

Abstract
Reaction of sulfur ylide with aldehyde, imine, and ketone functionality affords the desired three-membered heterocycle in excellent yield. The sulfur ylide is generated in situ upon decarboxylation of carboxymethylsulfonium betaine functionality. Of the seven carboxymethylsulfonium betaine derivatives surveyed, the highest level of conversion of π-acceptor to heterocycle was obtained having S-methyl and S-phenyl functionality bound to a thioacetate derivative. Methylene aziridinations and epoxidations involving the decarboxylation of carboxymethylsulfonium betaine functionality complements existing technologies with the advantages of the reaction protocol, levels of conversion and scope. While moderate levels of diastereocontrol were observed in the aziridination of imine functionality, the four oxiranes resolved using Jacobsen’s Co(II)-salen complex were obtained in both high yield and enantioselectivity. The isolated chiral non-racemic oxiranes constitute the formal synthesis of chelonin-B and combretastatin starting from 3-bromo-4-methoxybenzaldehyde and 3,4,5-trimethoxybenzaldehyde respectively.
1. Introduction
In the context of alkylidene transfer agents, a method which offers the direct and stereospecific assembly of small ring carbo- and heterocyclic building blocks represents an extremely attractive synthetic transformation.1 The crescendo being the synthesis of terminal (monosubstituted) three-membered rings of high stereoselectivity starting from a large variety of π-acceptors (Figure 1).2
Figure 1.

Formation of terminal three-membered rings.
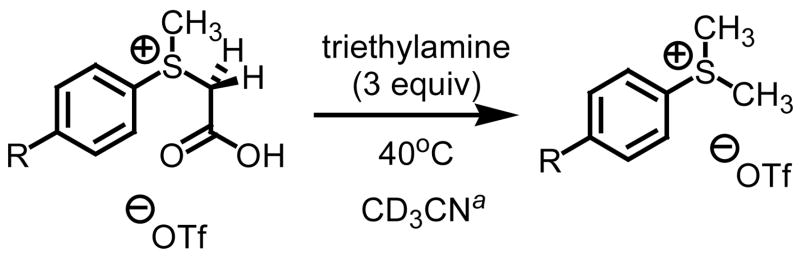
Nucleophilic alkylidene transfer agents have been shown to be extremely useful for the introduction of functionality into organic molecules.3 Of the many methods available, the sulfur ylide approach offers the most advantages when considering scope, atom efficiency and facility for stereocontrol. This method typically involves a stepwise sequence of 1) alkylation of a dialkyl sulfide to form a sulfonium salt and 2) treatment of the sulfonium salt with strong base to form the ylide.4 While highly enantio- and diastereoenriched systems can be prepared, limitations exist. The strength of our process involving the decarboxylation of carboxymethylsulfonium betaines is the trapping of aldehydes to form terminal epoxides and imines to form terminal aziridines (Scheme 1).5 Important to note is that the process does not require the use of strong and often pyrophoric bases such as butyl lithium or sodium hydride, does not require expensive metal catalysts and is compatible with ecologically benign solvent systems. The key step in our process is the decarboxylation of the carboxymethyl betaine functionality which generates the requisite sulfur ylide ii in situ.
Scheme 1.

While very satisfied with the chemistry, our methylene transfer technology has yet to reach its full potential when considering catalysis, scope and asymmetry. Efforts to address the latter two, additions to other π-acceptors and asymmetry, have been made and reported herein are our findings in the stereoselective assembly of heterocylic building blocks of aziridine and oxirane functionality.
2. Background
2.1. Carboxylmethyl Betaines
Previous communications from our lab focused on the process of generating nucleophilic alkylidene transfer agents using sulfur ylide technologies.5a,b To date, we have prepared and studied a total of seven sulfonium salts (Chart 1). While the preparation of each system started with the sulfenylation of either iodoacetic acid or bromoacetic acid,6 the efficacy of each salt to perform the role of alkylidene transfer agent varied greatly.
Chart 1.

As previously reported, added stability of the sulfur ylide and a more facile rate of decarboxylation resulted when switching from alkyl to aryl functionality.7 Sulfur ylide intermediate ii is best described as a semi-stabilized ylide when compared to thioacetate scaffolds consisting of not alkyl aryl substitution but alkyl alkyl substitution on sulfur. While traditional points of stabilization would focus on the alkylidene carbon, attachments onto the sulfur moiety and even solvation of the intermediate itself have also been shown to improve to the half-lives of sulfur ylides. The disappointingly low yields observed with the less electrophilic (electron rich) aldehydes were largely attributed to the instability of the S-ylide when working with alkyl, alkyl functionality bound to the thioacetate derivative (sulfonium salts 3 and 4).8,9
2.2. Rates of Decarboxylation
Based upon our kinetic studies, when adjusting the electronic environment about the sulfur atom, a significant change in the rate of decarboxylation of the betaine was observed (Table 1).5b This unfortunately resulted in a detrimental effect when considering mass throughput of aldehyde to oxirane. That is, all three sulfonium salts (2a–c) were ineffective at improving the levels of conversion from aldehyde to oxirane when compared to the results obtained with sulfonium salt 1.
