

Abstract
Abnormal glycosylation is one of the hallmarks of the cancer cell and is associated with tumour invasion and metastasis. The development of tumour associated carbohydrate antigen (TACA) vaccines has been problematic due to poor immunogenicity. However when appropriate targets can be identified, passive immunisation with monoclonal antibodies (mAbs) directed against TACAs have been shown to have anti-tumour activity. Fas ligand (FasL) is a transmembrane protein which induces apoptosis in cells expressing its receptor, Fas. When grafted into mice, FasL-expressing tumour cells break immunological tolerance to self-antigens and induce antibody mediated tumour immunity. Here, five IgM mAbs were produced from mice vaccinated with FasL-expressing B16F10 mouse melanoma cells. They recognise various syngeneic and allogeneic murine tumour cell lines. One mAb, TM10, recognises a range of human tumour cell lines including melanoma, prostate and ovarian cancer. It does not bind to untransformed cells. The epitopes recognised by all the mAbs were carbohydrates expressed on proteins. Using carbohydrate microarrays, the antigenic targets of TM10 were found to be high-mannose core structures of N-linked glycans. In normal cells high mannose clusters are hidden by extensive saccharide branching but they become exposed in cancer cells as a result of abnormal glycosylation pathways. Vaccination with FasL-expressing tumours therefore enables the immune system to break tolerance to self-antigens, allowing identification of novel TACAs that can form the basis of future humoral anti-cancer therapy.
Keywords: Fas ligand, tumour, monoclonal antibody, glycan
INTRODUCTION
Fas ligand (FasL, CD95L) is a transmembrane protein that induces apoptosis in cells bearing its receptor, Fas (CD95) (1, 2). Fas is expressed on almost all human tumour cells (3) and so the FasL/Fas pathway is an attractive target in cancer immunotherapy. However the systemic administration of FasL is not feasible due to unacceptable hepatotoxicity (3) and so other approaches have been explored. Perhaps the most interesting finding amongst these is that when grafted into mice, tumour cells forced to overexpress FasL are rapidly rejected, and this is followed by the development of a protective, antibody-mediated tumour immunity (4-6).
Approximately 80% of cell-surface proteins and 5% of lipids are glycosylated (7). Altered patterns of carbohydrate expression are one of the hallmarks of the cancer cell and changes include over- and under-expression of naturally occurring glycans, abnormal branching of glycoproteins and glycolipids, and neoexpression of sugars normally restricted to embryonic tissue (8, 9). Abnormal glycosylation is associated with tumour cell invasion, metastasis and angiogenesis (10, 11). Over 50% of cancers known to express the tumour associated carbohydrate antigens (TACAs) described so far, which include glycolipids (e.g. GM1, GM2, GD2 and GD3), Lewis antigens such as LeA, LeX and LeY, and Thomsen Friedenreich (TF) antigen (12).
In this paper we report on the generation of a panel of new monoclonal antibodies (mAb) from tumour-immune mice following vaccination with FasL-expressing mouse melanoma cells. One mAb, termed TM10, recognises a broad range of both murine and human tumour cells. Its carbohydrate epitope, high mannose core structures of N-linked glycans, is a novel tumour antigen and is described here for the first time.
MATERIALS AND METHODS
Cells and mice
Murine tumour cell lines used include B16F10 melanoma (syngeneic with the C57BL/6 mouse strain), K1735 melanoma (C3H), NS1 myeloma (BALB/c), MC57 fibrosarcoma (C57BL/6), CT26 colon carcinoma (BALB/c), methylcholanthrene (MCA)-transformed L-cell fibroblasts (C3H), P815 mastocytoma (DBA/2), and GM95 (ceramide specific glucosyltransferase deficient cells derived from B16F10 melanoma). FasL expressing B16F10 cells (B16FasL) were generated as described (6). Human cells used were primary prostate fibroblasts, dermal fibroblasts, 293T cells, melanoma (Trombelli, MM5), prostate (PC3, DU145, LN-CAP), breast (ZR75.1, MDA-MB 468), and ovarian cancer cell lines (PEA-1, PEO-1, SK-OV-3), kind gifts from Professor Jonathan Waxman, Dr Tahereh Kalamati, Professor Charles Coombes and Professor Hani Gabra, all of Imperial College London.
Female C57BL/6 mice, aged 5-7 weeks, were purchased from Harlan (Oxon, UK) and housed at the Central Biomedical Services of Imperial College London. Non-hybridoma cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated foetal calf serum (FCS), whilst hybridoma cells were supplemented with 20% batch-tested heat-inactivated FCS. Media were supplemented with 2mM L-glutamine, 100IU/ml penicillin, 100μg/ml streptomycin and, in the case of murine cells, 50μM 2-mercaptoethanol, 10mM HEPES, and 1% sodium pyruvate. All cells were incubated at 37°C with 5% CO2.
Generation and purification of mAbs
Murine hybridomas were generated by the fusion of splenocytes and myeloma cells as previously described (6). Briefly, 1×107 irradiated B16FasL melanoma cells were injected subcutaneously into female C57BL/6 mice which were then challenged three times, at monthly intervals, with 5×105 B16F10 cells. Anti-tumour cell antibody production was confirmed in mice which rejected these tumour challenges (‘protected mice’) by staining of B16F10 cells with 1:50 diluted serum, the minimum concentration previously determined by titration to provide optimal staining. Splenocytes from protected mice with a positive anti-tumour cell antibody response were fused to NS1 murine myeloma cells using polyethylene glycol (PEG) 1500 (Roche Diagnostics, Basel, Switzerland). Hybridomas were selected by culture in HAT (hypoxanthine, aminopterine, thymidine) containing medium, and then screened by incubating their neat cell culture supernatant with B16F10 followed by anti-mouse Ig PE (Dako, Glostrup, Denmark). Positive staining hybridomas were single cell cloned three times and the class and subclass of each mAb determined using Isostrips (Roche).
Cell culture supernatants from hybridoma colonies were purified over a protein L (Sigma-Aldrich, Dorset, UK) column, eluted with 0.1M glycine (pH 2.5) and dialysed in sterile PBS. mAb concentration was measured by spectrophotometer (280nm optical density). mAbs were titrated and used at 10μg/ml in the mircoarray and 20μg/ml in all other experiments.
