

Abstract
A number of missense mutations in the oxidative stress response protein DJ-1 are implicated in rare forms of familial Parkinsonism. The best-characterized Parkinsonian DJ-1 missense mutation, L166P, disrupts homodimerization and results in a poorly folded protein. The molecular basis by which the other Parkinsonism-associated mutations disrupt the function of DJ-1, however, is incompletely understood. In this study we show that three different Parkinsonism-associated DJ-1 missense mutations (A104T, E163K, and M26I) reduce the thermal stability of DJ-1 in solution by subtly perturbing the structure of DJ-1 without causing major folding defects or loss of dimerization. Atomic resolution X-ray crystallography shows that the A104T substitution introduces water and a discretely disordered residue into the core of the protein, E163K disrupts a key salt bridge with R145, and M26I causes packing defects in the core of the dimer. The deleterious effect of each Parkinsonism-associated mutation on DJ-1 is dissected by analysis of engineered substitutions (M26L, A104V, and E163K/R145E) that partially alleviate each of the defects introduced by the A104T, E163K and M26I mutations. In total, our results suggest that the protective function of DJ-1 can be compromised by diverse perturbations in its structural integrity, particularly near the junctions of secondary structural elements.
DJ-1 is a small (189 a.a.) conserved protein whose absence or inactivation results in Parkinsonism in humans (1) and is also a ras-dependent oncogene and has been implicated in the pathogenesis of several types of cancer (2). DJ-1 is a member of the large, eponymous DJ-1/PfpI superfamily, which has representatives in most organisms (3, 4). Human DJ-1 robustly protects cells from oxidative stress (5-14) and is found both in the cytoplasm (1) and the mitochondria (1, 5, 15). The precise biochemical role of DJ-1 is unknown, although it binds to multiple protein targets involved in transcriptional regulation (16-20), RNA binding (21), SUMOylation (22), protein folding (23), and apoptosis (24), suggesting that DJ-1 has multiple cellular functions. Furthermore, recent studies have shown that DJ-1 enhances the cellular oxidative stress response by regulating the activity of the antioxidant transcription factor Nrf2 (25) and by regulating the synthesis of glutathione (26). DJ-1 may also exert additional influence over cell fate thorough its involvement in the PTEN/Akt signalling pathway (27, 28). Considered in total, these results and studies in a variety of model organisms (8, 29-31) suggest that DJ-1 operates in multiple pathways to enhance cell survival in response to oxidative insult.
The cytoprotective function of DJ-1 can be disrupted by several missense mutations that have been discovered in patients with heritable Parkinsonism (32). The best characterized Parkinsonian missense mutation in DJ-1, L166P, disrupts homodimer formation and is rapidly degraded, thereby resulting in low steady-state levels of DJ-1 and accounting for the observed loss of protein function (33, 34). The crystal structure of wild-type DJ-1 suggests that the L166P substitution disrupts the penultimate α-helix G(35-39), and recent biochemical work has shown that the integrity of α-helix G is critical for DJ-1 dimer formation (40). In addition, several other homo- and heterozygous disease-associated point mutations in DJ-1 have been identified, including A104T (41), M26I (42), E64D (43) and E163K (44). These point mutations are distributed throughout the DJ-1 structure (Figure 1) and all have a less dramatic effect on cellular levels of the protein than the L166P substitution. Of these mutations, A104T and M26I are of particular interest since they reside in the core of the protein and therefore must exert their pathogenic effect by directly altering the properties of DJ-1. A very recent study showed that the M26I substitution leads to decreased thermal stability and an enhanced propensity for aggregation, although these defects are not as severe as those observed for L166P DJ-1 (45). The E64D missense mutation, which is located in the solvent exposed helix B of DJ-1, does not alter the structure of DJ-1 (46) and stabilizes the protein (43, 45). This suggests that E64D disrupts DJ-1 function through another mechanism that may involve interactions of DJ-1 with other macromolecules. In contrast, very little is known about the effects of the A104T and E163K point mutations on DJ-1. Furthermore, despite impressive progress made in the characterization of some pathological mutations in DJ-1, a detailed structural explanation for the observed deleterious effects of these disease-associated missense mutations on DJ-1 is still lacking.
Figure 1.

Location of three PD-associated mutations in the DJ-1 dimer. The DJ-1 dimer is represented with one monomer colored blue and the other gold. Three residues that are mutated in certain forms of familial Parkinsonism studied in this work are shown in both monomers of the DJ-1 dimer, with prime symbols indicating the symmetry-related residues. Both M26 and A104 are located in the core of dimeric DJ-1, while E163 is a surface-exposed residue in α-helix G. The figure was created with POVscript+(65).
In order to understand the impact of the A104T, E163K and M26I Parkinsonism-associated point mutations on DJ-1 structure and stability, we have determined the solution oligomerization state, thermal stabilities, CD spectral properties and atomic resolution crystal structures of each of these mutant proteins. Differential scanning calorimetry (DSC) indicates that all three Parkinsonian point mutations destabilize DJ-1 to varying degrees. In solution, these mutations do not significantly alter the secondary structural content or dimerization of DJ-1. Structural analysis indicates that each substitution compromises DJ-1 stability through a distinct structural mechanism that involves either the introduction of rotameric sidechain disorder and solvent into the DJ-1 core (A104T), disruption of a key stabilizing salt bridge (E163K), or the introduction of a packing defect into the core of the protein (M26I). Our results suggest that disease-associated point mutations compromise DJ-1 stability and function through multiple structural mechanisms.
Experimental Procedures
DJ-1 construct generation
Three different types of DJ-1 constructs were used in this study: one in which the DJ-1 gene was cloned between the NcoI and BamHI sites of pET15b and produces the native protein without a tag sequence (untagged DJ-1), a second in which the DJ-1 gene was cloned between the NdeI and XhoI sites of pET15b and produces a thrombin-cleavable N-terminal hexa-histidine tagged protein (tag-cleaved DJ-1), and a third in which the DJ-1 gene was cloned between the NdeI and XhoI sites of pET21a, producing a C-terminal hexa-histidine tagged protein (DJ-1-6xHis). All DJ-1 point mutations (A104T, A104V, E163K, E163K/R145E, M26I, and M26L) were generated by site-directed mutagenesis and the mutations were verified using the DNA sequencing service at the Genomics Core Research Facility at the University of Nebraska-Lincoln.
Protein expression and purification
Recombinant DJ-1 proteins were expressed in BL21(DE3) E. coli grown in LB media supplemented with 100 μg/ml of ampicilin at 37°C with shaking. Once the A600 of the culture reached 0.5-0.7, it was equilibrated at 20°C for 3 hours prior to induction of protein expression by the addition of IPTG to a final concentration of 0.1 mM. The induced culture was incubated at 20°C with shaking overnight and harvested by centrifugation the next day. Cell pellets were frozen and stored at −80°C until needed.
Untagged DJ-1 was purified using ammonium sulfate precipitation, hydrophobic interaction chromatography and anion exchange chromatography as previously described (47). The tag-cleaved and 6xHis-tagged DJ-1 proteins were purified using Ni2+-NTA metal affinity chromatography using standard procedures followed by subtractive purification using Hi-Q anion exchange resin (Bio-Rad). For DJ-1 proteins bearing a thrombin-cleavable N-terminal tag, Ni2+-NTA purified DJ-1 was incubated with thrombin (1 U thrombin/mg DJ-1) for 3-4 hours, followed by passage through a second Ni2+-NTA column to remove any protein that retained the hexa-histidine tag. Thrombin was removed from tag-cleaved DJ-1 by passage over benzamidine sepharose resin (GE Biosciences).
Purified DJ-1 was dialyzed against storage buffer (25 mM HEPES pH=7.5, 100 mM KCl, 2 mM DTT) for 4-12 hours at 4°C and concentrated to 20 mg/ml (ε280=4000 M−1 ml−1) using a 10 kDa MWCO centrifugal concentrator (Millipore). Purified DJ-1 ran as a single band on Biosafe Coomassie Blue (Bio-Rad) stained SDS-PAGE and was aliquoted, snap-frozen in liquid nitrogen, and stored at −80°C. The oxidation state of DJ-1 was determined by electrospray mass spectrometry (Redox Biology Center Mass Spectrometry Core Facility, University of Nebraska) to ensure that only reduced DJ-1 was used for the experiments in this study. Typically, samples of DJ-1 were >95% reduced, with less than 5% oxidized at C106 to cysteine-sulfinic acid. No evidence for other oxidized species of DJ-1 was seen in any sample.
Crystallization, data collection, and processing
For all crystallization experiments, DJ-1 at 20 mg/ml in storage buffer was crystallized using the hanging drop vapor diffusion method using drops containing 2 μl of protein and 2 μl of reservoir solution. Crystals of E163K DJ-1-6xHis in space group P3121 were grown at room temperature in 2-7 days using sodium citrate (1.3-1.6 M) as a precipitant buffered with 50 mM HEPES pH=7.5. E163K DJ-1-6xHis crystals were cryoprotected by serial transfer through solutions of incrementally higher concentrations of sodium malonate pH=7.4 to a final concentration of 3.4 M (48). The crystals were mounted in nylon loops and cryocooled by direct immersion into liquid nitrogen.
Crystals of A104T (6xHis), M26I (6xHis), A104V (tag-cleaved), E163K/R145E (tag-cleaved) and M26L (tag-cleaved) DJ-1 were grown using the hanging drop vapor diffusion method by mixing 2 μl of protein at 20 mg/ml with 2 μl of reservoir solution (20-25% PEG 3000, 100 mM HEPES pH=7.5, 200 mM NaCl) and equilibrating against the reservoir solution for 2-10 days at room temperature. The resulting bipyramidal (space group P3121) or javelin-shaped (space group P6522) crystals were cryoprotected by serial transfer through the reservoir solution that was supplemented with increasing concentrations of ethylene glycol to a final concentration of 30% v/v. Crystals were removed from the cryoprotectant in nylon loops and cryocooled by direct immersion into liquid nitrogen.