Table 1.
Rates of decarboxylation
 | ||
|---|---|---|
| entry | sulfonium salt | t1/2 (min) |
| 1 | 1 (R = H) | 13.3 |
| 2 | 2a (R = NO2) | 1.2 |
| 3 | 2b (R = CN) | 1.3 |
| 4 | 2c (R = OCH3) | 85.6 |
reactions monitored by 1H NMR
Use of strong electron deficient groups resulted in half lives for sulfonium salts 2a and 2b in less than a minute and a half as compared to just over 13 minutes for sulfonium salt 1 under identical reaction conditions. When switching to an electron releasing aryl substituent at position 4 of the aromatic ring, the half life of sulfonium salt (2c) exceeded one hour. To explain the drop in epoxide formation when working with sulfonium salts 2a–c we proposed that at higher temperatures (refluxing THF), the decarboxylation rate is high providing relatively high levels of ylide that can be trapped by carbonyl compounds, particularly the more electrophilic (electron deficient) ones. As the electrophilicity of the carbonyl compound decreases, the forward rate of epoxidation slows down and fails to compete effectively with ylide fragmentation and other side reactions. The data obtained clearly documented the need to recognize the factors associated with decarboxylation rates and competing side reactions when working with sulfur ylides as alkylidene transfer agents.
2.3. Asymmetry: Chiral Non-Racemic Imines
In our search for alternative mild protocols for S-ylide generation which might be capable of sustaining asymmetric methylene transfers, we were intrigued by reports from Stockman and co-workers using sulfinyl imines as chiral non-racemic π-acceptors in methylene transfer processes (Scheme 2).10 Using methylidene agents derived from trimethyl sulfonium iodide, a series of aliphatic, aromatic, and heterocyclic tert-butylsulfinyl aziridines were prepared. The Corey-Chaykovsky reaction of chiral non-racemic sulfinyl imines resulted in yields ranging from 63–84% and diastereomeric ratios as high as 98:2. Reaction times ranging between three and ten hours using NaH as base, DMSO as solvent at 20°C lead to the highest levels of conversion and diastereoselectivity.
Scheme 2.

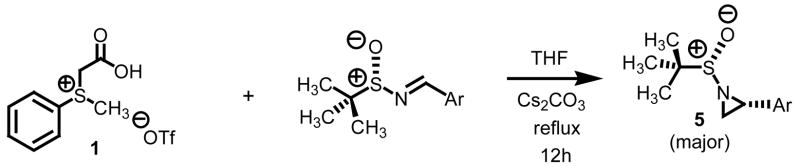
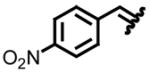
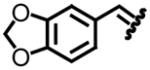
Our entry into stereoselective methylene transfer processes began with an aziridination study using our decarboxylation technology (Table 2).5c We chose to test a series of aryl substituted chiral non-racemic tert-butylsulfinyl imines against our sulfonium salt 1.
Table 2.
Methylene aziridinations using sulfonium salt 1.
 | |||||||
|---|---|---|---|---|---|---|---|
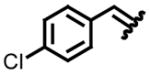
| entry | imine | yield (%)a | drb | entry | imine | yield (%)a | drb |
| 1, a |
|
97 | 87:13 | 5, e |
|
85 | 84:16 |
| 2, b |
|
96 | 78:22 | 6, f |
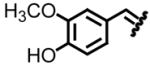
|
93 | 80:20 |
| 3, c |
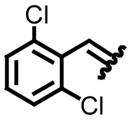
|
94 | 77:23 | 7, g |
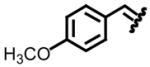
|
89 | 66:34 |
| 4, d |
|
95 | 74:26 | ||||
Isolated chromatographically pure material.
Determined by 1H NMR (crude reaction mixture).