Flow cytometry
Fc receptors on murine cells were blocked with rat anti-mouse CD16/32 (eBioscience, San Diego, CA) and human cells blocked with human serum (Gibco, Paisley, UK). In some experiments cells were fixed with 2% formaldehyde and then permeabilised using 0.5% saponin (Sigma-Aldrich). Secondary antibodies used were anti-mouse IgM FITC (Sigma-Aldrich) or anti-mouse Ig PE (Dako). A minimum of 2×104 cells were analysed per sample.
Immunohistochemistry
B16F10 cells were grown to confluence on 15mm glass coverslips and then fixed with 1% formaldehyde. Cells were blocked with 1:200 goat serum and, where indicated, permeabilised with 0.5% Triton X-100 (Sigma-Aldrich). They were incubated with mAbs (20μg/ml) and then anti-mouse IgM Alexa Fluor-568 (Molecular Probes, Invitrogen, Paisley, UK) or anti-mouse IgM FITC. Cells were mounted onto glass slides with Vectashield/DAPI (Vector Laboratories, Peterborough, UK) and examined by confocal fluorescent microscopy using a x63 objective (Zeiss LS510, Jena, Germany).
Immunoprecipitation
1×107 native or biotinylated (Biotin EZ-Link – Pierce, Cramlington, UK) B16F10 cells were lysed in 1ml ice-cold NP40 lysis buffer and, for biotinylated samples, 0.5% Mega-9 (Sigma-Aldrich). Samples were pre-cleared with Protein L (Sigma-Aldrich), blocked with 10% bovine serum albumin (BSA), and then incubated with 20μg/ml TM10 followed by 100μL 50% protein L. The immunoprecipitates were run on 12% gels using SDS-PAGE and developed with silver staining (Amersham Biosciences, Little Chalfont, UK) or analysed by Western Blot.
Western blot
1×107 B16F10 cells were lysed in ice-cold NP40 lysis buffer, spun supernatants were separated on 12% gels using SDS-PAGE, and then transferred onto Hybond C Extra nitrocellulose membrane (Amersham). After being blocking with 5% non-fat dry milk / PBS, the membrane was incubated with 20μg/ml TM10, and then with anti-mouse Ig HRP (Sigma-Aldrich). HRP was detected using ECL Western Blotting kit (Amersham).
Glycosylation inhibitors, competitors, and lectins
B16F10 cells were incubated at 37°C for 24 hours with 200ng/ml tunicamycin (Sigma-Aldrich) or for 72 hours with 1mM N-butyl-deoxynojirimycin (N-butyl-DNJ) or N-butyl-deoxygalactonojirimycin (N-butyl-DGJ) (Toronto Research Chemicals, Toronto, Canada). Alternatively 293T cells or peripheral blood lymphocytes (PBLs) were incubated overnight with 5μM kifunensine (a gift from Dr Veronica Chang, Institute of Molecular Medicine, Oxford).
Lectins, with or without conjugation to FITC, were derived from Sambucus Nigra (SNA), Maakia amurensis (MAA), Concanavalin A (ConA), Phaseolus vulgaris L (PHA-L), and Galanthus nivalis (GNA) (all Vector Laboratories, Burlingame, CA). D-(+)-mannose, D-(+)-galactose, D-(+)-glucose, N-acetylglucoasamine (GlcNAc), N-acetylgalactosamine (GluNAc) and mannose-6-phosphate (all Sigma-Aldrich) were used at 1mg/ml. In some experiments pre-incubation with lectins or saccharides were used to inhibit binding of mAbs. In other experiments pre-incubation with the mAbs was used to inhibit the binding of the lectins or saccharides.
Carbohydrate microarrays
Details of the protocol for the construction of carbohydrate microarrays have been previously published (13). Briefly, carbohydrate and lipid antigens were printed in triplicate onto microglass slides using a robotic array spotter. Lipids were used at 0.02-2.0μg/ml and carbohydrates at 0.5-1.0μg/μ initial concentration, and at a 1:5 dilution. Antibodies to murine IgM were also printed at given concentrations to serve as standard curves. The printed slides were blocked with BSA, incubated with the relevant mAb (10μg/ml) and then anti-mouse IgG or IgM FITC. Analysis was using ScanArray Express Microarray Scanner (PerkinElmer Life Science, Boston, MA). Fluorescence intensity values for each spot were calculated with QuantArray software (Parkard Bioscience, PerkinElmer).
RESULTS
Generation of mAbs from B16FasL vaccinated mice
FasL-expressing tumour cells induce development of antibody-mediated tumour immunity (4-6). mAbs were therefore produced from mice vaccinated with FasL-expressing B16F10 cells (B16FasL) by the fusion of their splenocytes and NS1 myeloma cells. In total 5 monoclonal IgMs (TM3, TM5, TM6, TM10 and TM12) were produced all of which demonstrated cell surface binding to B16F10 (Figure 1 – top row). The mAbs stained both B16FasL and B16F10 equally, implying that they are not directed against either FasL or green fluorescent protein (Figure 1 – bottom row).
FIGURE 1.
