysis.
Comparison of the inactive structure of monomeric, unphosphorylated CDK2 (20) with the structures of unphosphorylated CDK2 in complex with cyclin A (14), phosphorylated CDK2/cyclin A (15), and phosphorylated monomeric CDK2 (17) established that (i) that cyclin A association is necessary and sufficient to impose an active conformation on the C-helix of CDK2 (“C-helix in”) and pull the activation segment into a nearly active conformation [that displays ≈0.1% of full activity (40)], (ii) phosphorylation of T160 in monomeric CDK2 destabilizes the inactive conformation of the activation segment, and (iii) phosphorylation of T160 in CDK2/cyclin A refines the conformation of the activation segment so as to complete the substrate recognition site. The structure of a ternary T160pCDK2/cyclin A/peptide substrate complex showed that formation of a Michaelis complex with the substrate entailed little rearrangement of CDK2 beyond that already induced by cyclin association and phosphorylation of T160 (19).
In the case of CDK4/cyclin D3, however, it seems that association with the cyclin subunit does not rearrange the activation segment of CDK4. Fig. 2 compares the activation segment conformation of CDK4 in the CDK4/cyclin D3 complex with that in monomeric CDK2 and T160pCDK2/cyclin A. In monomeric CDK2 (Fig. 2A), the activation segment begins with a short αL12 helix, continues with a loop that contacts the glycine-rich loop in the N-terminal kinase domain, and then returns to the C-terminal domain at a short helical segment that contains the APE motif. In the T160pCDK2/cyclin A structure (Fig. 2C), αL12 is melted, directing the activation segment away from the core of the CDK2 molecule so as to contact a patch of cyclin A, formed from parts of the C-terminal CBF and the N-terminal helix of the cyclin. In this conformation, the C-terminal end of the activation segment presents a binding site for peptide substrates. By contrast, the activation segment of CDK4/cyclin D3 (Fig. 2B) differs markedly from an apparently active conformation. First, the peptide region that corresponds to αL12 has a helical conformation that directs the polypeptide chain away from the cyclin partner to form an additional α-helix. After this short helix, the structure rejoins the CDK fold, although the peptide-binding site is clearly malformed. As a result, the 3 arginine residues (R55, R139, and R163) that are predicted to be structurally equivalent to CDK2 residues R50, R126, and R150 are disparately located and do not form the spokes of a structural hub as observed in the T160pCDK2/cyclin A structure (Fig. S6). T172, the residue equivalent to CDK2 T160, does not appear to be phosphorylated in the CDK4/cyclin D3 structure and is solvent accessible.
Fig. 2 also illustrates the differences between monomeric CDK2, CDK4/cyclin D3, and T160pCDK2/cyclin A in the conformation of the C-helix. The striking observation is that, unlike cyclin A, cyclin D3 does not impose an active C-helix in conformation upon its cognate CDK (Fig. 2B). In CDK2, the “melting” of the short αL12 helix, located at the start of the activation segment, generates space that is required for the C-helix to adopt its active conformation (Fig. 2C). In CDK4 bound to cyclin D3, this peptide sequence retains an approximately helical conformation and its location and the effect on the CDK4 structure is more reminiscent of the equivalent sequence in monomeric CDK2 (Fig. 2A).
Comparison of the Structure of CDK4/Cyclin D3 with Other CDK/Cyclin Complexes.
The CDK4/cyclin D3 interface is far less extensive than that present within the T160pCDK2/cyclin A complex (Fig. 3). The CDK2/cyclin A interface is formed from the CDK N-lobe, C-helix, C-lobe, and activation segment, and the cyclin N-terminal helix, N-terminal CBF, and C-terminal CBF (Fig. 3 C and D). By contrast, the CDK4/cyclin D3 interface is dominated by contacts between the cyclin D3 N-terminal CBF and the CDK4 N-terminal fold, centered on the C-helix (Fig. 3 A and B). In this regard the structure of CDK4/cyclin D3 (Fig. 4A) is reminiscent of CDK9/cyclin T1 (Fig. S7B) that also has the cyclin subunit disposed so as to form an interface with the CDK that is relatively small and primarily mediated through interactions between the CDK N-terminal domain and the cyclin N-terminal CBF. Significantly it does not involve interactions between the CDK9 C-terminal domain and the cyclin N-terminal helix (24). Despite these differences however, key interactions between cyclin T1 α5 and the CDK9 C-helix are conserved and, as a result, CDK9 is in the C-helix in conformation and hence active (Fig. S7B).