A total of seven derivatives were surveyed. While the isolated yields were excellent, the diastereoselectivities observed in this series which is believed to be under kinetic control ranged from a low of 66:34 for electron releasing aryl imines to a high of 87:13 with electron withdrawing imines. While significantly higher levels of diastereocontrol could be obtained with our technology, it was at the expense of aziridine conversion (dr = 90:10 at 50% conversion for 5d). Albeit lower levels of diastereocontrol were observed overall, we were encouraged by the fact that extremely high levels of conversion were obtained with a process which does not rely on DMSO as solvent and strong bases such as NaH or nBuLi.
2.4. Asymmetry: Chiral Non-Racemic Sulfides
As previously stated, a number of groups have demonstrated and refined ingenious catalytic asymmetric approaches to chiral nonracemic aziridines, cyclopropanes and epoxides based on the generation of S-ylides.1 Aggarwal and co-workers via reaction of chiral sulfides with rhodium metallocarbenes have reported on a range of substituted heterocycles which can be generated in high yield and with excellent control of both absolute and relative stereochemistry. The same author has as well demonstrated a similar catalytic cycle using Simmons-Smith reagents (Et2Zn/ClCH2I) as S-ylide precursors to perform asymmetric methylene transfers (Scheme 3).11
Scheme 3.

A second example involving an asymmetric methylene epoxidation was reported two years later by Goodman and co-workers (Scheme 4).12 Using a D-mannitol derived chiral non-racemic sulfide, levels of enantiocontrol reached 57% in the formation of styrene oxide starting from benzaldehyde (54% yield). This is the highest level of enantiocontrol achieved to date when discussing methylene epoxidations.
Scheme 4.

The third and final example by Solladié-Cavallo and co-workers did not utilize a methylene epoxidation, rather they utilized paraformaldehyde and a chiral nonracemic benzyl substituted sulfonium salt (Scheme 5).13 As a result, extremely high levels of enantiocontrol have been achieved in the asymmetric preparation of terminal oxiranes (98%). While only two 2-aryloxiranes were reported in this communication, the oxathiane scaffold derived from (+)-(R)-pulegone is a clever design in sulfonium salt formation. That is, the 1,3-heteroatomic disposition within the fused bicycle effectively exploits the differences in lone pair basicity on sulfur resulting in a highly diastereoenriched sulfur ylide via the sulfonium salt.
Scheme 5.

2.5. Asymmetry: Resolutions
Recognizing the shortcomings of a protocol which relies on refluxing THF for complete conversion of π-acceptor to heterocycle, we sought alternative asymmetric processes in the assembly of chiral non-racemic oxiranes. When considering scope and highly stereoselective processes yielding terminal oxiranes, the two-step process starting with an asymmetric dihydroxylation followed by ring closure to afford the desired oxirane is the method of choice.14 With asymmetric dihydroxylations, a host of monosubstituted alkenes consistently yield chiral non-racemic diols with exceptionally high enantioselectivities and conversion. If an even higher level of enantioenrichment is desired (>99%), options do exist. One option being the kinetic resolution of chiral racemic materials using conventional or enzymatic processes.1,15 The first step allows for the recovery of enantiopure oxirane from diol via a hydrolytic kinetic resolution. Once separated, the second step consisting of ring closure of the diol furnishes the enantiomeric oxirane. Logistically, one key advantage to this strategy is that when both stereoisomers are needed, returning to an earlier synthetic step using an epimeric complex is not needed.
3. Results and Discussion
3.1. Methylene Epoxidations
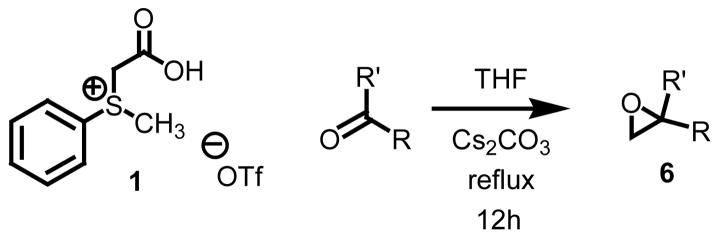
While the three communications by Aggarwal, Goodman, and Solladié-Cavallo all demonstrate elegant strategies in the chemistry of asymmetric transfers to furnish terminal oxiranes, each has limitations when considering catalysis, scope, levels of enantiocontrol, and conversion. Coupling hydrolytic kinetic resolutions with a very effective method capable of furnishing a host of monosubstituted oxiranes in high yield is an extremely viable strategy toward the preparation of chiral non-racemic terminal oxiranes. Accordingly, we chose to test sulfonium salt 1 against the same group of carbonyl derivatives used in our initial studies with the anticipation that the oxiranes generated can be resolved.