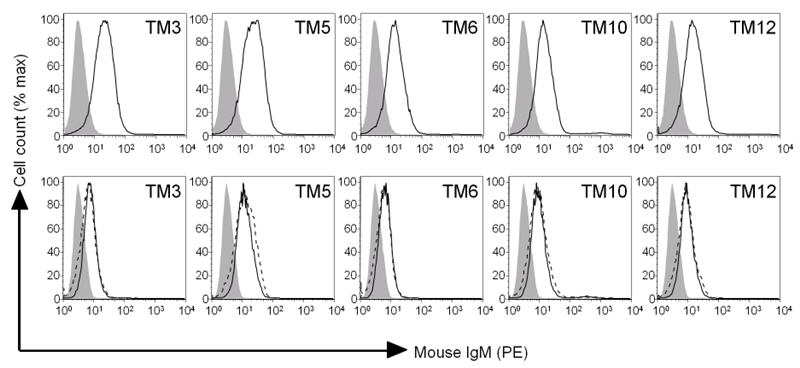
mAbs were generated from ‘protected’ mice vaccinated with 1×107 irradiated B16FasL cells and which had rejected at least three subsequent challenges with 5×105 B16F10 cells. Top row: B16F10 were stained with the mAbs indicated and analysed by flow cytometry. Open line - staining with mAb indicated. Bottom row: B16F10 (black line) and B16FasL (dashed line) were stained with the mAbs indicated. In all figures, closed line = isotype control staining of B16F10.
mAbs recognise a range of murine and human tumours
The mAbs generated recognised both syngeneic (MC57 fibrosarcoma from a C57BL/6 background) and allogeneic (K1735 melanoma and MCA-transformed L cells, both C3H background) murine tumour cells (Figure 2). Only one mAb (TM12) recognised P815 mastocytoma, and there was no significant staining of CT26 colon carcinoma by any of the mAbs. The latter negative results rule out the possibility that the mAbs are just recognising foetal calf serum in the culture medium or MHC antigens on the tumour cells. There was no staining of B16F10 or B16FasL with irrelevant murine IgM antibodies or with conjugated secondary antibodies alone (data not shown).
FIGURE 2.
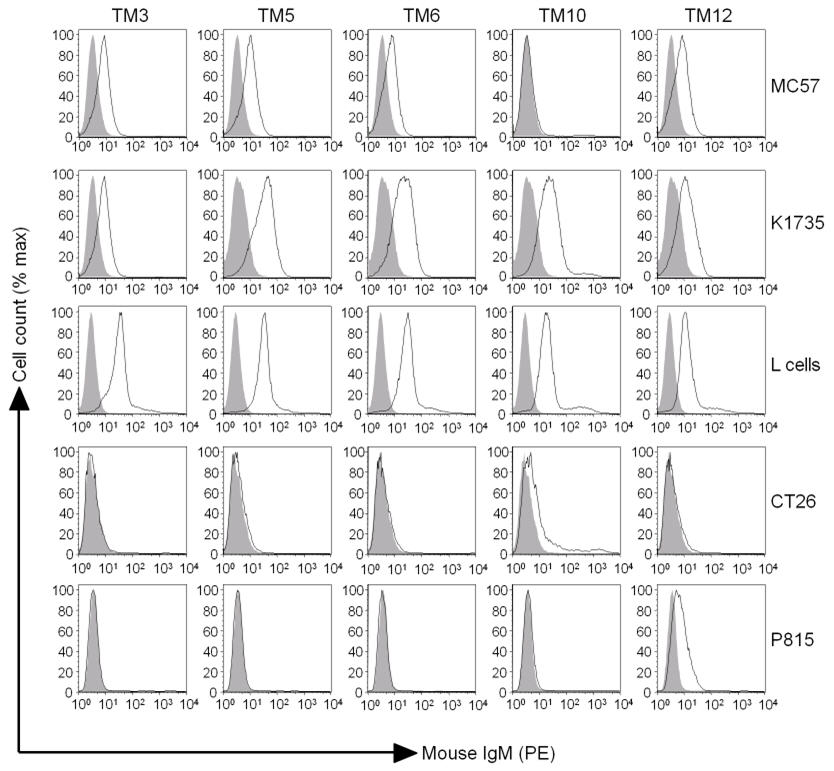
mAbs recognise both syngeneic and allogeneic murine tumours. The cell lines indicated were stained with the relevant mAb and analysed by flow cytometry. Closed line = isotype control, open line = mAb staining.
Since our mAbs recognised a range of murine tumour cells and because human melanoma-associated antigens can be highly homologous to their murine counterparts (14), binding of the mAbs to human tumour cells was investigated. Two mAbs – TM10 and TM12 – recognised a range of human cancer cell lines (Figure 3). TM10 stained human melanoma (Trombelli, MM5), prostate (PC3, LN-CAP, DU145), ovarian (PEA-1, PEO-1, SK-OV-3) and, to a lesser extent, breast cancer lines (MDA-MB 468 and ZR75.1). TM12 stained one prostate cancer cell line (PC3).
FIGURE 3.
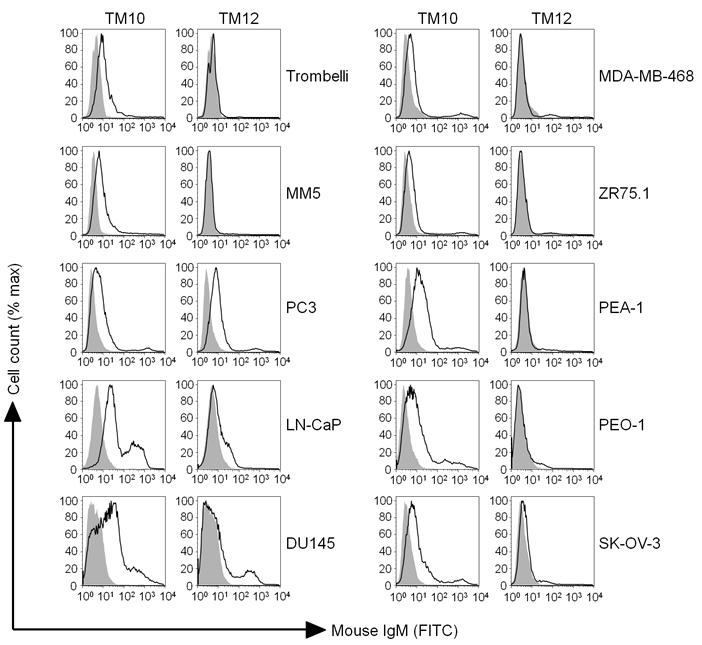
TM10 and TM12 recognise human cancer cells. Human melanoma (Trombelli, MM5), prostate (PC3, LN-CAP, DU145), ovarian (PEA-1, PEO-1, SK-OV-3) and breast (MDA-MB468, ZR75.1) cancer cells were stained with the relevant mAb and analysed by flow cytometry. Closed line = isotype control, open line = mAb staining.