X-ray diffraction data for E163K DJ-1-6xHis were collected at the Advanced Photon Source (APS), BioCARS beamline 14 BM-C. Data for A104T and M26I DJ-1-6xHis were collected at the Stanford Synchrotron Radiation Laboratory (SSRL), beamline 11-1. Single crystals maintained at 100 K were used for the collection of each dataset, and the data were collected in separate high and a low resolution passes with differing exposure times and detector distances in order to avoid overloaded pixels for intense low resolution reflections. Because C106 is sensitive to radiation-induced damage, the beam intensity was attenuated and the crystals were exposed to X-rays for 10 seconds or less per 1° oscillation. Diffraction data for A104V, M26L and E163K/R145E tag-cleaved DJ-1 were collected at 120 K on a MicroMax-007 Cu rotating anode source (Rigaku) operating at 40 kV and 20 mA with Osmic Blue confocal optics and a Raxis IV++ detector (Rigaku). All diffraction data were integrated and scaled using HKL2000 (49) and final data statistics for each data set are provided in Table 1.
Table 1.
Crystallographic Data and Model Statistics
| Data Statistics | A104T | E163K | M26I | A104V | M26L | E163K/R145E |
|---|---|---|---|---|---|---|
| Wavelength (Å) | 0.980 | 0.900 | 0.827 | 1.542 | 1.542 | 1.542 |
| Space Group | P3121 | P3121 | P3121 | P6522 | P3121 | P3121 |
| Unit Cell | ||||||
| a (Å) | 74.78 | 74.93 | 74.77 | 66.84 | 74.82 | 74.99 |
| c (Å) | 74.99 | 75.15 | 75.06 | 175.85 | 75.26 | 75.11 |
| Resolution limits | 50-1.05 Å | 50-1.15 Å | 50-1.15 Å | 50-1.85 Å |
50- 1.5Å |
50-1.5Å |
| Unique reflections | 110,339 | 86,963 | 86,085 | 20,778 | 39,440 | 39,554 |
| Completeness (%)a | 97.8 (94.5) | 100 (100) | 99.9 (99.5) |
99.8 (100) |
99.9 (99.9) |
100 (100) |
| Mean redundancya | 5.7 (3.8) | 10.7 (9.4) | 6.5 (4.7) | 13.6 (14.5) |
9.8 (8.9) |
10.3 (9.8) |
| Rmerge (%)a, b | 4.3 (43.7) | 7.9 (81.9) | 6.8 (56.0) | 9.9 (75.0) | 6.9 (16.0) |
4.8 (26.1) |
| 〈 I 〉/〈σ(I)〉a | 33.9 (2.6) | 34.1 (2.7) | 23.4 (2.3) | 31.1 (3.5) | 36.8 (13.0) |
46.1 (8.4) |
| Model Statistics | A104T | E163K | M26I | A104V | M26L | E163K/R145E |
| Molecules in ASU | 1 | 1 | 1 | 1 | 1 | 1 |
| Number of modeled residues |
187 | 186 | 187 | 191 | 187 | 187 |
| Number of solvent molecules |
229 | 178 | 255 | 189 | 211 | 212 |
| Number of residues in dual conformations |
12 | 10 | 6 | 2 | 11 | 4 |
| Rwork (%)c | 13.2 | 15.0 | 12.9 | 18.1 | 16.7 | 18.2 |
| Rwork (%) for FO>4σ(FO)c,d |
11.4 | 12.3 | 11.0 | N.A. | N.A. | N.A. |
| Rfree (%)e | 15.0 | 17.7 | 15.6 | 22.6 | 17.7 | 20.3 |
| Rfree (%) for FO>4σ(FO)e,d |
12.9 | 14.6 | 13.6 | N.A. | N.A. | N.A. |
| Rall (%)f | 13.3 | 15.0 | 12.9 | 18.3 | 16.7 | 18.3 |
| Rall (%) for FO>4σ(FO)d |
11.4 | 12.3 | 10.9 | N.A. | N.A. | N.A. |
| Mean protein Beq (Å2) |
14.2 | 16.6 | 13.4 | 19.7 | 11.0 | 15.8 |
| Mean protein anisotropyg |
0.48 | 0.53 | 0.47 | 1 | 1 | 1 |
| Mean solvent Beq (Å2) |
32.4 | 34.0 | 31.6 | 33.2 | 27.6 | 31.6 |
| Mean solvent anisotropyg |
0.39 | 0.42 | 0.38 | 1 | 1 | 1 |
| RMS bond length deviation (Å) |
0.02 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 |
| RMS angle length deviation (Å)d |
0.03 | 0.03 | 0.03 | N.A. | N.A. | N.A. |
| RMS bond angle deviation (degrees)h |
N.A. | N.A. | N.A. | 1.3 | 1.2 | 1.2 |
| RMS chiral volume deviation (Å3) |
0.11 | 0.09 | 0.09 | 0.09 | 0.07 | 0.08 |
Values in parenthesis indicate the statistics in the highest resolution shells. For the M26I and E163K DJ-1 datasets, this range is 1.19-1.15 Å. For the A104T DJ-1 dataset, the highest resolution shell is 1.09-1.05 Å. For the M26L and E163K/R145E datasets, it is 1.55-1.50 Å and for the A104V dataset, it is 1.92-1.85 Å.
, where i is the ith observation of a reflection with indices h,k,l and angle brackets indicate the average over all i observations.
, where Fchkl is the calculated structure factor amplitude with index h,k,l and Fohkl is the observed structure factor amplitude with index h,k,l.
A statistic that is reported for structures refined in SHELXL; N.A., not applicable.
Rfree is calculated as Rwork, where the Fohkl are taken from a test set comprising 5% of the data that were excluded from the refinement(51).
Rall is calculated as Rwork, where the Fohkl include all measured data (including the Rfree test set).
Anisotropy is calculated as the ratio of the smallest to the largest eigenvalues of the refined ADP tensor and is calculated using PARVATI (64).
A statistic that is reported for structures refined in REFMAC5; N.A., not applicable.
Crystal structure refinement
SHELX-97 was used for the refinement of coordinates and anisotropic displacement parameters for A104T, E163K, and M26I DJ-1. Refinement was performed against an intensity-based, weighted least squares residual target function that included stereochemical and displacement parameter restraints (50) and excluded a test set of 5% of randomly chosen reflections that were sequestered and used for the calculation of the Rfree value (51). The same reflections were chosen for inclusion in the test set for each of the three mutant structures. All measured data were used in the refinement and a bulk solvent correction was employed to allow the inclusion of the low resolution reflections. Initial rigid body refinement at 2.5 Å resolution using the model of human DJ-1 (PBD code 1P5F) (39) was followed by multiple cycles of stereochemically restrained refinement of coordinates and isotropic B-factors at 1.5 Å resolution using a stepwise increase in resolution (STIR instruction). Manual adjustments to the model, including construction of the initial solvent model, were made by inspection of 2mFo-DFc and mFo-DFc electron density maps in the program COOT (52). Manual adjustments to the model were followed by additional cycles of conjugate gradient refinement and inclusion of all data using the STIR instruction. Upon convergence of the refinement using a model with isotropic B-factors, anisotropic atomic displacement parameters (ADPs) were introduced, resulting in a 3-5% decrease in both R and Rfree. The final cycles of refinement were performed with riding hydrogen atoms (excluding the hydrogen atoms on Oγ of serine, O of tyrosine, Oγ1 of threonine, and Nδ1 of histidine), followed by combination of the working and test set data during the final cycles of refinement.
The structures of A104V, M26L, and E163K/R145E DJ-1 were refined in REFMAC5(53), part of the CCP4 suite of programs(54). A test set comprising 5% of the reflections were chosen at random and sequestered for the calculation of Rfree. A104V DJ-1 crystallizes in spacegroup P6522, which has not been previously observed for DJ-1. Therefore, the structure was solved by molecular replacement using PHASER (55, 56) with wild-type DJ-1 (PDB 1P5F) as a search model. For each structure, conjugate gradient minimization refinement against a maximum likelihood, amplitude-based target function that included stereochemical and B-factor restraints was followed by manual adjustments to the models guided by inspection of 2mFo-DFc and mFo-DFc electron density maps in COOT(52). Anisotropic scaling and a bulk solvent model were included in the refinement. The geometric and stereochemical quality of each model was validated using both the MolProbity server (57) and the validation tools within COOT (52). Final model statistics are shown in Table 1.
Differential Scanning Calorimetry
DSC experiments were performed using untagged wild-type, A104T, M26I and E163K DJ-1 at 10-20 μM concentration in a VP-DSC microcalorimeter (Microcal) over a temperature range of 10-100°C at a scan rate of 60°C/hour in passive feedback mode. Reduced DJ-1 was dialyzed against vacuum degassed DSC buffer (10 mM HEPES pH=7.5) supplemented with 1mM β-mercaptoethanol at 4°C. All DJ-1 samples were diluted to 0.01-0.02 mM in dialysis buffer and the protein concentration was verified by A280 using an extinction coefficient of ε280=4000 M−1 cm−1. Prior to loading the sample, 5-10 reference buffer-buffer scans were collected and the protein sample was loaded “in cycle” to avoid instrument thermal hysteresis that is invariably observed in DSC for the first scan of a series. All thermograms were collected with freshly prepared samples and the data were processed by reference thermogram subtraction, concentration normalization, and progress baseline subtraction in the ORIGIN software package for DSC analysis (Microcal, Northhampton, MA). All the DJ-1 proteins studied here denature irreversibly upon heating, as judged by the absence of an unfolding endotherm during a second 10-100°C scan. This second 10-100°C scan was performed after cooling from 100-10°C at a rate of 60°C/hour followed by a 15 minute rest period at 10°C. Tm values differ by 1°C or less in replicate DSC experiments on a representative DJ-1 sample.