The group of carbonyl derivatives ranged from electron deficient aryl aldehydes to less reactive ketone substrates. While electron deficient aryl aldehydes reacted the quickest yielding the dpesired oxirane in less than three hours, electron releasing aryl aldehydes required reaction times up to 12h. The data from the study is presented below in Table 3.
Table 3.
Methylene epoxidations using sulfonium salt 1.
 | |||||
|---|---|---|---|---|---|
| entry | carbonyl derivative | yield (%)a | entry | carbonyl derivative | yield (%)a |
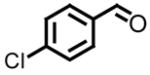
| 1, a |
|
97 | 6, f |
|
86 |
| 2, b |
|
97 | 7, g |
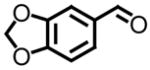
|
92 |
| 3, c |
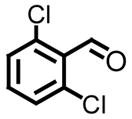
|
98 | 8, h |
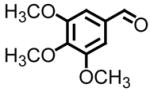
|
91 |
| 4, d |
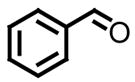
|
95 | 9, i |

|
95 |
| 5, e |
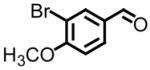
|
93 | 10, j |
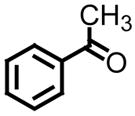
|
86 |
Isolated chromatographically pure material.
All reactions reported here were clean as judged by GC and NMR analysis of the crude reaction mixture. The analysis of the crude reaction mixtures revealed no by-products other than desired product and based upon the isolated yields, very little unreacted starting material. A comparison of this data with previous reports from our lab reveals several interesting trends. While moderate improvements, for comparative purposes, were observed for the electron deficient aryl aldehydes (entries 1–3), significant improvements were observed for benzaldehyde (95% (entry 4) versus 77% (reference 5b)) and 4-methoxybenzaldehyde (86% (entry 6) versus 25% (reference 5b)). Use of an alkyl aldehyde (95% yield (entry 9)) and ketone (86% yield (entry 10)) confirms the breadth of this technology.
Three additional electron releasing aryl aldehydes were surveyed (entries 5, 7, and 8). As evident in the isolated yields reported, excellent levels of conversion were obtained with this class of aldehydes which is a significant achievement in the area of methylene epoxidations.16 Our approach complements existing technologies toward the preparation of chiral racemic terminal oxiranes and offers the added advantages of scope, mild reaction conditions, ease in workup, and levels of conversion using a unified protocol. While reaction is observed when working with trimethylsulfonium salts and a host of aliphatic and aryl aldehydes, reports vary greatly on the efficacy of methylene transfer using trimethylsulfonium salts onto electron releasing aryl aldehydes.17 One solution toward the fluxional levels of conversion observed has been to utilize a biphasic system. The advantage with having a solvent partitioned protocol is the aqueous medium offers a more hospitable environment for the unstabilized ylide. However, utilizing this approach with the scope of carbonyl derivatives presented in Tables 2 and 3 would result in hydrolysis of the respective imine and oxirane functionality when working with electron deficient aryl imines and aldehydes.18 Furthermore, self-condensation reactions when working with enolizable carbonyl derivatives is a possibility.
3.2. Hydrolytic Kinetic Resolutions
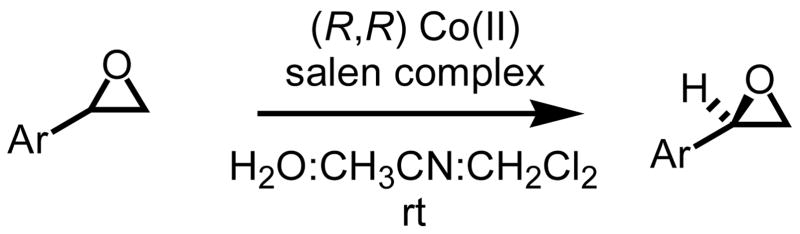
As stated above, while very satisfied with the chemistry, our approach toward methylene transfers has yet to reach its full potential when considering catalysis, scope and asymmetry. When working with chiral non-racemic sulfinyl imines, we, not surprisingly, documented an inverse relationship with temperature and stereoselectivity.5c Reaction temperatures ranging from room temperature to refluxing THF resulted in diastereoselectivities ranging from 90% to 65% respectively. The unacceptably low levels of conversion observed when operating at room temperature present an interesting challenge when wanting to pursue an asymmetric protocol. While we are determined to resolve this foreseeable limitation, options when wanting chiral non-racemic material do exist. One option being the hydrolytic kinetic resolution of chiral racemic oxiranes using Jacobsen’s Co(II)-salen complex (Figure 2).19
Figure 2.