Fig. 3.

Comparison of interfaces involved in the structure of CDK4/cyclin D3 with those involved in the T160pCDK2/cyclin A structure. (A and B) CDK4/cyclin D3. (C and D) T160pCDK2/cyclin A. Shown are the origins of residues on each subunit that compose the CDK/cyclin interface. Peeling the 2 molecules apart reveals that the CDK2/cyclin A and CDK4/cyclin D3 interfaces are very different. Whereas the CDK2/cyclin A interface is extensive and involves residues from both the CDK2 and cyclin A N- and C-terminal folds (C and D), the CDK2 C-helix (C) and activation segment (C) and the cyclin A N-terminal helix (D), that of CDK4/cyclin D3 (A and B) is much more confined and involves a more limited set of residues from the CDK4 and cyclin D3 N-terminal folds and the cyclin D3 N-terminal helix. The CDK N- and C-terminal folds are colored light gray and yellow, respectively, with the C-helix in cyan, and the activation segment in red. The cyclin N- and C- terminal CBFs are colored green and blue, respectively, and the N-terminal helix is colored gray and the C-terminal is helix dark gray. T160pCDK2/cyclin A is PDB code 1QMZ.
Fig. 4.
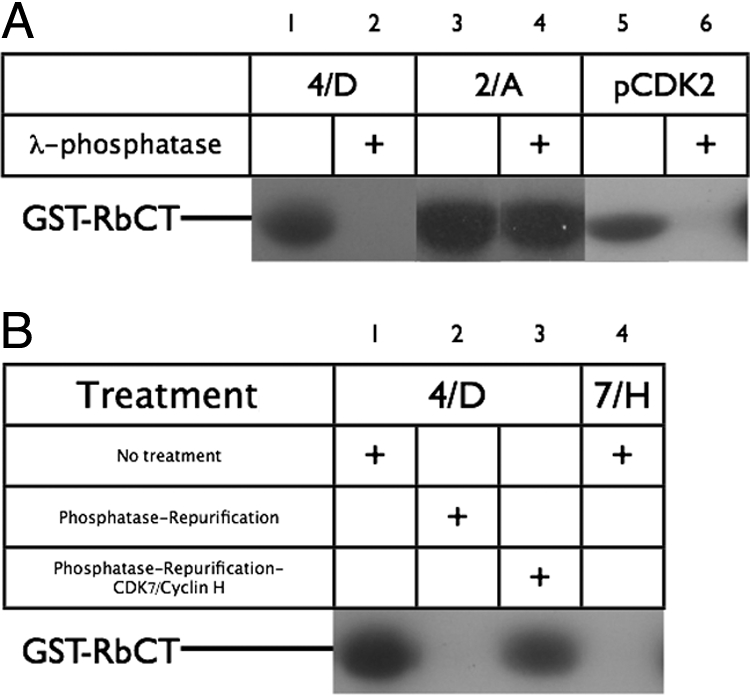
Modulation of CDK complex activity by λ-phosphatase and CDK7/cyclin H. (A) Kinase activity of different CDK-containing species toward GST-pRbCT was assayed by monitoring 32P incorporation either before (lanes 1, 3, and 5) or after (lanes 2, 4, and 6) treatment with λ-phosphatase. See Materials and Methods for further details. (B) Reactivation of repurified dephosphorylated CDK4/cyclin D3 by CDK7/cyclin H. Partially phosphorylated preparations of CDK4/cyclin D3 were assayed either as purified from insect cells (lane 1) or as repurified after treatment with λ-phosphatase (lanes 2 and 3). Repurified material was either not treated (lane 2) or treated (lane 3) with CDK7/cyclin H. Potential GST-pRbCT kinase activity in the CDK7/cyclin H is controlled against in lane 4.