Sedimentation Equilibrium Ultracentrifugation
Sedimentation equilibrium ultracentrifugation was performed at 20°C using a Beckman Coulter XL-I analytical ultracentrifuge (Beckman-Coulter, CA) employing a Ti-50 rotor and absorbance optics. Tag-cleaved protein samples were thawed on ice and buffer exchanged by rapid dialysis for 2 to 3 hours at 4° C against 25 mM HEPES, pH=7.5, 100 mM KCl and 1 mM DTT. After dialysis, the samples and buffer were centrifuged to remove particulates and diluted to the required concentration (1.0, 1.5 or 2.0 mg/ml) in the dialysis buffer. Samples (110 μl) and buffer (125 μl) were loaded in a six sector CFE sample cell fitted with quartz windows, allowing the monitoring of three different concentrations of sample simultaneously. Sedimentation equilibrium ultracentrifugation was performed at three different speeds: 1.7×104, 2.0×104 and 2.4×104 rpm, and the absorbance of each sample was monitored at 270 and 277 nm as a function of radius. Absorbance scans were collected after 20 hours and 22 hours of centrifugation for every sample and compared to ensure that equilibrium had been attained, after which the speed was increased and the same procedure was followed. The partial specific volume of the protein samples and the solvent density were calculated using the program SedNTerp(58), using the partial specific volume of Tris as a substitute for the HEPES buffer that was used in this experiment. The data were analyzed using the Origin 6.0 data analysis software provided by the manufacturer. A variety of self-association models were fit to the data and the quality of fit was judged by both the magnitude and absence of systematic trends in the fit residuals. A model containing a single dimeric species for each sample fit the data adequately, and more complex models that included monomer-dimer or dimer-tetramer self-association did not improve the fit. The general equation used to fit the dimeric model to the data was:
Where A(r) is the absorbance as a function of radial position in the sector cell, A(r0) is the absorbance at the reference position r0, M is the molecular weight of the protein dimer in g mol−1, ν̅ is the partial specific volume of the protein (ml g−1), ρ is the density of the solvent (g ml−1), ω is the angular velocity of the rotor (rad s−1), r is radial position in the sector cell, r0 is the reference position, R is the gas constant, T is temperature (K), and C is the baseline offset.
Circular Dichroism (CD) Spectroscopy
CD spectra between 260 and 190 nm were collected at room temperature using a Jasco J-810 CD spectrometer employing a grating with 3400 lines cm−1. Tag cleaved DJ-1 proteins were diluted to 10 μM in 10 mM potassium phosphate buffer, pH=7.5 and spectra were measured in continuous scanning mode at 50 nm min−1 with a 2 nm bandwidth and an accumulation of five scans per spectrum. The instrument response time was 1 second and the data pitch was 1 nm. Spectra were normalized to units of mean molar residue ellipticity (deg cm2 dmol−1 residue−1) using protein concentrations determined by A280 (ε280=4000 M−1 cm−1) according to the following equation: where [θ(λ)] is the mean residue molar ellipticity as a function of wavelength, n is the number of residues in the protein, c is the concentration of the protein (M), l is the sample path length (cm), and θobs(λ) is the observed ellipticity as a function of wavelength (nm). Approximate estimates of secondary structural content for the measured CD spectra were obtained using a Kohonen neural network trained against a set of 18 CD spectra collected from model proteins as implemented in the program K2D (59)
Results
DJ-1 is destabilized by each PD-associated missense mutation
The most common effect of disease-associated missense mutations is the reduction of protein stability(60). As each of the three PD-associated DJ-1 mutations have the potential to destabilize DJ-1, the thermal stabilities of wild-type and the three PD-associated mutants in their fully reduced forms were determined by differential scanning calorimetry (DSC). The denaturation of DJ-1 is irreversible under our experimental conditions, therefore no thermodynamic analysis of the unfolding transitions was performed. The apparent melting temperatures (Tm), however, still provide a useful measure of the relative stabilities of the proteins. The wild-type protein has a Tm of 66.2°C, which is reduced by each PD mutation to varying extents; M26I (63.4°C), A104T (58.7°C) and E163K (55.1°C). The unfolding endotherm for each sample is unimodal, allowing a meaningful Tm to be determined for each unfolding transition (Figure 2). Our results show that M26I is the least disruptive of the three PD-associated mutations studied here, while A104T and E163K both substantially reduce the melting temperature of DJ-1.
Figure 2.

All three PD-associated mutations thermally destabilize DJ-1. Differential scanning calorimetry (DSC) of wtDJ-1 (filled circles), M26I (open squares), A104T (filled squares) and E163K (grey triangles) shows that each pathogenic mutation reduces the apparent melting temperature (Tm) of DJ-1. The molar heat capacity at constant pressure (Cp) is calculated from the observed endotherms (ordinate), but cannot be subjected to thermodynamic analysis due to the irreversibility of DJ-1 denaturation. The Tm for each sample reported in the text is taken from the fit of a non-two state unfolding transition to the measured endotherm.
Dimerization and secondary structure of DJ-1 in solution is unchanged by three PD-associated mutations
DJ-1 exists as a homodimer and buries 2600 Å2 of surface area at the dimer interface(39). One well-characterized PD-associated missense mutation, L166P, disrupts the DJ-1 dimer and results in loss of protective activity(33, 34). As DJ-1 appears to be an obligate homodimer, disruption of the dimer interface might be one possible mechanism by which other PD-associated mutations impair DJ-1 function. Sedimentation equilibrium ultracentrifugation indicates that this is not the case for A104T, E163K and M26I DJ-1, all of which exist as dimers in solution (Figure 3A-D). Adequate fits to the observed data could be obtained with a model that assumes that only the dimeric species is present (Figure 3), with no evidence of either monomeric or higher order oligomeric states of the protein. Consequently, we made no attempt to characterize the Kd of the monomer-dimer equilibrium for these proteins, although this was done in a thorough previous study(45). Molecular weights for the dimeric species of each sample determined from the sedimentation equilibrium ultracentrifugation experiment shown in Figure 3 are provided in Supporting Information Table S1.
Figure 3.

Sedimentation equilibrium ultracentrifugation of DJ-1 and PD-associated mutants. Each panel represents the measured absorbance at 277 nm as a function of radius (lower plots), and the residuals after fitting to a dimeric model for each sample (upper plots) after sedimentation equilibrium ultracentrifugation. The best-fit curve is shown as a solid line in the lower plots for each sample. Shown in panel A is wtDJ-1, panel B is A104T, panel C is E163K and panel D is M26I. Representative data for an experiment conducted at 2×104 rpm and 25°C is shown. Other self-association models resulted in no improvement in the residuals and thus the simple dimer model was retained.
The secondary structural content of DJ-1 in solution is also unaffected by the A104T, E163K and M26I mutations, as shown by comparison of far-UV CD spectra (Figure 4). The secondary structural content of DJ-1 in solution is approximately 35% α-helix, 18% β-strand and 47% coil/turn/other, as determined using the program K2D(59) (see Experimental Procedures). These values agree reasonably well with the secondary structural content of DJ-1 calculated from the crystal structure using the PDB annotation for secondary structure in 1P5F (35% α-helix, 27% β-strand, 38% other), indicating that none of the PD-associated mutants significantly alter the secondary structure of DJ-1 in solution. We note that these results differ from those of a previous study that reported reduced secondary structural content for M26I DJ-1(45). Some possible reasons for this discrepancy are provided in the Discussion.
Figure 4.

The secondary structural content of DJ-1 in solution is unaffected by three PD-associated mutations. Circular dichroism (CD) spectra were collected at 20°C, pH=7.5 for 10μM wtDJ-1 (filled circles), M26I (open squares), A104T (filled squares) and E163K (grey triangles) and show no significant differences in the mean residue molar ellipicity ([Θ]; ordinate) as a function of wavelength.
The A104T mutation introduces disorder and water into the hydrophobic core of DJ-1
A104 is very highly conserved in homologues of DJ-1 and is located near the C-terminal end of β-strand 5 near a sharp turn that contains the functionally critical C106 residue. A104 resides in a tightly packed hydrophobic environment and the small size of the A104 sidechain allows for efficient packing with bulkier surrounding residues (L72, P109, and L112) in the central parallel β-sheet of DJ-1. The 1.05 Å resolution crystal structure of A104T DJ-1 shows that this substitution causes ∼0.4 Å displacements of L72 and L112 to alleviate steric clashes with the bulkier sidechain of T104 (Figure 5A). Apart from these local perturbations near the site of the A104T substitution, there are no major changes in either the structure or degree of order in other regions of the protein (Supporting Information Figures S2 and S3A).
Figure 5.
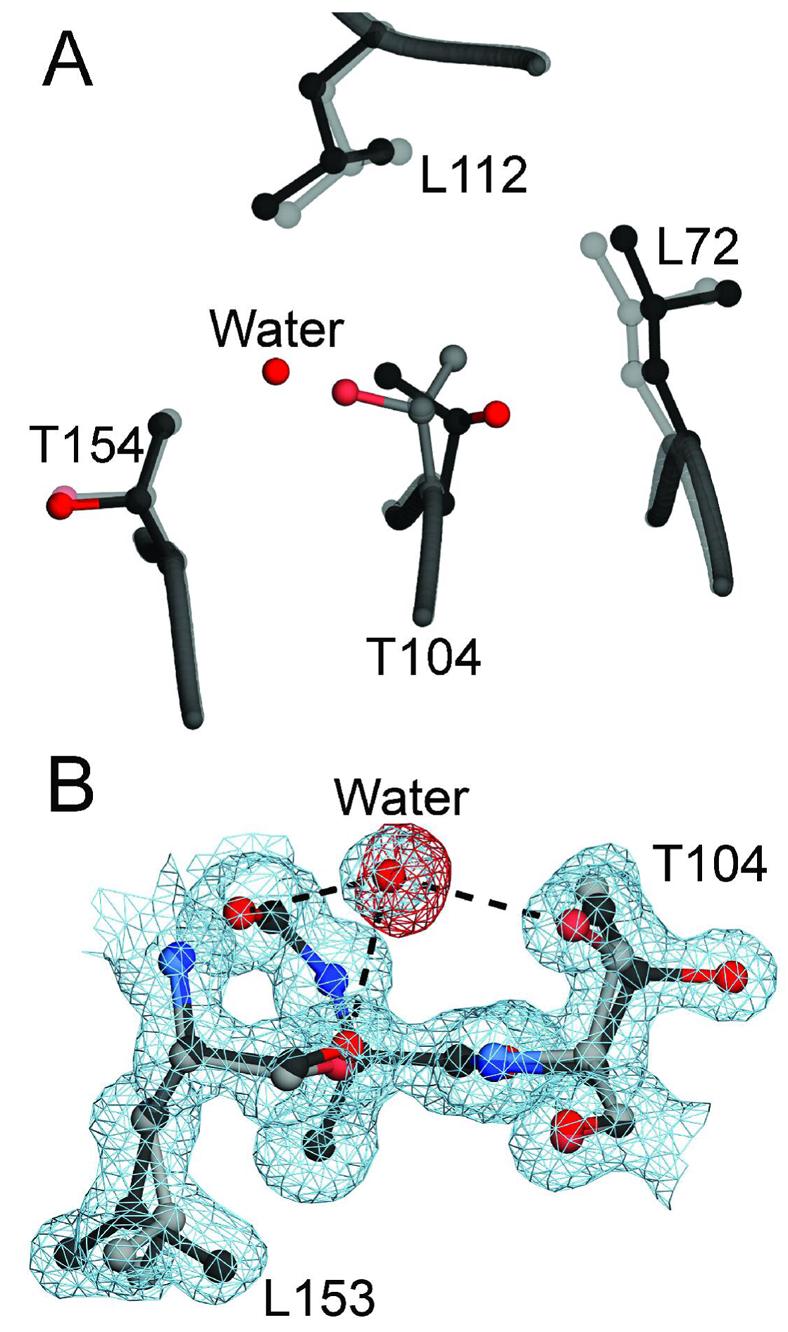
Buried solvent and sidechain rotameric disorder in A104T DJ-1. Panel A shows a superposition of wild-type (1SOA (5); semi-transparent grey) and A104T DJ-1 (dark grey) in the region around the site of the substitution. The introduction of a bulkier and disordered sidechain at residue 104 results in structural perturbations of the surrounding residues L72 and L112. Panel B shows a 1.05 Å resolution view of the discrete rotameric disorder at T104 and the partially occupied water molecule. Inspection of 2mFO-DFC electron density contoured at 1σ (blue) shows that the buried water molecule forms three hydrogen bonds (dashed lines) with surrounding residues, including the Oγ atom of one conformation of T104. Negative difference (mFO-DFC) electron density contoured at −3σ (red) indicates that the buried water molecule is partially occupied. This negative difference electron density disappeared when the occupancy of the water was reduced to 0.5 in the final model. The figure was created with POVscript+(65).