TM10 binds to the intracellular compartment, but not surface, of untransformed cells
Given the broad range of recognition of both murine and human tumour cells, the mAb TM10 was selected for further investigation. Using indirect immunofluorescence, TM10 was found to have a punctate surface staining pattern of B16F10 cells (Figure 4A). At flow cytometry there was no cell surface binding of TM10 to a range of untransformed cells including murine splenocytes and human PBLs (Figure 4B), prostatic fibroblasts and dermal fibroblasts (data not shown). However when permeabilised with Triton X-100 there was now strong intracellular staining of both normal and cancer cells (Figures 4A and 4B). This suggests that all cells have an intracellular reserve of the antibody's epitope but it is only tumour cells that express it on their surface. There was no staining of any cells by isotype control antibody using direct immunofluorescence or flow cytometry.
FIGURE 4.
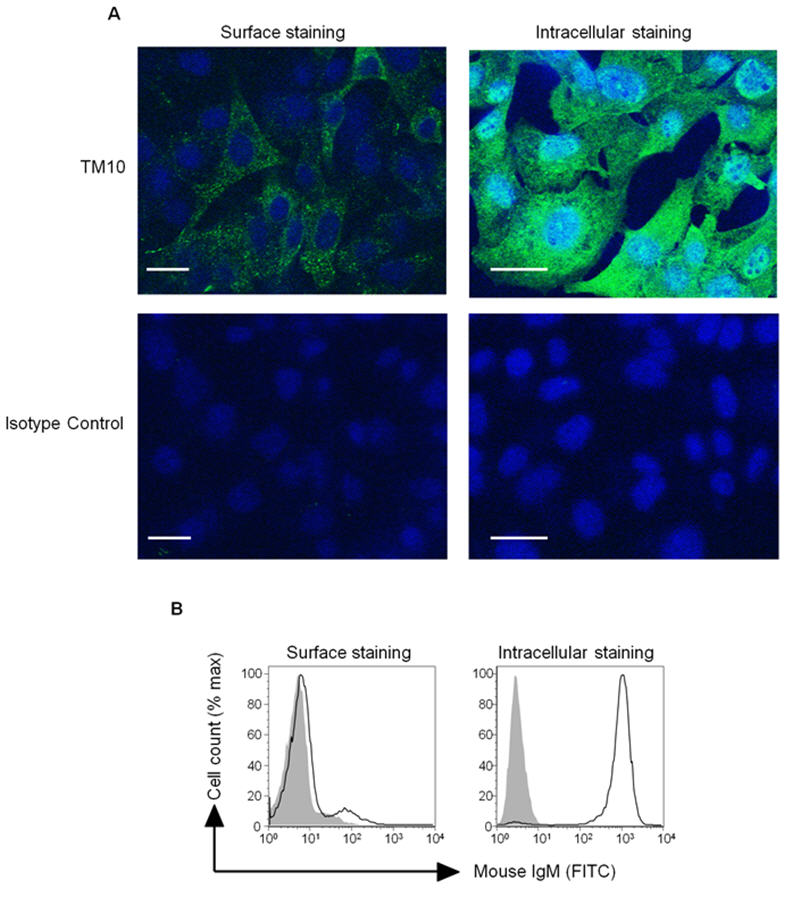
TM10 binds to the surface of tumour but not to untransformed cells, and there is a large intracellular reserve of its epitope in all cells. (A) Immunofluorescent microscopy (objective x63) of B16F10 cells surface stained and intracellular stained (permeabilised with Triton X-100) with TM10 or isotype control. Bar = 10μm. (B) Surface and intracellular (permeabilised with saponin) staining of human PBLs with TM10. Closed line - isotype control, open line = mAb staining.
TM10 recognises a carbohydrate, not protein, epitope
A protein epitope for our mAbs was initially sought. However Western blots and immunoprecipitations from native, surface biotinylated or 35S-methionine labelled B16F10 repeatedly revealed multiple protein bands (Supplemental Figure 1). This raised the possibility that our mAbs were in fact recognising sugars expressed on more than one glycoprotein or glycolipid. Experiments using glycosylation inhibitors supported this. Tunicamycin, a mixture of homologous nucleoside antibiotics, is an inhibitor of N-glycoprotein synthesis (Supplemental Figure 2). When B16F10 cells were grown in the presence of tunicamycin, there was a reduction in staining by all the mAbs except TM6 (Figure 5A – top row). The imino sugar N-butyl-deoxynojirimycin (N-butyl-DNJ) is a non-toxic inhibitor of α-glucosidase and prevents N-glycosylation one step downstream of the effects of tunicamycin (Supplemental Figure 2). There was reduced binding of all mAbs (except TM5) when B16F10 cells were pre-treated with N-butyl-DNJ (Figure 5A – bottom row). As controls, tunicamycin also inhibited the binding of the lectin Con A that binds preferentially to mannose, and N-butyl-DNJ reduced binding of MAA, a lectin that preferentially recognises sialic acid which is one of the residues found on N-linked structures.
FIGURE 5.