Similarly, despite notable differences in the nature of the CDK–cyclin interface, CDK6 bound to a cyclin derived from herpesvirus saimiri (Vcyclin) is in the C-helix in active conformation and adopts an activation segment conformation compatible with peptide substrate binding despite not being phosphorylated (23). Thus, CDK6 in that complex differs from CDK4 in complex with cyclin D3, a difference that results either from differences between CDK4 and CDK6 (proteins closely related in sequence) or from peculiar structural properties of CDK6 when “hijacked” by a pathogenic activator.
By contrast, CDK6 in a ternary complex composed additionally of a Kaposi's sarcoma-associated herpesvirus D-type cyclin and the CDK inhibitor p18Ink4c adopts an inactive conformation (Fig. S7C and ref. 41). That CDK6 adopts an inactive conformation in this complex was proposed to result from the presence of the INK subunit that distorts the CDK ATP binding site and therefore weakens the CDK–cyclin interface, thus favoring the CDK6 “C-helix out” conformation (41). By analogy with the structure described here, however, such a conformation might be adopted in CDK6–cyclin complexes even when the INK subunit is not present. The inhibitory activity of the INK subunit would then result from its ability to bind to and stabilize this inactive conformation, thus preventing the transient adoption of an active conformation that we deduce must occur during the CDK4/6 catalytic cycle. In summary, and in contrast to all other binary CDK/cyclin complexes for which structures have been determined to date, cyclin D3 binding to CDK4 does not impose an active CDK4 conformation.
Dephosphorylation of T172 of CDK4/Cyclin D3 Can Be Readily Achieved by λ Phosphatase, Yielding an Entirely Inactive Complex.
As the CDK4/cyclin D3 complex used for crystallization trials had readily detectable kinase activity but the structure appeared to be that of a nonphosphorylated CDK4/cyclin D3 complex, we set out to establish the phosphorylation state of the CDK4/cyclin D3 present in the crystals. Crystals were harvested, extensively washed in mother liquor, and then dissolved before being subjected to thorough trypsinolysis and subsequent mass spectrometry. This analysis revealed the presence of a peptide with a molecular mass consistent with the predicted tryptic peptide that spans T172 in the activation segment. No evidence could be found for ions corresponding to the phosphorylated form of that peptide. Hence, the structure described here corresponds to that of CDK4/cyclin D3 that is not phosphorylated on the activation segment.
To characterize the phosphorylation state of the CDK4/cyclin D3 preparation used in crystallization trials we subjected it to analysis by NEpHGE (Fig. S3A). Incubation of the preparation with λ phosphatase caused a change in the pattern of bands apparent on the Coomassie-stained NEpHGE gel: the band corresponding to phosphorylated CDK4 disappeared, but not the band corresponding to unphosphorylated CDK4. We next undertook quantitative phosphorylation of CDK4 by coincubation with human CDK-activating kinase (CAK, CDK7/cyclin H) and ATP. This treatment brought about the anticipated redistribution of species apparent in the NEphGE gel: the band corresponding to unphosphorylated CDK4 disappeared, whereas the band that corresponded to phosphorylated CDK4 did not.
Having established that the activation segment phosphorylation state of CDK4 in the CDK4/cyclin D3 complex could be manipulated by using λ phosphatase and CDK7/cyclin H, we set out to determine the level of kinase activity associated with the fully phosphorylated and fully dephosphorylated species. Initially we looked for activity of the preparation against a fragment of pRb (GST-RbCT, residues 792–928) that contains 5 canonical CDK substrate sequences and a recruitment motif. T160pCDK2/cyclin A and monomeric T160pCDK2 were analyzed in a similar fashion to provide controls for which the behavior in such assays has been previously characterized. Duplicate samples of CDK4/cyclin D3, T160pCDK2/cyclin A, and monomeric T160pCDK2 were first either incubated with λ phosphatase or buffer. After phosphatase inactivation each sample was then either incubated with CDK7/cyclin H or buffer, before being assayed for their ability to phosphorylate GST-RbCT.