In addition, the T104 sidechain is discretely disordered and samples two rotameric conformations with refined occupancies of 0.54 and 0.46. The conformational disorder at T104 is correlated with the presence of a partially occupied water molecule that makes a hydrogen bond to Oγ in one conformation of T104 (Figure 5B). The 2mFO-DFC electron density for this water molecule is unambiguous (4.8 σ), however initial attempts to model this water molecule at full occupancy resulted in pronounced (−5.2 σ) negative mFO-DFC electron density (Figure 5B) after extensive positional and displacement parameter refinement in SHELX (see Experimental Proceedures). Therefore, the occupancy of this water molecule was reduced to 0.5 in order to eliminate this difference electron density, although this results in no significant change in the Rfree value. The reduced occupancy of this water molecule closely matches the refined occupancy (0.46) of the conformation of the discretely disordered T104 residue with which it interacts. This indicates that the presence of this water molecule in the core of DJ-1 is correlated with discrete disorder at T104. Further supporting this argument, this water molecule is absent in all other crystal structures of DJ-1. We propose that the A104T substitution destabilizes DJ-1 principally by burying a discretely disordered polar amino acid and an associated water molecule in a largely hydrophobic environment, thereby ferrying solvent into the core of the protein.
To establish to what degree the polar character of T104 influences the discrete disorder in the core of DJ-1 created by the A104T mutation, we engineered a non-polar A104V substitution and determined its structure using X-ray crystallography. The 1.85 Å resolution crystal structure of A104V DJ-1 shows that V104 is better ordered than T104, despite the very similar steric properties of these two residues (Figure 6A). A nearby peak in the difference electron density map (mFO-DFC) contoured at 3.0 σ suggests that V104 samples a second minor sidechain conformation (Figure 6A). The good agreement between the model and the 2mFO-DFC electron density indicates, however, that V104 exists predominantly in the modeled rotamer (Figure 6A). As expected, no buried water is observed near V104 in A104V DJ-1. The non-polar character of V104 eliminates its ability to hydrogen bond both to the buried water molecule observed in A104T DJ-1 and to the backbone carbonyl oxygen of L72 (Figure 6B), thereby reducing the stabilizing interactions that favor discrete disorder at T104. Therefore, we conclude that the discrete disorder observed in A104T DJ-1 is due largely (but not entirely) to the enhanced hydrogen bonding potential of T104.
Figure 6.
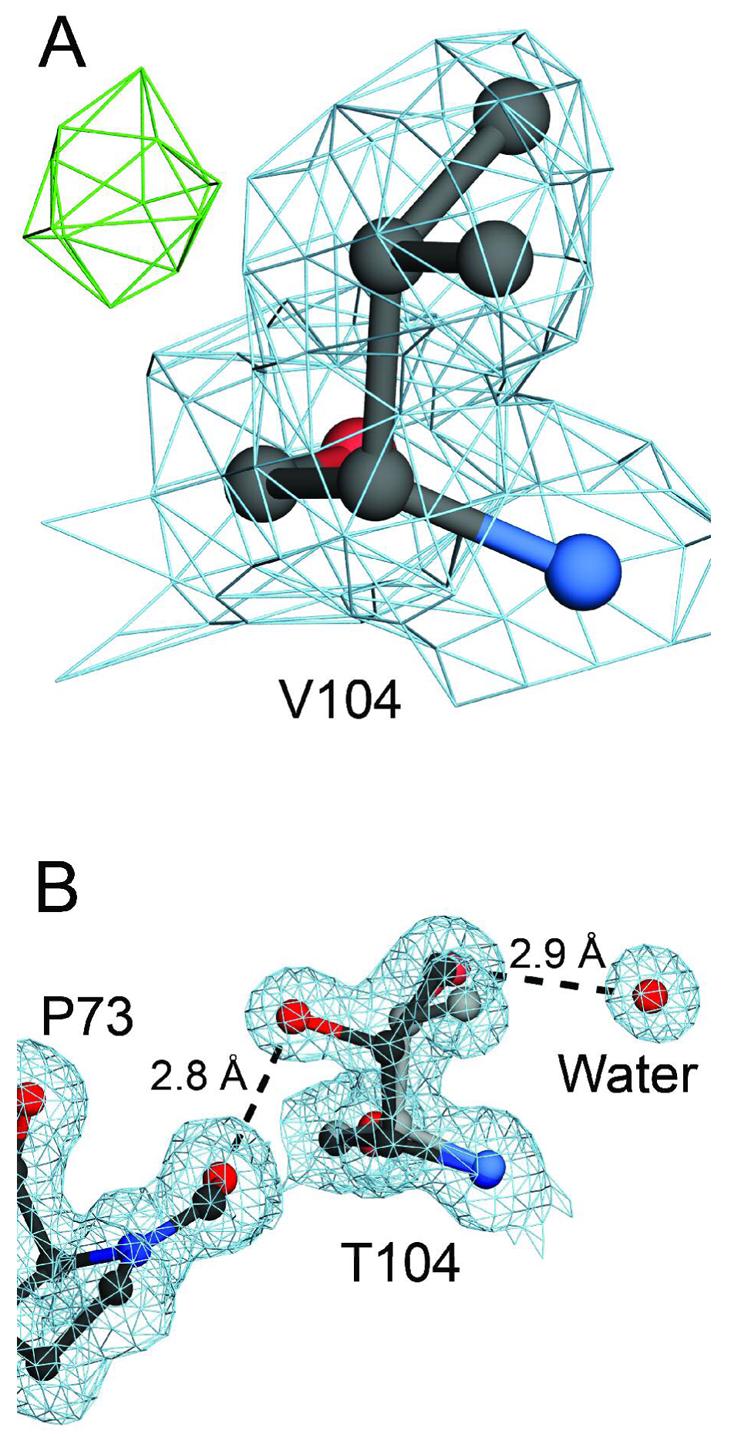
Hydrogen bonding stabilizes discrete disorder at T104 in A104T DJ-1. Panel A shows the environment of an engineered A104V substitution in DJ-1. Inspection of 1.85 Å resolution 2mFo-DFc electron density at 1.0σ (blue) shows that V104 is less disordered than T104, despite the steric similarity of these two residues. Positive difference (mFo-DFc) electron density contoured at 3.0σ (green), however, indicates that the residue likely samples a second minor conformation. In panel B, T104 makes two mutually exclusive hydrogen bonds that favor discrete disorder for this residue. The higher occupancy conformation for T104 is shown in the darker line, electron density (2mFo-DFc) is contoured at 1.0σ (blue), and hydrogen bonds are shown in dashed lines with their distances provided in Ångstroms. The orientation of residue 104 in panels A and B is the same. The figure was created with POVscript+(65).
E163K disrupts a key salt bridge in DJ-1
In wtDJ-1, the carboxylate sidechain of Glu163 makes a salt bridge with the guanidinium sidechain of R145, which in turn anchors the C-terminus of the other DJ-1 monomer at the dimer interface by donating two hydrogen bonds to the peptide carbonyl oxygen of V186 (Figure 7A). The very high degree of conservation of both E163 and R145 and the electrostatically conservative character of sequence variations at these two positions in homologues of DJ-1 suggests that this salt bridge interaction is likely important. The 1.15 Å resolution crystal structure of E163K DJ-1 shows that substitution of lysine for E163 disrupts this salt bridge and results in increased mobility of R145 due to electrostatic conflict and loss of hydrogen bonding potential between K163 and R145 (Figure 7B). The principal structural change in E163K DJ-1 is that the sidechain of K163 swings away from R145 to alleviate repulsive electrostatic interactions resulting from the E163K mutation.
Figure 7.