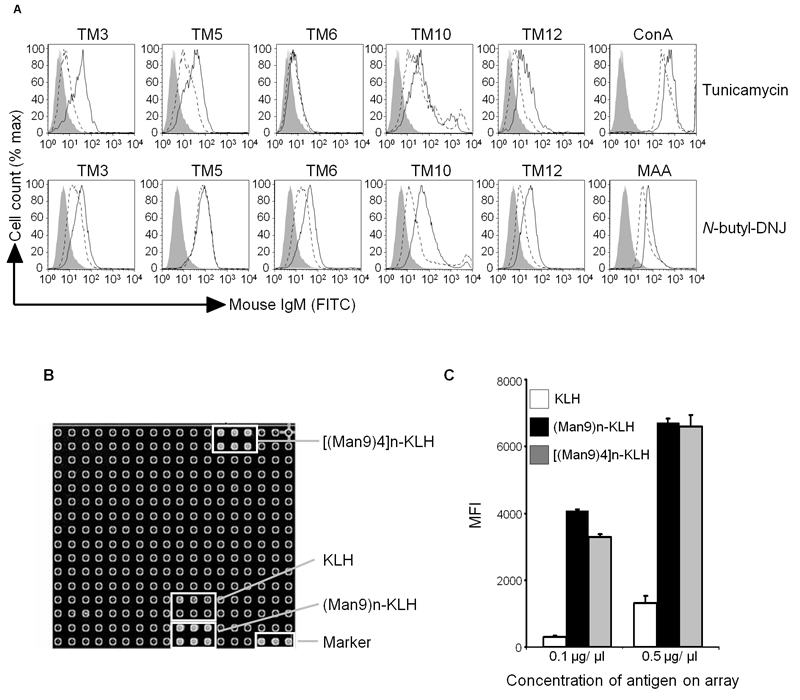
The epitope for TM10 is the carbohydrate, high mannose clusters. (A) B16F10 cells were grown in the presence of (Top row) tunicamycin (inhibitor of N-linked glycosylation) for 18 hours, or (Bottom row) n-butyl-DNJ (inhibitor of α-glucosidase) for 72 hours. They were then stained with the mAbs indicated and analysed by flow cytometry. ConA - concanavalin A agglutinin. MAA - maakia amurensis agglutinin. Closed line = isotype control; black line = staining of untreated cells; dashed line = staining following incubation with relevant glycosylation inhibitor. (B) Carbohydrate microarray analysis of TM10 (10μg/ml) showing specific binding to (Man9)n-KLH and [(Man9)4]n-KLH, but not to keyhole limpet haemocyanin (KLH) alone. (C) Graph of mean fluorescence intensity (MFI) of triplicate results from the array shown in B. Error bars represent SD.
The carbohydrates recognised by the mAbs appeared to be restricted to glycoproteins and are not expressed on glycolipids. Incubation of B16F10 with another imino sugar, N-butyl-deoxygalactonojirimycin (N-butyl-DGJ) which inhibits ceramide specific glucosyltransferase and so glycolipid but not glycoprotein formation, had no effect on mAbs binding (data not shown). Furthermore GM95, a B16F10 derived cell line that has reduced levels of ceramide specific glucosyltransferase and so impaired glycolipid expression, showed the same level of surface staining by all the mAbs when compared to B16F10 (data not shown).
The epitope for TM10 is a high-mannose cluster
With results pointing strongly towards carbohydrate epitopes, we screened our mAbs on a microarray of carbohydrates and glycolipids. TM10 bound strongly to two of these antigens (Figures 5B and 5C), both high-mannose clusters, displayed on the array as (Man9)n – which represents the mannose-core of N-glycoproteins – and [(Man9)4]n which mimics the mannose clusters displayed by the gp120 glycoprotein of HIV-1 (15, 16). Both mannose clusters were bound to the carrier keyhole limpet haemocyanin (KLH) but there was only weak binding of TM10 to KLH alone (Figure 5B). Indeed, when the mean fluorescent intensities (MFIs) are compared at 0.1 μg/μl, (Man9)n-KLH and [(Man9)4]n-KLH gave a 14- and 11-fold increase in signal compared to that of KLH alone (Figure 5C). It remains possible that the TM10 mAb may also have low affinity for some sugar epitopes on KLH. TM10 did not bind to any other antigens on the array, and there was no significant binding to the array of any of the other mAbs screened (data not shown).
To confirm the array finding of a high-mannose cluster epitope for TM10, we used the α-mannosidase inhibitor kifunensine which prevents normal glycoprotein synthesis and leads to an accumulation of Man9 complexes (Supplementary Figure 2). Treatment of 293T cells with kifunensine increased the binding of TM10 confirming that the antibody is recognising mannose clusters (Figure 6A). As expected the expression of sialic acid residues, detected through binding of MAA, was reduced in the presence of kifunensine. Inhibition experiments using D-(+)-mannose, D-(+) galactose, D-(+) glucose, D-(+) fucose, N-acetylglucosamine (GlcNAc), N-acetylgalactosamine (GluNAc) or mannose-6-phosphate to reduce binding of TM10 to B16F10 were negative (data not shown), suggesting that the antibody is recognising a more complex structure than just these simple saccharide units.
FIGURE 6.
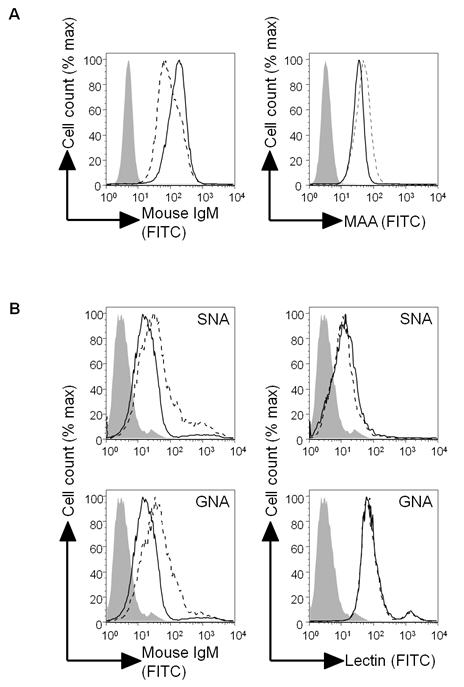
TM10 recognises high mannose clusters but the epitope is different to that recognised by lectins. (A) 293T cells were cultured for 24 hours with or without 5μM kifunensine and then stained with TM10 or MAA as indicated. Dashed line = plain culture medium; solid line = kifunsensine. (B) B16F10 cells alone (left-hand graphs, solid line), or pre-incubated with SNA (top left graph, dashed line) or GNA (bottom left graph, dashed line), were stained with TM10. Alternatively B16F10 cells alone (right-hand graphs, solid line) or pre-incubated with TM10 (right-hand graphs, dashed line) were stained with SNA (top graphs) or GNA (bottom graphs). In all figures closed line = unstained B16F10 cells. MAA = maakia amurensis agglutinin, SNA = sambucus nigra agglutinin, GNA = galanthus nivalus agglutinin.