Monomeric T160pCDK2 could be inactivated by phosphatase treatment (Fig. 4A, lanes 5 and 6). In the structure of monomeric T160pCDK2, the activation segment, including T160, is both flexible and accessible (17). T160pCDK2/cyclin A proved to be resistant to phosphatase treatment (Fig. 4A, lane 4). This result was expected given that phosphorylated T160 is embedded in a network of interactions within the T160pCDK2/cyclin A structure (15), and hence not accessible to phosphatase. Treatment with λ phosphatase fully dephosphorylates CDK4/cyclin D3 (Fig. S3) and the dephosphorylated form is completely inactive (Fig. 4A, lane 2). Thus, T172p must be accessible to λ phosphatase in the T172pCDK4/cyclin D3 complex present in this sample. This result further suggests that unphosphorylated CDK4/cyclin D3, unlike unphosphorylated CDK2/cyclin A (40), lacks any detectable activity.
In contrast to mock-treated, unphosphorylated CDK1/cyclin B, phosphatase-treated CDK4/cyclin D3 did not appear to recover GST-RbCT kinase activity after treatment with CDK7/cyclin H (Fig. S8). To explore this phenomenon further, we modified the experimental protocol to include a gel-filtration step between incubation with phosphatase and incubation with CDK7/cyclin H, so as to separate residual phosphatase activity and small molecular contaminants such as ADP from the dephosphorylated CDK4/cyclin D3 preparation. Under these conditions, we found that treatment of dephosphorylated CDK4/cyclin D3 with CDK7/cyclin H was sufficient to activate CDK4/cyclin D3 as a GST-RbCT kinase (Fig. 4B).
Discussion
The above observations suggest that the paradigm for CDK activation established by structures of CDK2/cyclin A, CDK2/cyclin E, and CDK5/p25 does not extend to CDK4. In particular, the structure suggests that the imposition of an active conformation on the C-helix of CDK4 is not a direct consequence of cyclin D3 association. Although this conformation might be suggested to arise when cyclin binding is accompanied by T172 phosphorylation, a structure of phosphorylated CDK4/cyclin D1 described in the accompanying article by Day et al. (50) confirms that C-helix realignment is not achieved even upon phosphorylation. Hence realignment of the C-helix, which is a prerequisite of the establishment of a catalytically competent active site configuration, must occur transiently when phosphorylated CDK4/cyclin D3 forms a Michaelis complex with ATP and protein substrates. That the active conformation of CDK4 depends on substrate binding in addition to cyclin binding and activation segment phosphorylation may explain why CDK4 appears to have such strict substrate requirements in vivo: only binding of appropriate substrates might induce an active CDK4 conformation. Notably, our comparison of the activities of CDK2/cyclin A with CDK4/cyclin D3 confirms that the latter is significantly more dependent on the presence of an intact recruitment motif in substrates.
This model of CDK4 activation would predict that phosphorylated CDK4/cyclin D complexes retain a relatively high degree of dynamic behavior. Consistent with this prediction, we found that phosphorylated CDK4/cyclin D3, unlike phosphorylated CDK2/cyclin A, is a good substrate for λ phosphatase, and hence has a readily accessible activation segment. Notably, however, we have demonstrated that dephosphorylated CDK4/cyclin D3 can be activated by phosphorylation on T172 by CDK7/cyclin H in the absence of any additional factors. This observation suggests that the role of HSP90/cdc37 in forming an active CDK4/cyclin D complex must be other than to contribute to phosphorylation of that complex by CDK7/cyclin H, or to any subsequent rearrangement of the phosphorylated CDK4/cyclin D complex toward an active conformation.
Materials and Methods
Protein Expression and Purification.