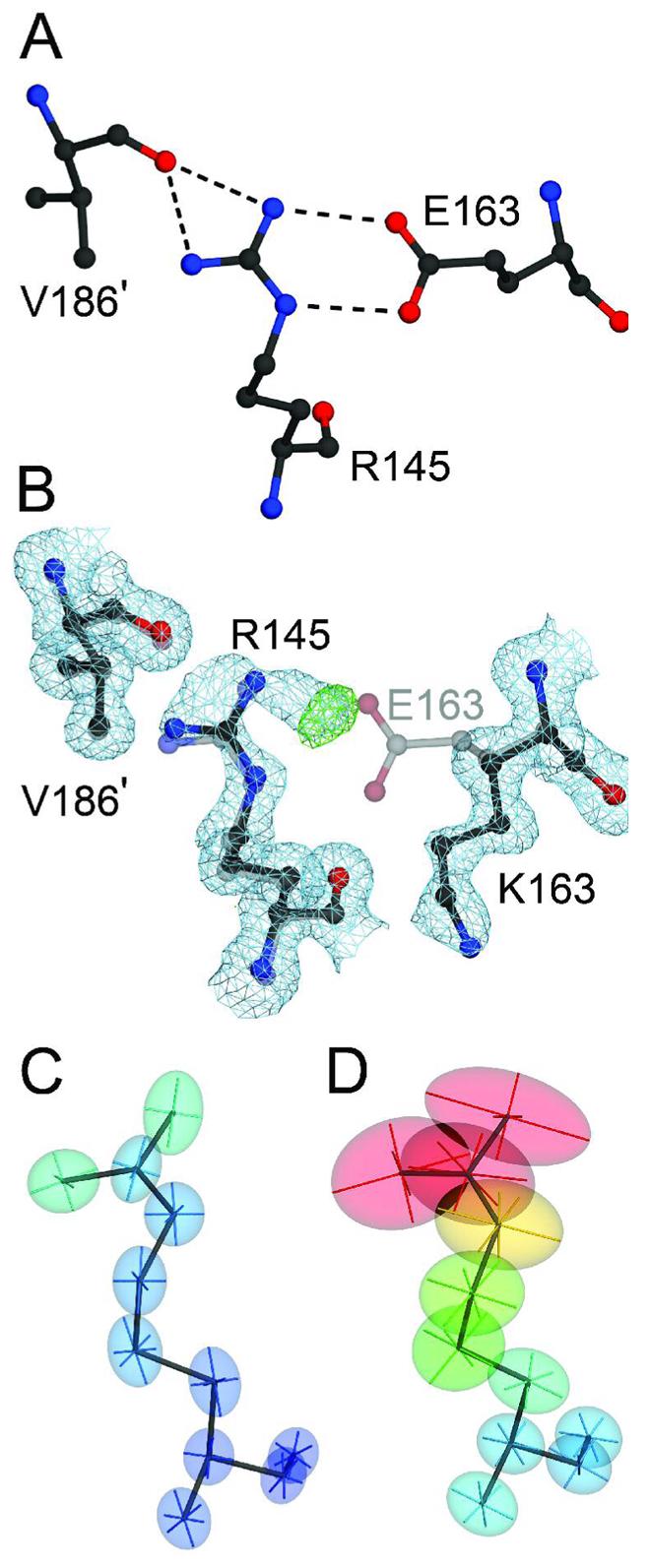
The E163K substitution disrupts a critical salt bridge in DJ-1. Panel A shows the network of hydrogen bonds (dashed lines) that extend from the E163-R145 salt bridge to V186 in the other monomer of the DJ-1 dimer (marked with a prime). Panel B shows a superposition of wild-type (1SOA(5); semi-transparent grey) and the 1.15 Å resolution crystal structure of E163K DJ-1 (dark grey), showing that the substitution of a lysine at position 163 disrupts the salt bridge with R145 and leads to reorientation of the K163 sidechain. Electron density (2mFO-DFC) contoured at 1σ (blue) and positive difference mFO-DFC electron density contoured at 4σ (green) calculated from the E163K DJ-1 model indicate that loss of the E163-R145 salt bridge leads to disorder at Arg145 in E163K DJ-1. The increased mobility of R145 is evident by comparison of the thermal ellipsoids calculated from the refined ADPs of wtDJ-1 (Panel C) and E163K DJ-1 (Panel D). The thermal ellipsoids with their principal axes are shown at the 50% probability level and are colored to indicate the magnitude of the total atomic displacement (blue; 6 Å2, red; 40 Å2). The figure was created with POVscript+(65).
A comparison of refined anisotropic atomic displacement parameters (ADPs) for R145 in both wild-type and E163K DJ-1 shows that R145 becomes more disordered as a result of the E163K mutation, as demonstrated by the greatly elevated ADPs for the guanidinium sidechain of R145 (Figure 7C,D). The residue averaged B-factor for R145 increases from 10.4 Å2 in wtDJ-1 to 25.6 Å2 in E163K DJ-1 and B-factors throughout helices G and H (residues 159–189) in E163K DJ-1 are also elevated, demonstrating that loss of the R145-E163 salt bridge leads to increased disorder in this region of DJ-1 (Supporting Information Figure S3C). We propose that the E163K substitution destabilizes DJ-1 through disruption of a key salt bridge between E163 in α-helix G and R145, located in the turn between β-strands 8 and 9. The disruption of this salt bridge leads to a loss of anchoring hydrogen bonds between R145 and the C-terminus of the other monomer in the DJ-1 dimer, increasing the mobility of R145 and thereby weakening a network of hydrogen bonds that spans the dimer interface.
The salt bridge between R145 and E163 in wtDJ-1 is stabilized by both charge complementarity and hydrogen bonding. The E163K mutation eliminates both types of interaction, leading to destabilization of the protein. To determine the relative contribution of electrostatics and hydrogen bonding to the stability of the 145-163 salt bridge, an engineered E163K/R145E double mutant was created to restore the electrostatic interaction between residues 145 and 163. The 1.5 Å resolution crystal structure of E163K/R145E DJ-1 shows that the sidechain of K163 moves to form a salt bridge with E145 (Figure 8) in the engineered double mutant DJ-1. Despite the restoration of the salt bridge, the sidechain of K163 is partially disordered as indicated by weak 2mFO-DFC electron density, prominent negative difference mFO-DFC electron density, and elevated B-factors (Figure 8). The disorder at K163 in E163K/R145E DJ-1 indicates that hydrogen bonding between the guanidinium sidechain of R145 and the carboxylate of E163 in wtDJ-1, which is absent in the engineered E163K/R145E double mutant, is a critical contributor to stability of the interaction between residues 145 and 163 in DJ-1. Further support for this conclusion is provided by the robust precipitation of E163K/R145E DJ-1 that occurs during unsuccessful attempts to measure its Tm by DSC.
Figure 8.
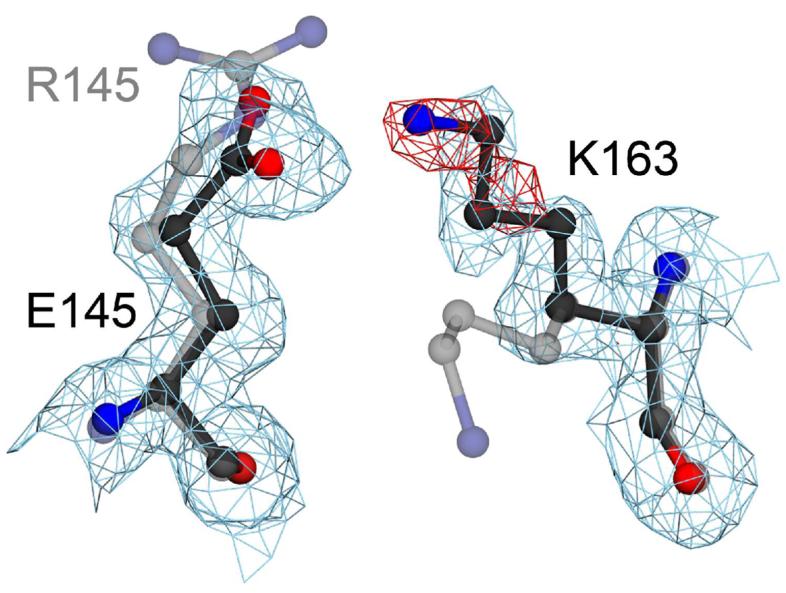
An engineered E163K/R145E double mutation creates a poorly ordered salt bridge. A superposition of E163K (semi-transparent grey) and an engineered E163K/R145E DJ-1 double mutant (darker line) shows that restoring electrostatic complementarity creates a salt bridge with the poorly ordered sidechain of K163. Electron density (2mFO-DFC) calculated from the 1.5 Å resolution crystal structure of E163K/R145E DJ-1 is contoured at 1σ (blue) and shows that K163 moves to interact with E145. Prominent negative difference (mFO-DFC) electron density contoured at −3.5σ (red) indicates that K163 is poorly ordered and that native hydrogen bonding interactions between R145 and E163 in wtDJ-1 are required for optimal interactions in this region of the protein. The figure was created with POVscript+(65).
M26I causes packing defects in the core of the DJ-1 dimer
M26 is a conserved residue in α-helix A that is located in the core of the protein. Although M26 is near the dimer interface, it does not directly participate in dimer-spanning contacts (Figure 1). The 1.15 Å resolution crystal structure of M26I DJ-1 reveals that this point mutation is the least structurally disruptive of the Parkinsonism-associated mutations studied here (Figure 9A). The modest disruption of hydrophobic packing contacts around I26 is a consequence of introducing a β-branched amino acid in the tightly packed hydrophobic core of the protein. The steric conflict between the Cγ2 atom of I26 and the sidechain of I31 results in a ∼0.7 Å displacement of I31 away from I26. In addition, the M26I substitution creates a small (∼30 Å3) cavity with consequent loss of optimal packing contacts in the interior of the protein due to the loss of the Cε and Sδ atoms of M26.
Figure 9.