(R,R)-Co(II)-salen complex.
With both enantiomeric complexes commercially available and ample literature precedence on the process, we chose to resolve a series of aryl substituted oxiranes, four in all, using Jacobsen’s Co(II)-salen complex.20 The data from the study is presented below in Table 4.
Table 4.
Hydrolytic kinetic resolutions using Co(II) salen complex.21
 | ||||
|---|---|---|---|---|
| entry | oxirane | recovery (%)a | ee (%)b | [α]D |
| 1 |
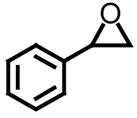
|
46 | >99 | −24.0 (−23.7 (lit)) |
| 2 |
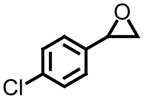
|
45 | >99 | −17.2 (+19.3 (lit (ent))) |
| 3 |
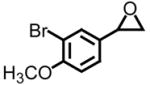
|
42 | >99 | −9.8 |
| 4c |
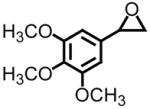
|
43 | >89d | +13.8 (+15.5 (lit)) |
Isolated chromatographically pure material. Monitored by 1H NMR.
HPLC (Regis (R,R)-Whelko-O1 column
(S,S) Co(II) salen complex
Based upon specific rotaton; unable to resolve enantiomers by HPLC
Using styrene oxide as our benchmark standard (entry 1), we were successful in the resolution of both electron deficient (entry 2) and electron releasing (entries 3 and 4) 2-aryloxiranes. The successful resolution of these systems adds to an already impressive body of work where highly enantioenriched terminal oxiranes have been isolated using this technology. Using HPLC (Whelko-O1), we were unable to observe any of the minor isomer for entries 1–3. While we were not successful in separating the racemic mixture of 2-(3,4,5-trimethoxyphenyl)oxirane by HPLC (entry 4), our evidence for the isolation of highly enantioenriched material was furnished by comparing the specific rotation obtained with literature data.21c Additional evidence for the formation of highly enantioenriched material for all four systems includes 1) the analysis of resolved oxirane using the enantiomeric Co(II)-salen complex and 2) the efficiency of this process when analyzing the levels of conversion and specific rotations of isolated diol.22
3.3. Chelonin B and Combretastatin
Entries 5 and 8 from Table 3 are two of 17 examples which illustrate the scope of sulfonium mediated methylene transfers onto π-acceptors using sulfonium salt 1. Isolated yields of 2-(3-bromo-4-methoxyphenyl)oxirane and 2-(3,4,5-trimethoxyphenyl)oxirane starting from 3-bromo-4-methoxybenzaldehyde and 3,4,5-trimethoxybenzaldehyde were 93% and 91% respectively. Both systems as racemates were successfully resolved. While oxirane 6e (2-(3-bromo-4-methoxyphenyl)oxirane) was isolated in >99% ee, our unsuccessful attempts in separating the racemate 2-(3,4,5-trimethoxyphenyl)oxirane (6h) by HPLC using the Whelko-O1 column resulted in the recording of an 89% ee using data obtained by a polarimeter. While we have no reason to believe the enantioenrichment of this material is in excess of 99%, our evidence is based upon literature data as shown in Table 4, entry 4.
Synthesized oxiranes 6e and 6h as chiral non-racemic materials are interesting examples in that they constitute the formal syntheses of two systems of biological and medicinal significance (Figure 3).
Figure 3.

Retrosynthetic analyses for chelonin B, bromochelonin B, and combretastatin.
Starting from 3-bromo-4-methoxybenzaldehyde, Lawrence and Bushell prepared (S)-(+)-chelonin B in less than ten steps.23 The key step in the preparation of the marine natural product was an asymmetric dihydroxylation of the monosubstituted styrene derivative obtained from the Wittig methylenation of 3-bromo-4-methoxybenzaldehyde. The corresponding diol was next cyclized to afford chiral non-racemic oxirane in 76% overall yield (three synthetic transformations) and 96% ee based upon HPLC analysis. A direct comparison of our approach with theirs has an almost identical overall yield (78%24 versus 76%) and slight improvement in the level of enantioenrichment (>99% versus 96%). While a clear disadvantage is the mass throughput when considering the resolution, advantages include the number of synthetic steps needed to obtain chiral non-racemic oxirane 6e (two versus three), the type and use of materials needed, and the fact that both enantiomers are available for further use when performing a hydrolytic kinetic resolution.