Human CDK1, CDK4, and cyclin D3 were all expressed in insect cells. CDK1 was expressed as a GST fusion and purified as described (32). CDK4 and cyclin D3 were cloned into pFastBac1 (Invitrogen) with, respectively, GST and PreScission protease recognition sequences and a FLAG tag at the amino terminus, and they were coexpressed in Sf-9 cells and purified by exploiting the CDK4 GST tag. A detailed protein purification protocol is provided in SI Text. CDK2 phosphorylated on T160 (T160pCDK2/cyclin A) (19), monomeric T160pCDK2 (17), CDK7/cyclin H (42), cyclin B (43), GST-RbCT (encoding residues 792–928 of the retinoblastoma protein) (19), and GST-p107spacer mutants (encoding p107 residues 618–672) (32, 44) were prepared as described. His-tagged p27Kip1 was expressed and purified from recombinant Escherichia coli cells by using sequential affinity and size-exclusion chromatography.
Multiangle Laser Light Scattering.
The solution molecular mass of the CDK4/cyclin D3 complex was determined by using on-line multiangle laser light scattering coupled with size exclusion chromatography (SEC-MALLS) (Fig. S5C). Further experimental details are provided in SI Text.
Mass Spectrometry.
CDK4/cyclin D3 crystals were harvested into mother liquor and extensively washed by sequential transfer. Dissolved crystals were analyzed by SDS/PAGE using NuPAGE 4–12% Bis-Tris gels (Invitrogen), and Mops SDS running buffer. After Coomassie blue staining, the protein bands (under these conditions CDK4 and cyclin D3 comigrated) were excised. Samples were subjected to tryptic digestion followed by LC/ MS/MS using a Thermo LTQ Orbitrap mass spectrometer coupled to a Dionex Ultimate 3000 nano HPLC system.
Kinase Assays.
CDK4/cyclin D3 and T160pCDK2/cyclin A or T160pCDK2 (100 ng unless otherwise stated) were mixed with 5 μg of substrate (Histone H1, GST-RbCT, or GST-p107spacer), 0.1 mM ATP, 1 μCi [32P]γ-ATP, 5 mM MgCl2, 50 mM Tris·HCl (pH 7.5) (final volume 10 μL) at 20 °C. After 7.5 min, the reactions were stopped by the addition of SDS and samples were analyzed by SDS/PAGE and autoradiography (32). p27Kip1 was included as appropriate.
λ-Phosphatase Treatment.
For analytical phosphatase treatment (Fig. 4A) 4–400 units of λ phosphatase, (New England Biolabs) was incubated with CDK4/cyclin D3, T160pCDK2/cyclin A, or monomeric T160pCDK2 in the kinase assay buffer for 30 min at 30 °C. The phosphatase reaction was suspended by addition of 20 mM sodium vanadate. For preparative phosphatase treatment (Fig. 4B), phosphatase treatment (10,000 units of λ phosphatase; New England Biolabs) was performed on 100 μg of CDK4/cyclin D3 for 30 min at 30 °C. The phosphatase was separated from the kinase by using size exclusion chromatography (Superdex 200 10/30; GE Healthcare). The kinase peak fraction (100 ng) was then assayed by using GST-RbCT as a substrate, as above, with and without the addition of 100 ng of GST-CDK7/cyclin H.
Crystallization, X-Ray Crystallography, Data Collection, and Processing.
CDK4/cyclin D3 crystals were grown at 4 °C by using the sitting drop method by mixing equal volumes (0.2 or 0.5 μL) of CDK4/cyclin D3 (3.5 mg/mL supplemented with 1 mM AMP-PNP) and well solution [0.1 M Hepes (pH 6.6), ≈15% PEG3350, ≈10% Tacsimate]. All crystals were cryoprotected in the crystallization solution supplemented with 25% PEG 200 and were flash-cooled in liquid nitrogen before data collection. Although the majority of crystals diffracted to <3.5-Å resolution, we were able to collect a complete dataset to 3.0-Å resolution from a single crystal of CDK4/cyclin D3 by screening ≈60 samples at the synchrotron. All collected data were integrated in MOSFLM (45) and scaled by using SCALA (46). The crystals contain 2 copies of CDK4/cyclin D3 per asymmetric unit. Data collection statistics are given in Table S1.