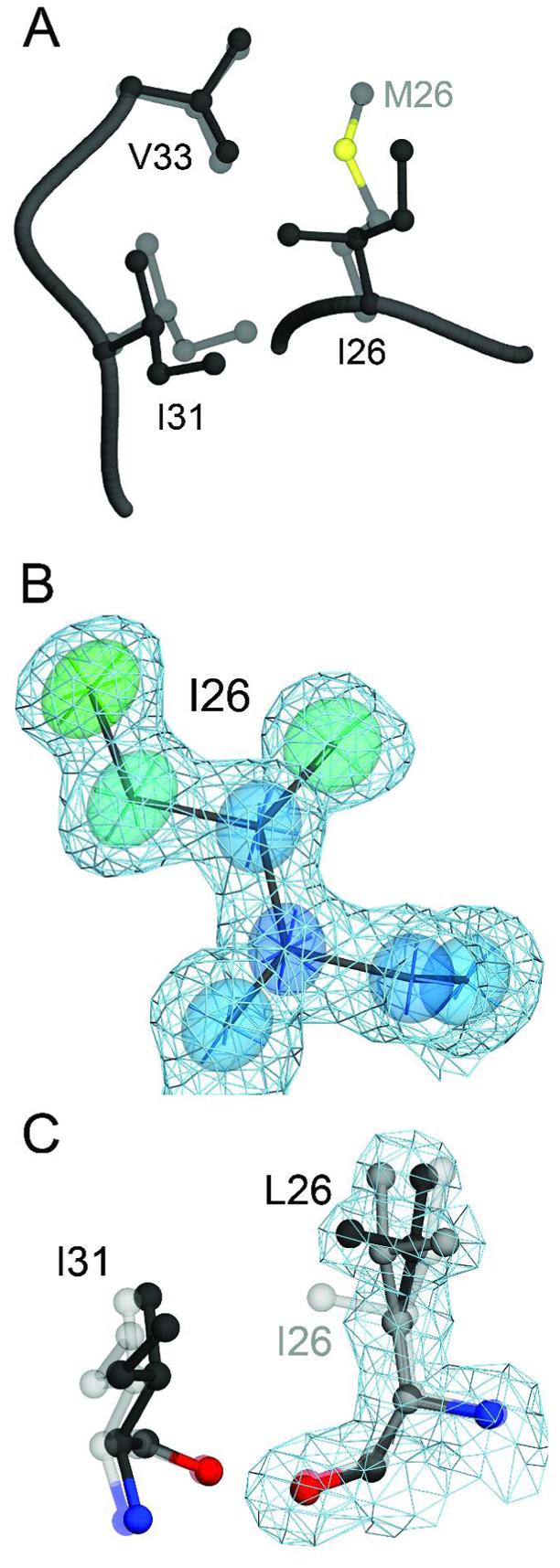
The M26I substitution results in packing defects in the core of DJ-1. Panel A shows a superposition of wild-type (1SOA(5); semi-transparent grey) and M26I DJ-1 (dark grey) in the region around the site of the substitution. The M26I substitution results in a ∼0.7 Å displacement of I31 to accommodate the Cγ2 atom of I26. In addition, the loss of the Sδ and Cε atoms of M26 creates a minor packing defect in the hydrophobic core of the protein. In Panel B, 2mFO-DFC electron density contoured at 1σ (blue) is shown with thermal ellipsoids calculated from the refined ADPs of I26, indicating that this residue is well-ordered in the crystal. The thermal ellipsoids are shown at the 60% probability level with their principal axes and are colored to indicate the magnitude of the total atomic displacement (blue; 6 Å2, red; 15 Å2). Panel C shows a superposition of M26I (semi-transparent grey) and the engineered M26L substitution, with alternate conformations for L26 shown in black and dark grey. The cavity created by the M26L substitution allows L26 to sample two rotameric conformations in the core of DJ-1, as shown by 2mFO-DFC electron density contoured at 1σ (blue) calculated from the 1.5 Å resolution crystal structure of M26L DJ-1. The figure was created with POVscript+((65).
Unlike A104T and E163K DJ-1 (see above), the M26I mutation does not result in a significant change in sidechain conformation or degree of order at the site of the substitution. I26 has a mean sidechain B-factor of 9.3 Å2, which is only slightly elevated compared to the mean sidechain B-factor of 7.0 Å2 for M26 in oxidized wtDJ-1 that was refined at comparable resolution (PDB accession code 1SOA)(5). We note that the original 1.1 Å resolution crystal structure (1P5F) displays elevated B-factors and reduced anisotropy for most atoms as a result of radiation damage that was incurred during data collection(39). Therefore, the ADPs from a less damaged crystal structure of C106-sulfinic acid oxidized wtDJ-1 (1SOA) are used for comparison with the ADPs from the mutant DJ-1 crystal structures reported in this study(5). The ADPs and 2mFO-DFC electron density for I26 indicate that the residue does not show a pronounced directional bias in atomic mobility (mean residue anisotropy=0.59) and exists in a single rotameric state in the crystal (Figure 9B). Analysis of the crystal structure suggests that the modest thermal destabilization of M26I DJ-1 observed in DSC is due to minor core packing defects created by the branched sidechain of I26 and the loss of favorable packing contacts with the Cε and Sδ atoms of M26. These results are in broad agreement with previous molecular dynamics simulations that showed no major structural changes in response to the M26I missense mutation(61).
An engineered M26L substitution was created that eliminates the van der Waals conflict between I26 and I31 in M26I DJ-1. The 1.5 Å resolution crystal structure of M26L DJ-1 shows that, as expected, I31 returns to the conformation observed in wtDJ-1 (Figure 9C). Unexpectedly, the sidechain of L26 is discretely disordered and samples two conformations in the hydrophobic core of the protein (Figure 9C). This disorder is the result of the small cavity (∼30 Å3) created by the loss of the Cε and Sδ atoms of M26, which permits two rotameric states of L26 to be accommodated without steric clashes. Although similar disorder is not observed in the pathogenic M26I mutation, the alternate conformations of L26 confirm that the cavity formed by removal of the M26 sidechain permits greater conformational flexibility in the core of DJ-1 and likely contributes to the modest thermal destabilization of M26I DJ-1 observed by DSC (Figure 2).
Discussion
In this study, a combination of DSC, CD spectroscopy, and sedimentation equilibrium ultracentrifugation was used to show that the A104T, E163K and M26I PD-associated mutations in DJ-1 variably reduce the stability of the protein without causing major structural changes or loss of dimerization in solution. Atomic resolution X-ray crystallography provides a structural rationale for the differing effects of the A104T, E163K, and M26I substitutions on the thermal stability of reduced DJ-1. The A104T point mutation introduces a discretely disordered residue into a hydrophobic environment near the functionally critical residue C106. A partially occupied water molecule that is absent in structures of wtDJ-1 is hydrogen bonded to one of the two rotameric conformations of T104, and the burial of a disordered polar residue and its accompanying water molecule is proposed to account for much of the destabilization of A104T DJ-1. The E163K point mutation disrupts a critical salt bridge to R145, which leads to elevated mobility for the guanidinium moiety of R145 and consequently interferes with a network of hydrogen bonds that involves L186 and spans the dimer interface. We note that the homozygous E163K mutation in DJ-1 was originally isolated from patients that also carried mutations in the promoter region of the gene, complicating the interpretation of the structural result in the context of the unusual clinical pathology of E163K DJ-1(44). The M26I mutation results in packing defects in the core of the protein due to both steric conflict with I31 and the creation of a small cavity resulting from the replacement of the bulkier methionine sidechain with isoleucine. Considered in total, our structural analysis suggests that even minor perturbations to the structure of DJ-1 can result in impairment of function sufficient to cause disease. This observation lends strong support to previous suggestions (45) that compounds that increase DJ-1 stability may make attractive targets for possible use in the pharmacological enhancement of DJ-1 function. Our results suggest that the solvent-exposed region between α-helix G and β-strand 10, where the E163-R145 salt bridge is located, provides a particularly attractive target for future investigation.
The results described here differ in some respects from a previous report on the solution properties of M26I DJ-1(45). In particular, our CD spectroscopic and X-ray crystallographic data show that M26I DJ-1 is well-structured in solution and in the crystal, while Hulleman et al. show that M26I DJ-1 has reduced secondary structural content and displays a pronounced tendency to aggregate. We propose that this discrepancy is due to the differing oxidation states of our respective preparations of recombinant DJ-1. DJ-1 is subject to oxidation of its cysteine and methionine residues and this oxidation is relevant for the function of DJ-1 in vivo(5, 6, 62). We used reduced recombinant DJ-1 in this study, while Hulleman et al. used more oxidized samples that contained a mixture of C106-sulfinic and C106-sulfonic acid, as well as other oxidative modifications(45). Previous studies have shown that oxidation of C106 to the cysteine-sulfonic acid leads to a partial loss of secondary structure, diminished stability, and enhanced aggregation (47), possibly accounting for the reduced CD signal for M26I DJ-1 observed by Hulleman et al. (45). The differences in the properties of our reduced M26I DJ-1 and the more oxidized sample studied by Hulleman et al. suggest that the M26I mutation may preferentially destabilize more extensively oxidized forms of DJ-1 (i.e. DJ-1 containing cysteine-sulfonic acid and methionine sulfoxide), thereby enhancing the loss of properly folded DJ-1 under conditions of cellular oxidative stress. In support of the in vivo relevance of oxidation-induced changes in DJ-1 stability, Hulleman et al. note that all of their DJ-1 samples exhibit oxidative modifications that are similar to those observed in DJ-1 isolated from brain tissue(45).
Each of the three PD-associated mutations studied here causes a distinct type of defect, though none results in large-scale structural changes in DJ-1. One feature that these three PD-associated mutations share is that they are all located near the junctions between secondary structural elements in DJ-1. The structural integrity of the termini of secondary structural elements and the junctions between them is known to exert substantial influence on protein folding and unfolding kinetics (63). Therefore, mutation-induced changes in folding efficiency of DJ-1 may contribute to the pathogenicity of these mutations by reducing the amount of properly folded protein in the cell. This proposal is speculative, however, because detailed studies of the folding and conformational dynamics of DJ-1 and its pathogenic variants are currently lacking. A detailed biophysical characterization of DJ-1 will aid in understanding the impact of disease-associated mutations on the protective function of the protein and promises to be an active field of future study.
Supplementary Material
Acknowledgements
We thank the staff of BioCARS 14-BMC and of SSRL beamline 11-1 for beamline support, Dr. Hideaki Moriyama of the UNL Structural Biology Core Facility for assistance with rotating anode data collection, Dr. Donald Becker for assistance with the sedimentation equilibrium centrifugation experiments, and Dr. Todd Holyoak (University of Kansas Medical Center) for collection of the CD spectra. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under Contract No. W-31-109-Eng-38. Use of the BioCARS Sector 14 was supported by the National Institutes of Health, National Center for Research Resources, under grant number RR07707. Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences.