An almost identical approach was used in the preparation of combretastatin.21c Starting from not the bromo methoxy but rather the trimethoxy derivative of benzaldehyde, ring opening of chiral non-racemic oxirane 6h with an organometallic reagent (bromine lithium exchange of an aryl bromide) afforded the secondary alcohol in 75% yield. Aside from reporting the specific rotation of oxirane 6h, the communication reports an enantioenrichment of 97% of the TBDMS derivative shown in Figure 3 by HPLC analysis. Starting from 3,4,5-trimethoxybenzaldehyde, preparation of the key intermediate, chiral non-racemic oxirane 6h, required four synthetic transformations (Wittig methylenation, asymmetric dihydroxylation, formation of a tosyl derivative, and ring closure). The overall yield was 88%. A direct comparison again of the two approaches reveals a lower overall yield (78% versus 88%) and assuming the optical purity of oxirane 6h obtained upon dihydroxylation of the styrene derivative is identical to the secondary alcohol, a slight improvement from 97% to >99% ee is observed. As was the case with chelonin B, the amount of materials needed to prepare oxirane 6h alone far exceeds that required using methylene transfer technology.
As for absolute stereochemistry, by virtue of resolving the racemates into highly enantioenriched oxiranes and diols, both enantiomeric lines are generated and thus confirming the formal synthesis of chelonin B and combretastatin. Research in our group continues in the areas of 1) understanding the kinetics of the reaction, 2) increasing the scope using modified sulfonium salts, and 3) the development of a decarboxylative protocol involving the use of chiral, non-racemic sulfide promoter. The results from these studies will be reported in due course.
4. Conclusion
A host of existing protocols all demonstrate elegant strategies in the chemistry of methylene transfers. Each has limitations when considering scope, levels of enantiocontrol, and conversion. Our unified two-step approach complements existing protocols with several key advantages. Methylene transfers starting from a carboxymethylsulfonium betaine scaffold allows for the efficient conversion of both imine and aldehyde functionality to the corresponding aziridine and oxirane. Coupling hydrolytic kinetic resolutions with a very effective method capable of furnishing a host of monosubstituted oxiranes in high yield is an extremely viable strategy toward the preparation of highly enantioenriched terminal oxiranes.
5. Experimental
5.1. General Experimental Considerations
1H NMR (300 MHz) and 13C NMR (75 MHz) spectra were obtained as solutions in CDCl3. Chemical shifts were reported in parts per million (ppm) and referenced to δ 7.27 (1H NMR) and δ 77.00 (13C NMR). Infrared spectra were recorded using a FT IR and reported in wavenumbers (cm−1). Analytical high pressure liquid chromatography (HPLC) was performed with a built-in photometric detector using a (R,R)-Whelk-O 1 column (4.6 mm × 25cm). Solvents for HPLC analyses were of spectroscopic grade and filtered before use. GC analyses were performed using a ZB-5 with guardian (30 meter + 5 meter guardian end, 0.25 mm ID, and 0.25 μm film thickness). Optical rotations were recorded on a digital polarimeter and are reported as follows: [α]T (c, solvent) where c = g/100mL. TLC analyses were performed on flexible aluminium backed TLC plates with a fluorescent indicator. Detection was conducted by UV absorption (254 nm) followed by charring with 10% KMnO4 in water. Solutions were concentrated in vacuo using a rotary evaporator. The resulting residue was purified using a neutral alumina column (150 mesh, 58Ǻ) unless specified otherwise. All chemicals used for synthetic procedures were reagent grade or better.
5.2. Representative procedure for methylene epoxidations
An oven-dried 25 mL round-bottomed flask was equipped with a stir bar, septum, and water-jacketed condenser. The system was next charged with aldehyde (1.0 mmol, 1.0 equiv), cesium carbonate (2.0 equiv) and THF (3.0 mL). To this slurry was added a solution of betaine (2.0 equiv) in THF (2.0 mL) via syringe in two equal portions at 6 h intervals. The system was externally heated to 80 °C (sand bath) and the reaction mixture was allowed to stir for a period of 12 h. After cooling to room temperature, the reaction mixture was filtered over a pad of Celite, concentrated in vacuo and then immediately purified by chromatography (neutral alumina (150 mesh, 58 Ǻ)) using a gradient eluent system of hexanes and ethyl acetate to afford analytically pure oxirane.