Structure Solution and Refinement.
The structure was solved by molecular replacement using the program PHASER (47). Search ensembles were created from selected CDK2 [Protein Data Bank (PDB) ID codes 1HCK and 1QMZ], CDK6 (PDB ID code 1G3N), and cyclin structures (cyclins A, B, E, and H; PDB ID codes 1QMZ, 2JGZ, 1W98, and 1KXU, respectively). Cyclins were pruned back to the core CBFs. The structure was refined with tight noncrystallographic restraints and with TLS parameters assigned to each polypeptide chain, using phenix.refine (48) and REFMAC5 (49).
Supplementary Material
Acknowledgments.
We thank the staff at the European Synchrotron Radiation Facility (Grenoble, France), Diamond Light Source (Harwell, UK), and E. Lowe for data collection; G. Lolli (Istituto di Ricerche di Biologia Molecolare P. Angeletti, Rome) for CDK7/cyclin H; J. Nettleship (Oxford Protein Production Facility) and B. Thomas (Department of Pathology, University of Oxford) for assistance with mass spectrometry; T. Walter (Wellcome Trust Centre for Human Genetics, Oxford) and P. Roversi and S. Lea (Department of Pathology, University of Oxford) for assistance in setting up the crystallization trials; I. Taylor (National Institute for Medical Research, Mill Hill, U.K.) for assistance in protein characterization; and I. Taylor for technical support. This work was supported by Cancer Research UK (T.T. and T.H.), the Kanae Foundation for the Promotion of Medical Science (T.T.), Framework Program 6 of the European Commission (PROKINASE Project; A.E.), and the Medical Research Council (N.B., J.A.E. and M.E.M.N.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The CDK4/cyclin D3 structure atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 3G33).
This article contains supporting information online at www.pnas.org/cgi/content/full/0809674106/DCSupplemental.
References
- 1.Kozar K, Sicinski P. Cell cycle progression without cyclin D–CDK4 and cyclin D–CDK6 complexes. Cell Cycle. 2005;4:388–391. doi: 10.4161/cc.4.3.1551. [DOI] [PubMed] [Google Scholar]
- 2.Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18:2699–2711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- 3.Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci. 2005;30:630–641. doi: 10.1016/j.tibs.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 4.Sherr CJ, Roberts JM. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 5.Macaluso M, Montanari M, Giordano A. Rb family proteins as modulators of gene expression and new aspects regarding the interaction with chromatin remodeling enzymes. Oncogene. 2006;25:5263–5267. doi: 10.1038/sj.onc.1209680. [DOI] [PubMed] [Google Scholar]
- 6.Zarkowska T, Mittnacht S. Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J Biol Chem. 1997;272:12738–12746. doi: 10.1074/jbc.272.19.12738. [DOI] [PubMed] [Google Scholar]
- 7.Lees JA, Buchkovich KJ, Marshak DR, Anderson CW, Harlow E. The retinoblastoma protein is phosphorylated on multiple sites by human cdc2. EMBO J. 1991;10:4279–4290. doi: 10.1002/j.1460-2075.1991.tb05006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kitagawa M, et al. The consensus motif for phosphorylation by cyclin D1–Cdk4 is different from that for phosphorylation by cyclin A/E–Cdk2. EMBO J. 1996;15:7060–7069. [PMC free article] [PubMed] [Google Scholar]
- 9.Matsushime H, et al. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell. 1992;71:323–334. doi: 10.1016/0092-8674(92)90360-o. [DOI] [PubMed] [Google Scholar]
- 10.Matsuura I, et al. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature. 2004;430:226–231. doi: 10.1038/nature02650. [DOI] [PubMed] [Google Scholar]