This work was supported in part by a grant to M.A.W. from the American Parkinson's Disease Association as well as a grant from the National Institutes of Health (P20RR17675). The XL-I analytical centrifuge was funded by NSF grant DBI-0619764.
ABBREVIATIONS
- ADP
anisotropic atomic displacement parameter
- CD
circular dichroism
- DSC
differential scanning calorimetry
- DTT
dithiothreitol
- HEPES
(4-(2-hydroyethyl)-1-piperazineethansulfonic acid
- PD
Parkinson's disease
- PEG
polyethylene glycol
- rpm
revolution per minute
- wt
wild-type
Footnotes
Experimental diffraction data and refined model coordinates have been deposited with the Protein Data Bank with accession codes 2RK3 (A104T DJ-1), 2RK4 (M26I DJ-1), 2RK6 (E163K DJ-1), 3B36 (M26L DJ-1), 3B38 (A104V DJ-1), and 3B3A (E163K/R145E DJ-1).
Supporting Information Available
Table S1 shows a comparison of the calculated molecular weights of the DJ-1 PD-associated mutants with those determined by fitting to the sedimentation equilibrium ultracentrifugation data shown in Figure 3. Figures S2 and S3 show the distribution of refined B-factors for wtDJ-1 and each of the PD-associated mutants studied in this work. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 2.Nagakubo D, Taira T, Kitaura H, Ikeda M, Tamai K, Iguchi-Ariga SM, Ariga H. DJ-1, a novel oncogene which transforms mouse NIH3T3 cells in cooperation with ras. Biochem. Biophys. Res. Commun. 1997;231:509–513. doi: 10.1006/bbrc.1997.6132. [DOI] [PubMed] [Google Scholar]
- 3.Bandyopadhyay S, Cookson MR. Evolutionary and functional relationships within the DJ1 superfamily. BMC Evol. Biol. 2004;4 doi: 10.1186/1471-2148-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lucas JI, Marin I. A New Evolutionary Paradigm for the Parkinson Disease Gene DJ-1. Mol. Biol. Evol. 2006;24:551–561. doi: 10.1093/molbev/msl186. [DOI] [PubMed] [Google Scholar]
- 5.Canet-Aviles RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S, Baptista MJ, Ringe D, Petsko GA, Cookson MR. The Parkinson's disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc. Natl. Acad. Sci. USA. 2004;101:9103–9108. doi: 10.1073/pnas.0402959101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi J, Sullards MC, Olzmann JA, Rees HD, Weintraub ST, Bostwick DE, Gearing M, Levey AI, Chin LS, Li L. Oxidative damage of DJ-1 is linked to sporadic Parkinson and Alzheimer diseases. J. Biol. Chem. 2006;281:10816–10824. doi: 10.1074/jbc.M509079200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li HM, Taira T, Maita C, Ariga H, Iguchi-Ariga SM. Protection against nonylphenol-induced cell death by DJ-1 in cultured Japanese medaka (Oryzias latipes) cells. Toxicology. 2006;228:229–238. doi: 10.1016/j.tox.2006.08.040. [DOI] [PubMed] [Google Scholar]
- 8.Meulener MC, Xu K, Thomson L, Ischiropoulos H, Bonini NM. Mutational analysis of DJ-1 in Drosophila implicates functional inactivation by oxidative damage and aging. Proc. Natl. Acad. Sci. USA. 2006;103:12517–12522. doi: 10.1073/pnas.0601891103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mitsumoto A, Nakagawa Y. DJ-1 is an indicator for endogenous reactive oxygen species elicited by endotoxin. Free Radic. Res. 2001;35:885–893. doi: 10.1080/10715760100301381. [DOI] [PubMed] [Google Scholar]
- 10.Park J, Kim SY, Cha GH, Lee SB, Kim S, Chung J. Drosophila DJ-1 mutants show oxidative stress-sensitive locomotive dysfunction. Gene. 2005;361:133–139. doi: 10.1016/j.gene.2005.06.040. [DOI] [PubMed] [Google Scholar]
- 11.Sekito A, Koide-Yoshida S, Niki T, Taira T, Iguchi-Ariga SM, Ariga H. DJ-1 interacts with HIPK1 and affects H2O2-induced cell death. Free Radic. Res. 2006;40:155–165. doi: 10.1080/10715760500456847. [DOI] [PubMed] [Google Scholar]
- 12.Shendelman S, Jonason A, Martinat C, Leete T, Abeliovich A. DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. Plos Biology. 2004;2:1764–1773. doi: 10.1371/journal.pbio.0020362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taira T, Saito Y, Niki T, Iguchi-Ariga SM, Takahashi K, Ariga H. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 2004;5:213–218. doi: 10.1038/sj.embor.7400074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takahashi-Niki K, Niki T, Taira T, Iguchi-Ariga SM, Ariga H. Reduced anti-oxidative stress activities of DJ-1 mutants found in Parkinson's disease patients. Biochem Biophys Res Commun. 2004;320:389–397. doi: 10.1016/j.bbrc.2004.05.187. [DOI] [PubMed] [Google Scholar]
- 15.Zhang L, Shimoji M, Thomas B, Moore DJ, Yu SW, Marupudi NI, Torp R, Torgner IA, Ottersen OP, Dawson TM, Dawson VL. Mitochondrial localization of the Parkinson's disease related protein DJ-1: implications for pathogenesis. Hum. Mol. Genet. 2005;14:2063–2073. doi: 10.1093/hmg/ddi211. [DOI] [PubMed] [Google Scholar]
- 16.Shinbo Y, Taira T, Niki T, Iguchi-Ariga SM, Ariga H. DJ-1 restores p53 transcription activity inhibited by Topors/p53BP3. Int J Oncol. 2005;26:641–648. [PubMed] [Google Scholar]
- 17.Taira T, Iguchi-Ariga SM, Ariga H. Co-localization with DJ-1 is essential for the androgen receptor to exert its transcription activity that has been impaired by androgen antagonists. Biol Pharm Bull. 2004;27:574–577. doi: 10.1248/bpb.27.574. [DOI] [PubMed] [Google Scholar]
- 18.Takahashi K, Taira T, Niki T, Seino C, Iguchi-Ariga SM, Ariga H. DJ-1 positively regulates the androgen receptor by impairing the binding of PIASx alpha to the receptor. J Biol Chem. 2001;276:37556–37563. doi: 10.1074/jbc.M101730200. [DOI] [PubMed] [Google Scholar]
- 19.Xu J, Zhong N, Wang H, Elias JE, Kim CY, Woldman I, Pifl C, Gygi SP, Geula C, Yankner BA. The Parkinson's disease-associated DJ-1 protein is a transcriptional co-activator that protects against neuronal apoptosis. Hum Mol Genet. 2005;14:1231–1241. doi: 10.1093/hmg/ddi134. [DOI] [PubMed] [Google Scholar]
- 20.Zhong N, Kim CY, Rizzu P, Geula C, Porter DR, Pothos EN, Squitieri F, Heutink P, Xu J. DJ-1 transcriptionally up-regulates the human tyrosine hydroxylase by inhibiting the sumoylation of pyrimidine tract-binding protein-associated splicing factor. J. Biol. Chem. 2006;281:20940–20948. doi: 10.1074/jbc.M601935200. [DOI] [PubMed] [Google Scholar]
- 21.Hod Y, Pentyala SN, Whyard TC, El-Maghrabi MR. Identification and characterization of a novel protein that regulates RNA-protein interaction. J. Cell. Biochem. 1999;72:435–444. [PubMed] [Google Scholar]
- 22.Shinbo Y, Niki T, Taira T, Ooe H, Takahashi-Niki K, Maita C, Seino C, Iguchi-Ariga SM, Ariga H. Proper SUMO-1 conjugation is essential to DJ-1 to exert its full activities. Cell Death Differ. 2006;13:96–108. doi: 10.1038/sj.cdd.4401704. [DOI] [PubMed] [Google Scholar]
- 23.Jin J, Li GJ, Davis J, Zhu D, Wang Y, Pan C, Zhang J. Identification of novel proteins interacting with both a-synuclein and DJ-1. Mol Cell Proteomics. 2006;6(5):845–859. doi: 10.1074/mcp.M600182-MCP200. 2007. [DOI] [PubMed] [Google Scholar]
- 24.Junn E, Taniguchi H, Jeong BS, Zhao X, Ichijo H, Mouradian MM. Interaction of DJ-1 with Daxx inhibits apoptosis signal-regulating kinase 1 activity and cell death. Proc. Natl. Acad. Sci. USA. 2005;102:9691–9696. doi: 10.1073/pnas.0409635102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clements CM, McNally RS, Conti BJ, Mak TW, Ting JP. DJ-1, a cancer- and Parkinson's disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl. Acad. Sci. USA. 2006;103:15091–15096. doi: 10.1073/pnas.0607260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou W, Freed CR. DJ-1 up-regulates glutathione synthesis during oxidative stress and inhibits A53T alpha-synuclein toxicity. J Biol Chem. 2005;280:43150–43158. doi: 10.1074/jbc.M507124200. [DOI] [PubMed] [Google Scholar]
- 27.Kim RH, Peters M, Jang Y, Shi W, Pintilie M, Fletcher GC, DeLuca C, Liepa J, Zhou L, Snow B, Binari RC, Manoukian AS, Bray MR, Liu FF, Tsao MS, Mak TW. DJ-1, a novel regulator of the tumor suppressor PTEN. Cancer Cell. 2005;7:263–273. doi: 10.1016/j.ccr.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 28.Tang B, Xiong H, Sun P, Zhang Y, Wang D, Hu Z, Zhu Z, Ma H, Pan Q, Xia JH, Xia K, Zhang Z. Association of PINK1 and DJ-1 confers digenic inheritance of early-onset Parkinson's disease. Hum Mol Genet. 2006;15:1816–1825. doi: 10.1093/hmg/ddl104. [DOI] [PubMed] [Google Scholar]
- 29.Bai Q, Mullett SJ, Garver JA, Hinkle DA, Burton EA. Zebrafish DJ-1 is evolutionarily conserved and expressed in dopaminergic neurons. Brain Res. 2006;1113:33–44. doi: 10.1016/j.brainres.2006.07.057. [DOI] [PubMed] [Google Scholar]