5.3. Representative procedure for the kinetic resolution of aryl oxiranes
To a 25 mL round bottom flask with stir bar, was added oxirane (12.5 mmol), H2O (7.5 mmol), a 1:1 mixture of CH2Cl2 and CH3CN (0.25 ml), and the colbalt(II) salen complex (0.3 mol%). The reaction mixture was allowed to stir at room temperature. During that time, the reaction was monitored by 1H NMR using as markers the diagnostic peaks associated with oxirane and diol. Upon observing 50% conversion, the reaction was judged as complete. Workup involved filtering the reaction mixture through a plug of anhydrous Mg2SO4. The resulting organic solution was then concentrated in vacuo. Prior to HPLC analysis the reaction mixture was purified by column chromatography using neutral alumina (hexanes). All HPLC analyses were performed using a (R,R)-Whelk-O 1 column (25 cm × 4.6 mm, 5μm). The mobile phase was hexanes:IPA at a flow rate ranging between 0.9 mL/min to 1.0 mL/min with a detector wavelength of 220nm. Prior to and post each HPLC analysis, runs using chiral racemic aryl oxiranes were performed. In addition to HPLC analyses, optical rotations of chiral non-racemic material were obtained and compared to published values when possible.
Supplementary Material
Supporting Information Available
Experimental and spectral data in the preparation of compounds 6a–j and resolution of compounds 6a, 6d, 6e, and 6h.
Acknowledgments
DCF would like to thank NIGMS (NIH NIGMS 1R15GM085936), NSF (CHE 0514004), and the Camille and Henry Dreyfus Foundation (TH-06-008) for partial funding of this research. SAP would like to acknowledge financial support through the Alabama Space Grant Scholars Program. SCP and SAP would as well like to acknowledge financial support through the University of South Alabama (UCUR and University Honors Program).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Yudin A, editor. Aziridines and Epoxides in Organic Synthesis. Wiley; New York: 2006. [Google Scholar]
- 2.(a) McGarrigle EM, Myers EL, Illa O, Shaw MA, Riches SL, Aggarwal VK. Chem Rev. 2007;107:5841. doi: 10.1021/cr068402y. [DOI] [PubMed] [Google Scholar]; (b) Dalko PI, Moisan L. Angew Chem, Int Ed. 2004;43:5138. doi: 10.1002/anie.200400650. [DOI] [PubMed] [Google Scholar]; (c) Dai LX, Hou XL, Zhou YG. Pure and Applied Chemistry. 1999;71:369. [Google Scholar]
- 3.(a) Trost BM, Melvin LS., Jr . Sulfur Ylides Emerging Synthetic Intermediates. Vol. 31 Academic Press; New York: 1975. [Google Scholar]; (b) Clark JS, editor. Nitrogen, Oxygen and Sulfur Ylides Chemistry: A Practical Approach. Oxford University Press; Oxford: 2002. [Google Scholar]
- 4.(a) Blot V, Brier JF, Davoust M, Miniere S, Reboul V, Metzner P. Phosphorus, Sulfur, and Silicon and the Related Elements. 2005;180:1171. [Google Scholar]; (b) Aggarwal VK. In: Comprehensive Asymmetric Catalysis. Jacobsen EN, Pfaltz A, Yamamoto H, editors. II. Springer-Verlag; Heidelberg: 1999. pp. 679–693. [Google Scholar]; (c) Li AH, Dai LX, Aggarwal VK. Chem Rev. 1997;97:2341. doi: 10.1021/cr960411r. [DOI] [PubMed] [Google Scholar]; (d) Stockman RA. Ann Rep Prog Chem, Sec B. 2004;100:149. [Google Scholar]
- 5.(a) Forbes DC, Standen MC, Lewis DL. Org Lett. 2003;5:2283. doi: 10.1021/ol034612a. [DOI] [PubMed] [Google Scholar]; (b) Forbes DC, Amin SR, Bean CJ, Standen MC. J Org Chem. 2006;71:8287. doi: 10.1021/jo061370u. [DOI] [PubMed] [Google Scholar]; (c) Forbes DC, Bettigeri SV, Amin SR, Bean CJ, Law AM, Stockman RA. doi: 10.1080/00397910802654898. (submitted) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ratts KW, Yao AN. J Org Chem. 1966;31:1185. [Google Scholar]