- 11.Ewen ME, et al. Functional interactions of the retinoblastoma protein with mammalian D-type cyclins. Cell. 1993;73:487–497. doi: 10.1016/0092-8674(93)90136-e. [DOI] [PubMed] [Google Scholar]
- 12.Kato J, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 1993;7:331–342. doi: 10.1101/gad.7.3.331. [DOI] [PubMed] [Google Scholar]
- 13.Blain SW, Montalvo E, Massague J. Differential interaction of the cyclin-dependent kinase (Cdk) inhibitor p27Kip1 with cyclin A-Cdk2 and cyclin D2-Cdk4. J Biol Chem. 1997;272:25863–25872. doi: 10.1074/jbc.272.41.25863. [DOI] [PubMed] [Google Scholar]
- 14.Jeffrey PD, et al. Mechanism of CDK activation revealed by the structure of a cyclinA–CDK2 complex. Nature. 1995;376:313–320. doi: 10.1038/376313a0. [DOI] [PubMed] [Google Scholar]
- 15.Russo AA, Jeffrey PD, Pavletich NP. Structural basis of cyclin-dependent kinase activation by phosphorylation. Nat Struct Biol. 1996;3:696–700. doi: 10.1038/nsb0896-696. [DOI] [PubMed] [Google Scholar]
- 16.Russo AA, Jeffrey PD, Patten AK, Massague J, Pavletich NP. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A–Cdk2 complex. Nature. 1996;382:325–331. doi: 10.1038/382325a0. [DOI] [PubMed] [Google Scholar]
- 17.Brown NR, et al. Effects of phosphorylation of threonine 160 on cyclin-dependent kinase 2 structure and activity. J Biol Chem. 1999;274:8746–8756. doi: 10.1074/jbc.274.13.8746. [DOI] [PubMed] [Google Scholar]
- 18.Welburn JP, et al. How tyrosine 15 phosphorylation inhibits the activity of cyclin-dependent kinase 2-cyclin A. J Biol Chem. 2007;282:3173–3181. doi: 10.1074/jbc.M609151200. [DOI] [PubMed] [Google Scholar]
- 19.Brown NR, Noble ME, Endicott JA, Johnson LN. The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nat Cell Biol. 1999;1:438–443. doi: 10.1038/15674. [DOI] [PubMed] [Google Scholar]
- 20.De Bondt HL, et al. Crystal structure of cyclin-dependent kinase 2. Nature. 1993;363:595–602. doi: 10.1038/363595a0. [DOI] [PubMed] [Google Scholar]
- 21.Adams PD, et al. Retinoblastoma protein contains a C-terminal motif that targets it for phosphorylation by cyclin–cdk complexes. Mol Cell Biol. 1999;19:1068–1080. doi: 10.1128/mcb.19.2.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tarricone C, et al. Structure and regulation of the CDK5–p25(nck5a) complex. Mol Cell. 2001;8:657–669. doi: 10.1016/s1097-2765(01)00343-4. [DOI] [PubMed] [Google Scholar]
- 23.Schulze-Gahmen U, Kim SH. Structural basis for CDK6 activation by a virus-encoded cyclin. Nat Struct Biol. 2002;9:177–181. doi: 10.1038/nsb756. [DOI] [PubMed] [Google Scholar]
- 24.Baumli S, et al. The structure of P-TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. EMBO J. 2008;27:1907–1918. doi: 10.1038/emboj.2008.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsushime H, et al. D-type cyclin-dependent kinase activity in mammalian cells. Mol Cell Biol. 1994;14:2066–2076. doi: 10.1128/mcb.14.3.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsuoka M, Kato JY, Fisher RP, Morgan DO, Sherr CJ. Activation of cyclin-dependent kinase 4 (cdk4) by mouse MO15-associated kinase. Mol Cell Biol. 1994;14:7265–7275. doi: 10.1128/mcb.14.11.7265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kato JY, Matsuoka M, Strom DK, Sherr CJ. Regulation of cyclin D-dependent kinase 4 (cdk4) by cdk4-activating kinase. Mol Cell Biol. 1994;14:2713–2721. doi: 10.1128/mcb.14.4.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pearl LH. Hsp90 and Cdc37-a chaperone cancer conspiracy. Curr Opin Genet Dev. 2005;15:55–61. doi: 10.1016/j.gde.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 29.LaBaer J, et al. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997;11:847–862. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- 30.Cheng M, et al. The p21(Cip1) and p27(Kip1) CDK “inhibitors” are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999;18:1571–1583. doi: 10.1093/emboj/18.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stevenson-Lindert LM, Fowler P, Lew J. Substrate specificity of CDK2-cyclin A. What is optimal? J Biol Chem. 2003;278:50956–50960. doi: 10.1074/jbc.M306546200. [DOI] [PubMed] [Google Scholar]