- 30.Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, Costa C, Tong Y, Martella G, Tscherter A, Martins A, Bernardi G, Roth BL, Pothos EN, Calabresi P, Shen J. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron. 2005;45:489–496. doi: 10.1016/j.neuron.2005.01.041. [DOI] [PubMed] [Google Scholar]
- 31.Kim RH, Smith PD, Aleyasin H, Hayley S, Mount MP, Pownall S, Wakeham A, You-Ten AJ, Kalia SK, Horne P, Westaway D, Lozano AM, Anisman H, Park DS, Mak TW. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc. Natl. Acad. Sci. USA. 2005;102:5215–5220. doi: 10.1073/pnas.0501282102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abou-Sleiman PM, Healy DG, Wood NW. Causes of Parkinson's disease: genetics of DJ-1. Cell Tissue Res. 2004;318:185–188. doi: 10.1007/s00441-004-0922-6. [DOI] [PubMed] [Google Scholar]
- 33.Miller DW, Ahmad R, Hague S, Baptista MJ, Canet-Aviles R, McLendon C, Carter DM, Zhu PP, Stadler J, Chandran J, Klinefelter GR, Blackstone C, Cookson MR. L166P mutant DJ-1, causative for recessive Parkinson's disease, is degraded through the ubiquitin-proteasome system. J. Biol. Chem. 2003;278:36588–36595. doi: 10.1074/jbc.M304272200. [DOI] [PubMed] [Google Scholar]
- 34.Moore DJ, Zhang L, Dawson TM, Dawson VL. A missense mutation (L166P) in DJ-1, linked to familial Parkinson's disease, confers reduced protein stability and impairs homo-oligomerization. J. Neurochem. 2003;87:1558–1567. doi: 10.1111/j.1471-4159.2003.02265.x. [DOI] [PubMed] [Google Scholar]
- 35.Honbou K, Suzuki NN, Horiuchi M, Niki T, Taira T, Ariga H, Inagaki F. The crystal structure of DJ-1, a protein related to male fertility and Parkinson's disease. J Biol Chem. 2003;278:31380–31384. doi: 10.1074/jbc.M305878200. [DOI] [PubMed] [Google Scholar]
- 36.Huai Q, Sun YJ, Wang HC, Chin LS, Li L, Robinson H, Ke HM. Crystal structure of DJ-1/RS and implication on familial Parkinson's disease. FEBS Letters. 2003;549:171–175. doi: 10.1016/s0014-5793(03)00764-6. [DOI] [PubMed] [Google Scholar]
- 37.Lee SJ, Kim SJ, Kim IK, Ko J, Jeong CS, Kim GH, Park C, Kang SO, Suh PG, Lee HS, Cha SS. Crystal structures of human DJ-1 and Escherichia coli Hsp31, which share an evolutionarily conserved domain. J. Biol. Chem. 2003;278:44552–44559. doi: 10.1074/jbc.M304517200. [DOI] [PubMed] [Google Scholar]
- 38.Tao X, Tong L. Crystal structure of human DJ-1, a protein associated with early onset Parkinson's disease. J. Biol. Chem. 2003;278:31372–31379. doi: 10.1074/jbc.M304221200. [DOI] [PubMed] [Google Scholar]
- 39.Wilson MA, Collins JL, Hod Y, Ringe D, Petsko GA. The 1.1-angstrom resolution crystal structure of DJ-1, the protein mutated in autosomal recessive early onset Parkinson's disease. Proc. Natl. Acad. Sci. USA. 2003;100:9256–9261. doi: 10.1073/pnas.1133288100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gorner K, Holtorf E, Waak J, Pham TT, Vogt-Weisenhorn DM, Wurst W, Haass C, Kahle PJ. Structural determinants of the C-terminal helix-kink-helix motif essential for protein stability and survival promoting activity of DJ-1. J Biol Chem. 2007;282:13680–13691. doi: 10.1074/jbc.M609821200. [DOI] [PubMed] [Google Scholar]
- 41.Clark LN, Afridi S, Mejia-Santana H, Harris J, Louis ED, Cote LJ, Andrews H, Singleton A, Wavrant De-Vrieze F, Hardy J, Mayeux R, Fahn S, Waters C, Ford B, Frucht S, Ottman R, Marder K. Analysis of an early-onset Parkinson's disease cohort for DJ-1 mutations. Mov Disord. 2004;19:796–800. doi: 10.1002/mds.20131. [DOI] [PubMed] [Google Scholar]
- 42.Abou-Sleiman PM, Healy DG, Quinn N, Lees AJ, Wood NW. The role of pathogenic DJ-1 mutations in Parkinson's disease. Ann Neurol. 2003;54:283–286. doi: 10.1002/ana.10675. [DOI] [PubMed] [Google Scholar]
- 43.Gorner K, Holtorf E, Odoy S, Nuscher B, Yamamoto A, Regula JT, Beyer K, Haass C, Kahle PJ. Differential effects of Parkinson's disease-associated mutations on stability and folding of DJ-1. J. Biol. Chem. 2004;279:6943–6951. doi: 10.1074/jbc.M309204200. [DOI] [PubMed] [Google Scholar]
- 44.Annesi G, Savettieri G, Pugliese P, D'Amelio M, Tarantino P, Ragonese P, La Bella V, Piccoli T, Civitelli D, Annesi F, Fierro B, Piccoli F, Arabia G, Caracciolo M, Ciro Candiano IC, Quattrone A. DJ-1 mutations and parkinsonism-dementia-amyotrophic lateral sclerosis complex. Ann Neurol. 2005;58:803–807. doi: 10.1002/ana.20666. [DOI] [PubMed] [Google Scholar]
- 45.Hulleman JD, Mirzaei H, Guigard E, Taylor KL, Ray SS, Kay CM, Regnier FE, Rochet JC. Destabilization of DJ-1 by familial substitution and oxidative modifications: implications for Parkinson's disease. Biochemistry. 2007;46:5776–5789. doi: 10.1021/bi7001778. [DOI] [PubMed] [Google Scholar]
- 46.Hering R, Strauss KA, Tao X, Bauer A, Woitalla D, Mietz EM, Bauer P, Schaible JB, Muller T, Schols L, Klein C, Berg D, Meyer PT, Schulz JB, Wollnik B, Tong L, Kruger R, Riess O. Novel homozygous p.E64D mutation in DJ1 in early onset Parkinson disease (PARK7) Human Mutation. 2004;24:321–329. doi: 10.1002/humu.20089. [DOI] [PubMed] [Google Scholar]
- 47.Zhou W, Zhu M, Wilson MA, Petsko GA, Fink AL. The oxidation state of DJ-1 regulates its chaperone activity toward alpha-synuclein. J Mol Biol. 2006;356:1036–1048. doi: 10.1016/j.jmb.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 48.Holyoak T, Fenn TD, Wilson MA, Moulin AG, Ringe D, Petsko GA. Malonate: a versatile cryoprotectant and stabilizing solution for salt-grown macromolecular crystals. Acta Crystallogr. 2003;D59:2356–2358. doi: 10.1107/s0907444903021784. [DOI] [PubMed] [Google Scholar]
- 49.Otwinowski Z, Minor W. Methods in Enzymology. Academic Press; New York: 1997. Processing of X-ray diffraction data collected in oscillation mode; pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 50.Sheldrick GM, Schneider TR. SHELXL: High-resolution refinement. In: Carter C. W. J. a. S., R.M., editor. Methods in Enzymology. Academic Press; New York: 1997. pp. 319–343. [PubMed] [Google Scholar]
- 51.Brunger AT. Free R-Value - a Novel Statistical Quantity for Assessing the Accuracy of Crystal-Structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 52.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 53.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 54.Collaborative Computational Project, Number 4 The CCP4 Suite - Programs for Protein Crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 55.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr. 2005;D61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 56.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr. 2004;D60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 57.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Laue TM, Shah BD, Ridgeway TM, Pelletier SL. In: Analytical Ultracentrifugation in Biochemistry and Polymer Science. Harding SE, Rowe AJ, Horton JC, editors. The Royal Society of Chemistry; Cambridge: 1992. pp. 90–125. [Google Scholar]
- 59.Andrade MA, Chacon P, Merelo JJ, Moran F. Evaluation of secondary structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network. Protein Eng. 1993;6:383–390. doi: 10.1093/protein/6.4.383. [DOI] [PubMed] [Google Scholar]
- 60.Yue P, Li Z, Moult J. Loss of protein structure stability as a major causative factor in monogenic disease. J Mol Biol. 2005;353:459–473. doi: 10.1016/j.jmb.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 61.Herrera FE, Zucchelli S, Jezierska A, Lavina ZS, Gustincich S, Carloni P. On the oligomeric state of DJ-1 protein and its mutants associated with parkinson disease. A combined computational and in vitro study. J Biol Chem. 2007;282:24905–24914. doi: 10.1074/jbc.M701013200. [DOI] [PubMed] [Google Scholar]
- 62.Kinumi T, Kimata J, Taira T, Ariga H, Niki E. Cysteine-106 of DJ-1 is the most sensitive cysteine residue to hydrogen peroxide-mediated oxidation in vivo in human umbilical vein endothelial cells. Biochem. Biophys. Res. Commun. 2004;317:722–728. doi: 10.1016/j.bbrc.2004.03.110. [DOI] [PubMed] [Google Scholar]
- 63.Kapp GT, Richardson JS, Oas TG. Kinetic role of helix caps in protein folding is context-dependent. Biochemistry. 2004;43:3814–3823. doi: 10.1021/bi035683k. [DOI] [PubMed] [Google Scholar]
- 64.Merritt EA. Expanding the model: anisotropic displacement parameters in protein structure refinement. Acta Crystallogr. 1999;D55:1109–1117. doi: 10.1107/s0907444999003789. [DOI] [PubMed] [Google Scholar]
- 65.Fenn TD, Ringe D, Petsko GA. POVScript+: a program for model and data visualization using persistence of vision ray-tracing. J. Appl. Crystallogr. 2003;36:944–947. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.