Several different lectins known to bind to mannose-based carbohydrates were used to stain B16F10 and competitively inhibit TM10 binding. SNA (binding preferentially to sialic acid attached to terminal galactose), GNA (preferential binding to α1,3-mannose), and PHA-L (specific for Galβ1-4 GlcNAc structures) all stained B16F10, indicating that mannose-based epitopes are present on the cell surface (Figure 6B and data not shown). However there was no inhibition of lectin binding when cells were pre-incubated with TM10. Furthermore, when B16F10 cells were pre-incubated with lectins there was no abrogation, but instead an increase, in TM10 binding. Taken together these findings suggest that the epitopes recognised by TM10 and the lectins differ, but that the target for TM10 may also be expressed on the lectins themselves. Lectins are glycoproteins, and SNA for example contains 7.8% carbohydrates, principally mannose and glucosamine (17).
DISCUSSION
We have previously shown that FasL expressed on B16F10 murine melanoma leads to the development of antibody-mediated tumour immunity (6). To investigate this humoral response further, mAbs were produced from tumour immune mice that had been vaccinated with FasL-expressing melanoma cells (B16FasL). In total five monoclonal IgMs were generated all of which bound to the cell surface of both B16FasL and B16F10. One mAb, TM10, binds to both human and murine tumours and its epitope is a novel carbohydrate tumour antigen, high mannose clusters. The humoral nature of FasL-associated tumour immunity is important as antibody immunotherapy represents one of the success stories of immunology and has become established as proven treatment in the fields of oncology and haematology.
It is known that vaccination with whole tumour cells induces an IgM predominant response directed against repetitive carbohydrate structures on the cell surface (18). The relative absence of an IgG response is because the T cell help required for an isotype switch from IgM to IgG production is missing since carbohydrates are T cell independent antigens. This raises the question whether the mAbs generated here are just a product of melanoma cell vaccination as opposed to being specifically induced by the presence of FasL. Although we did not attempt to make mAbs from mice vaccinated with wild-type B16F10, there are three reasons why this is not the case. First, in the original description of the B16FasL vaccination model there was antibody binding to B16F10 from the serum of mice vaccinated with B16FasL but not from the serum of those vaccinated with wild-type melanoma (6). Secondly, vaccination with B16F10 was not capable of inducing protection from further tumour challenge as other mAbs have previously been shown to do. Finally, only transfer of serum from B16FasL vaccinated mice conferred tumour protection.
Using FasL expression on tumours as an adjuvant, there is an efficient generation of mAbs targeting TACAs. TM10 recognises high-mannose clusters which constitute the core of N-linked glycoproteins. The high-mannose clusters targeted appear not be a major component of glycolipids since there was no abrogation of TM10 binding in the presence of glycolipid inhibitors or to cells with impaired glycolipid expression (data not shown). In normal cells, high-mannose clusters are inaccessible on the surface of cells as they are masked by extensive N-acetylglucosamine (GlcNAc) branching and sialylation. Consequently TM10 does not bind to the surface of normal, untransformed cells such as murine splenocytes or human PBLs. In transformed cells abnormal glycosylation pathways (e.g. N-acetylaminyltransferase mutations) can prevent GlcNAc branching, allowing high-mannose clusters to become accessible and so facilitating TM10 binding to the tumour cell surface. In normal cells high-mannose clusters are an intermediary step in the production of N-linked glycoproteins and as such they exist in abundance in the endoplasmic reticulum. This intracellular pool is therefore present in all cells, both normal and transformed, and is seen in the strong TM10 staining of cells that have been permeabilised. This work is the first description of high-mannose clusters as defined tumour antigens. High mannose clusters have also been implicated in other pathological processes such as experimental allergic encephalomyelitis (EAE)1.
Changes in tumour cell glycosylation are associated with invasion and metastasis. Over-branching of N- and O-linked linked glycans, increased global sialylation and polysialic acid expression, presence of Lewis antigens and changes in glycolipids have been found to promote tumour invasiveness, metastasis, angiogenesis and drug resistance in a range of tumour types (7, 10, 11). Correspondingly such changes have been found to correlate with poorer prognosis in patients with cancer (8).
New theories have been put forward regarding the role of aberrant glycosylation in cancer. C-type lectin receptors (CLRs), pattern recognition receptors expressed on many cells including dendritic cells, recognise not only foreign glycan antigens but also self-antigens. In doing so they influence cell adhesion, migration and clearance of circulating glycoproteins (7). Recently it has been shown that antigen-presenting cells use their CLRs to adhere to carbohydrate structures on tumour cells but not on normal cells (19). It may be that this process inhibits DC maturation and so represents a mechanism employed by the tumour cell to evade the immune response. Indeed several pathogens such as HIV-1 and Lactobacillus have been shown to use sugars to promote the development of regulatory T cells and to inhibit the generation of an effective T cell response (7, 20).
Abnormal glycosylation on cancer cells makes carbohydrates attractive targets for tumour immunotherapy. However the development of carbohydrate vaccines has been problematic as glycans on tumours are insufficiently immunogenic, in part because they can also be expressed on embryonal tissue and normal tissue (9, 12). The assembly of multi-antigenic glycan vaccines, incorporation of carriers such as KLH, chemical modification of individual monosaccharides, and the use of endogenous adjuvants such as α-Gal antibodies have all been used to improve immunogenicity (9). It has been shown here and in previous studies (6, 21) that FasL expression on tumour cells is an effective adjuvant in the generation of carbohydrate recognising antibodies.
Antibodies directed against TACAs have therapeutic potential. mAbs raised against synthetically altered polysialic acid and the ganglioside GD3 controlled leukaemia cell metastases and protected against the development of human melanoma xenografts (22, 23). KM871, a chimeric IgG1 targeting GD3, induced clinical responses in patients with melanoma by mediating CDC (24). Meanwhile BR96, directed against the LeY antigen, has been conjugated to both chemotherapy and toxins although its effects in patients with metastatic breast and colon carcinomas have been disappointing, partly as a result of dose-limiting toxicity (25, 26). The presence of naturally occurring anti-GM2 antibodies in patients with melanoma are associated with an improved survival (27), although more recent studies have failed to confirm the clinical significance of the antibodies in other cancer types (28, 29).