- 7.(a) Cheng JP, Liu B, Zhang XM. J Org Chem. 1998;63:7574. doi: 10.1021/jo981129i. [DOI] [PubMed] [Google Scholar]; (b) Johnson AW, Amel RT. Can J Chem. 1968;46:461. [Google Scholar]
- 8.Trost BM, Bogdanowicz MJ. J Am Chem Soc. 1973;95:5298. [Google Scholar]
- 9.Ciaccio JA, Drahus AL, Meis RM, Tingle CT, Smrtka M, Geneste R. Syn Commun. 2003;33:2135. [Google Scholar]
- 10.(a) Chigboh K, Nadin A, Stockman RA. Synlett. 2007;18:2879. [Google Scholar]; (b) Morton D, Stockman RA. Tetrahedron. 2006;62:8869. [Google Scholar]; (c) Morton D, Pearson D, Field RA, Stockman RA. Org Lett. 2004;6:2377. doi: 10.1021/ol049252l. [DOI] [PubMed] [Google Scholar]; (d) Morton D, Pearson D, Field RA, Stockman RA. Synlett. 2003;13:1985. [Google Scholar]
- 11.Aggarwal VK, Coogan MP, Stenson RA, Jones RVH, Fieldhowse R, Blacker J. Eur J Org Chem. 2002:319. [Google Scholar]
- 12.Bellenie BR, Goodman JM. Chem Commun. 2004:1076. doi: 10.1039/b316653h. [DOI] [PubMed] [Google Scholar]
- 13.Solladié-Cavallo A, Diep-Vohuule A. J Org Chem. 1995;60:3494. [Google Scholar]
- 14.(a) Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem Rev. 1994;94:2483. [Google Scholar]; (b) Sharpless KB, Amberg W, Bennani YL, Crispino GA, Hartung J, Jeong KS, Kwong HL, Morikawa K, Wang ZM, Xu D, Zhang XL. J Org Chem. 1992;57:2768. [Google Scholar]; (c) Kolb HC, Sharpless KB. Tetrahedron. 1992;48:10515. [Google Scholar]
- 15.(a) Kim S, Jacobsen EN. Angew Chem Int Ed. 2004;43:3952. doi: 10.1002/anie.200460369. [DOI] [PubMed] [Google Scholar]; (b) Larrow JF, Jacobsen EN. Topics Organomet Chem. 2004;6:123. [Google Scholar]
- 16.Winn CL, Bellenie BR, Goodman JM. Tetrahedron Lett. 2002;43:5427. [Google Scholar]
- 17.(a) Alvarez M, Granados R, Lavilla R, Salas M. J Heterocyclic Chem. 1985;22:745. [Google Scholar]; (b) Haridas K, Dev S. Indian J Chem. 1987;26B:1018. [Google Scholar]; (c) Bouda H, Borredon ME, Delmas M, Gaset A. Syn Commun. 1987;17:503. [Google Scholar]; (d) Nudelman A, McCaully RJ. Eur J Med Chem Chemica Therapeutica. 1981;16:333. [Google Scholar]
- 18.Law AM. BS Honors Thesis. University of South Alabama; Mobile, AL: May, 2007. Alternative Technologies in Organic Synthesis. [Google Scholar]
- 19.Schaus SE, Brandes BD, Larrow JF, Tokunaga M, Hansen KB, Gould AE, Furrow ME, Jacobsen EN. J Am Chem Soc. 2002;124:1307. doi: 10.1021/ja016737l. [DOI] [PubMed] [Google Scholar]
- 20.(a) Tokunaga M, Larrow JF, Kakiuchi F, Jacobsen EN. Science. 1997;277:936. doi: 10.1126/science.277.5328.936. [DOI] [PubMed] [Google Scholar]; (b) Jacobsen EN. Acc Chem Res. 2000;33:421. doi: 10.1021/ar960061v. [DOI] [PubMed] [Google Scholar]; (c) Larrow JF, Hemberger KE, Jasmin S, Kabir H, Morel P. Tetrahedron Asymmetry. 2003;14:3589. [Google Scholar]
- 21.For the specific rotation of styrene oxide, see: White DE, Jacobsen EN. Tetrahedron Asymmetry. 2003;14:3633.For the specific rotation of 2-(4-chlorophenyl)oxirane, see: Pedragosa-Moreau S, Morisseau C, Baratti J, Zylber J, Archelas A, Furstoss R. J Org Chem. 1996;61:7402. doi: 10.1021/jo960558i.For the specific rotation of 2-(3,4,5-trimethoxyphenyl)oxirane, see: Ramacciotti A, Fiaschi R, Napolitano E. Tetrahedron Asymmetry. 1996;4:1101.
- 22.For yields and specific rotations using both (R,R)- and (S,S)-Co(II)salen complexes in oxirane recovery and diol formation, see Supporting Information.
- 23.Lawrence NJ, Bushell SM. Tetrahedron Lett. 2001;42:7671. [Google Scholar]
- 24.The 78% overall yield was based upon the observed 93% yield in oxirane formation and 50% theoretical recovery associated with the hydrolytic kinetic resolution.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available
Experimental and spectral data in the preparation of compounds 6a–j and resolution of compounds 6a, 6d, 6e, and 6h.