- 32.Brown NR, et al. Cyclin B and cyclin A confer different substrate recognition properties on CDK2. Cell Cycle. 2007;6:1350–1359. doi: 10.4161/cc.6.11.4278. [DOI] [PubMed] [Google Scholar]
- 33.Takeda DY, Wohlschlegel JA, Dutta A. A bipartite substrate recognition motif for cyclin-dependent kinases. J Biol Chem. 2001;276:1993–1997. doi: 10.1074/jbc.M005719200. [DOI] [PubMed] [Google Scholar]
- 34.Cheng KY, et al. The role of the phospho-CDK2/cyclin A recruitment site in substrate recognition. J Biol Chem. 2006;281:23167–23179. doi: 10.1074/jbc.M600480200. [DOI] [PubMed] [Google Scholar]
- 35.Peterlin BM, Price DH. Controlling the elongation phase of transcription with P-TEFb. Mol Cell. 2006;23:297–305. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 36.Michels AA, et al. MAQ1 and 7SK RNA interact with CDK9/cyclin T complexes in a transcription-dependent manner. Mol Cell Biol. 2003;23:4859–4869. doi: 10.1128/MCB.23.14.4859-4869.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garber ME, et al. The interaction between HIV-1 Tat and human cyclin T1 requires zinc and a critical cysteine residue that is not conserved in the murine CycT1 protein. Genes Dev. 1998;12:3512–3527. doi: 10.1101/gad.12.22.3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 39.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 40.Hagopian JC, et al. Kinetic basis for activation of CDK2/cyclin A by phosphorylation. J Biol Chem. 2001;276:275–280. doi: 10.1074/jbc.M007337200. [DOI] [PubMed] [Google Scholar]
- 41.Jeffrey PD, Tong L, Pavletich NP. Structural basis of inhibition of CDK-cyclin complexes by INK4 inhibitors. Genes Dev. 2000;14:3115–3125. doi: 10.1101/gad.851100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lolli G, Lowe ED, Brown NR, Johnson LN. The crystal structure of human CDK7 and its protein recognition properties. Structure (London) 2004;12:2067–2079. doi: 10.1016/j.str.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 43.Petri ET, Errico A, Escobedo L, Hunt T, Basavappa R. The crystal structure of human cyclin B. Cell Cycle. 2007;6:1342–1349. doi: 10.4161/cc.6.11.4297. [DOI] [PubMed] [Google Scholar]
- 44.Leng X, Noble M, Adams PD, Qin J, Harper JW. Reversal of growth suppression by p107 via direct phosphorylation by cyclin D1/cyclin-dependent kinase 4. Mol Cell Biol. 2002;22:2242–2254. doi: 10.1128/MCB.22.7.2242-2254.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data. Jnt CCP4/ESF-EACBM Newsl Prot Crystallogr. 1992;26:27–33. [Google Scholar]
- 46.Collaborative Computational Project No 4. The CCP4 Suite: Programs for protein crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 47.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adams PD, et al. PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr D. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 49.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 50.Day P, et al. Crystal structure of human CDK4 in complex with a D-type cyclin. Proc Natl Acad Sci USA. 2009;106 doi: 10.1073/PNAS.0809645106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.