Therapeutic mAbs can exert their anti-tumour effects through antibody dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) (30). IgMs are the most efficient isotype for complement activation (30) and, in data not presented here, TM10 was very effective at mediating CDC against B16F10 cells. Others have shown that mAbs targeting modified tumour-surface carbohydrates kill tumour cells by CDC both in vitro and in vivo (23, 31). TM10 was found not to be directly cytotoxic nor have any effect on tumour growth or proliferation (data not shown), supporting the notion that TACAs are involved in tumour invasion and metastasis, as opposed to roles in cell cycling or survival.
The in vivo anti-tumour effects of TM10 were investigated but were disappointing as it did not significantly protect mice from the development of new melanomas nor retard the growth of existing tumours (data not shown). This can be explained as IgM antibodies predominantly remain in the vasculature, have a shorter biological half-life and do not mediate ADCC. Indeed most therapeutically successful antibodies are IgGs as this isotype is the most efficient at mediating Fc domain-based functions such as ADCC. In a previous panel of mAbs, only the IgG antibody demonstrated anti-tumour activity in vivo, mediated at least in part by antibody dependent cellular phagocytosis (ADCP) (21). Future work with TM10 will involve generating an IgG isotype to optimise its in vivo potential.
Vaccination with FasL expressing tumours breaks immunological tolerance to self antigens and induces antibody-mediated tumour immunity (6). Previous work has shown that these antibodies are recognising carbohydrates on the tumour cell and, when of the correct isotype, possess anti-tumour in vivo activity (21). Here we focus on one such mAb, TM10, which is notable for binding to a range of both murine and human tumour cells. Its epitope, high mannose clusters, is a novel tumour antigen. In addition to mediating well established cytotoxic mechanisms such as CDC, TM10 may be interfering with CLR dependent tumour inhibition of antigen presenting cells. Further work will clarify its activity in vivo activity and its potential as a therapeutic antibody.
Supplementary Material
ACKNOWLEDGEMENTS
Thanks to Tahereh Kamalati for her assistance with confocal microscopy, to Veronica Chang for kifunensine and to Alison Cowper for general assistance. We also thank the Medical Research Council, Cancer Research UK and NIH for funding these experiments.
Grants and Funding: Thomas Newsom-Davis and Paul Chen were funded by Cancer Research UK. Katharina Simon is funded by the Association of International Cancer Research. Gavin Screaton is funded by the Medical Research Council; National Institute of Health Research (Biomedical Research Centre) and the Wellcome Trust. Work at Stanford University is supported in part by research grants R01NS055997 (Lawrence Steinman) and U01CA128416 (Denong Wang) from National Institute of Health.
ABBREVIATIONS
- ADCC
Antibody-dependent cellular cytotoxicity
- CDC
Complement-dependent cytotoxicity
- FasL
Fas ligand
- GNA
Galanthus nivalis agglutinin
- GlcNAc
N-acetylglucosamine
- KLH
Keyhole limpet haemocyanin
- mAb
Monoclonal antibody
- PHA-L
Phaseolus vulgaris Leucoagglutinin
- SNA
Sambucus nigra agglutinin
- TACA
Tumour associated carbohydrate antigen
Footnotes
Conflicts of Interest: None declared
L Steinman – unpublished observations.
REFERENCES
- 1.Itoh N, Yonehara S, Ishii A, et al. The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell. 1991;66:233–43. doi: 10.1016/0092-8674(91)90614-5. [DOI] [PubMed] [Google Scholar]
- 2.Suda T, Takahashi T, Golstein P, Nagata S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell. 1993;75:1169–78. doi: 10.1016/0092-8674(93)90326-l. [DOI] [PubMed] [Google Scholar]
- 3.Peter ME, Budd RC, Desbarats J, et al. The CD95 receptor: apoptosis revisited. Cell. 2007;129:447–50. doi: 10.1016/j.cell.2007.04.031. [DOI] [PubMed] [Google Scholar]
- 4.Seino K, Kayagaki N, Okumura K, Yagita H. Antitumor effect of locally produced CD95 ligand. Nat Med. 1997;3:165–70. doi: 10.1038/nm0297-165. [DOI] [PubMed] [Google Scholar]
- 5.Shimizu M, Fontana A, Takeda Y, Yagita H, Yoshimoto T, Matsuzawa A. Induction of antitumor immunity with Fas/APO-1 ligand (CD95L)-transfected neuroblastoma neuro-2a cells. J Immunol. 1999;162:7350–7. [PubMed] [Google Scholar]
- 6.Simon AK, Gallimore A, Jones E, Sawitzki B, Cerundolo V, Screaton GR. Fas ligand breaks tolerance to self-antigens and induces tumor immunity mediated by antibodies. Cancer Cell. 2002;2:315–22. doi: 10.1016/s1535-6108(02)00151-4. [DOI] [PubMed] [Google Scholar]
- 7.Aarnoudse CA, Garcia Vallejo JJ, Saeland E, van Kooyk Y. Recognition of tumor glycans by antigen-presenting cells. Curr Opin Immunol. 2006;18:105–11. doi: 10.1016/j.coi.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Hakomori S. Aberrant glycosylation in tumors and tumor-associated carbohydrate antigens. Adv Cancer Res. 1989;52:257–331. doi: 10.1016/s0065-230x(08)60215-8. [DOI] [PubMed] [Google Scholar]
- 9.Dube DH, Bertozzi CR. Glycans in cancer and inflammation--potential for therapeutics and diagnostics. Nat Rev Drug Discov. 2005;4:477–88. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- 10.Yoshimura M, Ihara Y, Matsuzawa Y, Taniguchi N. Aberrant glycosylation of E-cadherin enhances cell-cell binding to suppress metastasis. J Biol Chem. 1996;271:13811–5. doi: 10.1074/jbc.271.23.13811. [DOI] [PubMed] [Google Scholar]
- 11.Reddy BV, Kalraiya RD. Sialilated beta1,6 branched N-oligosaccharides modulate adhesion, chemotaxis and motility of melanoma cells: Effect on invasion and spontaneous metastasis properties. Biochim Biophys Acta. 2006;1760:1393–402. doi: 10.1016/j.bbagen.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 12.Slovin SF, Keding SJ, Ragupathi G. Carbohydrate vaccines as immunotherapy for cancer. Immunol Cell Biol. 2005;83:418–28. doi: 10.1111/j.1440-1711.2005.01350.x. [DOI] [PubMed] [Google Scholar]
- 13.Wang R, Liu S, Shah D, Wang D. A practical protocol for carbohydrate microarrays. Methods Mol Biol. 2005;310:241–52. doi: 10.1007/978-1-59259-948-6_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bowne WB, Srinivasan R, Wolchok JD, et al. Coupling and uncoupling of tumor immunity and autoimmunity. J Exp Med. 1999;190:1717–22. doi: 10.1084/jem.190.11.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang LX, Ni J, Singh S, Li H. Binding of high-mannose-type oligosaccharides and synthetic oligomannose clusters to human antibody 2G12: implications for HIV-1 vaccine design. Chem Biol. 2004;11:127–34. doi: 10.1016/j.chembiol.2003.12.020. [DOI] [PubMed] [Google Scholar]
- 16.Ni J, Song H, Wang Y, Stamatos NM, Wang LX. Toward a carbohydrate-based HIV-1 vaccine: synthesis and immunological studies of oligomannose-containing glycoconjugates. Bioconjug Chem. 2006;17:493–500. doi: 10.1021/bc0502816. [DOI] [PubMed] [Google Scholar]
- 17.Kaku H, Peumans WJ, Goldstein IJ. Isolation and characterization of a second lectin (SNA-II) present in elderberry (Sambucus nigra L.) bark. Arch Biochem Biophys. 1990;277:255–62. doi: 10.1016/0003-9861(90)90576-k. [DOI] [PubMed] [Google Scholar]
- 18.Ragupathi G. Carbohydrate antigens as targets for active specific immunotherapy. Cancer Immunol Immunother. 1996;43:152–7. doi: 10.1007/s002620050316. [DOI] [PubMed] [Google Scholar]
- 19.van Gisbergen KP, Aarnoudse CA, Meijer GA, Geijtenbeek TB, van Kooyk Y. Dendritic cells recognize tumor-specific glycosylation of carcinoembryonic antigen on colorectal cancer cells through dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin. Cancer Res. 2005;65:5935–44. doi: 10.1158/0008-5472.CAN-04-4140. [DOI] [PubMed] [Google Scholar]
- 20.Hodges A, Sharrocks K, Edelmann M, et al. Activation of the lectin DC-SIGN induces an immature dendritic cell phenotype triggering Rho-GTPase activity required for HIV-1 replication. Nat Immunol. 2007;8:569–77. doi: 10.1038/ni1470. [DOI] [PubMed] [Google Scholar]
- 21.Simon AK, Newsom-Davis T, Frayne ME, Ch'en PF, McMichael AJ, Screaton GR. Generation of tumour-rejecting anti-carbohydrate monoclonal antibodies using melanoma modified with Fas ligand. Int Immunol. 2008;20:525–34. doi: 10.1093/intimm/dxn011. [DOI] [PubMed] [Google Scholar]
- 22.Zhang GX, Gran B, Yu S, et al. Induction of experimental autoimmune encephalomyelitis in IL-12 receptor-beta 2-deficient mice: IL-12 responsiveness is not required in the pathogenesis of inflammatory demyelination in the central nervous system. J Immunol. 2003;170:2153–60. doi: 10.4049/jimmunol.170.4.2153. [DOI] [PubMed] [Google Scholar]
- 23.Liu T, Guo Z, Yang Q, Sad S, Jennings HJ. Biochemical engineering of surface alpha 2-8 polysialic acid for immunotargeting tumor cells. J Biol Chem. 2000;275:32832–6. doi: 10.1074/jbc.C000573200. [DOI] [PubMed] [Google Scholar]
- 24.Scott AM, Liu Z, Murone C, et al. Immunological effects of chimeric anti-GD3 monoclonal antibody KM871 in patients with metastatic melanoma. Cancer Immun. 2005;5:3. [PubMed] [Google Scholar]
- 25.Posey JA, Khazaeli MB, Bookman MA, et al. A phase I trial of the single-chain immunotoxin SGN-10 (BR96 sFv-PE40) in patients with advanced solid tumors. Clin Cancer Res. 2002;8:3092–9. [PubMed] [Google Scholar]
- 26.Saleh MN, Sugarman S, Murray J, et al. Phase I trial of the anti-Lewis Y drug immunoconjugate BR96-doxorubicin in patients with lewis Y-expressing epithelial tumors. J Clin Oncol. 2000;18:2282–92. doi: 10.1200/JCO.2000.18.11.2282. [DOI] [PubMed] [Google Scholar]
- 27.Livingston PO, Wong GY, Adluri S, et al. Improved survival in stage III melanoma patients with GM2 antibodies: a randomized trial of adjuvant vaccination with GM2 ganglioside. J Clin Oncol. 1994;12:1036–44. doi: 10.1200/JCO.1994.12.5.1036. [DOI] [PubMed] [Google Scholar]
- 28.Perez CA, Ravindranath MH, Soh D, Gonzales A, Ye W, Morton DL. Serum anti-ganglioside IgM antibodies in soft tissue sarcoma: clinical prognostic implications. Cancer J. 2002;8:384–94. doi: 10.1097/00130404-200209000-00009. [DOI] [PubMed] [Google Scholar]
- 29.Ravindranath MH, Muthugounder S, Presser N, Ye X, Brosman S, Morton DL. Endogenous immune response to gangliosides in patients with confined prostate cancer. Int J Cancer. 2005;116:368–77. doi: 10.1002/ijc.21023. [DOI] [PubMed] [Google Scholar]
- 30.Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat Biotechnol. 2005;23:1147–57. doi: 10.1038/nbt1137. [DOI] [PubMed] [Google Scholar]
- 31.Bajorin DF, Chapman PB, Wong G, et al. Phase I evaluation of a combination of monoclonal antibody R24 and interleukin 2 in patients with metastatic melanoma. Cancer Res. 1990;50:7490–5. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.