

Abstract
Human apolipoprotein (ApoE) genotype influences the development of Alzheimer's disease and cerebral amyloid angiopathy (CAA). Specific mutations within the amyloid-β protein (Aβ) peptide have been identified that cause familial forms of CAA. However, the effect of APOE genotype on accumulation of CAA mutant Aβ in brain is not well understood. In the present study, we determined how human ApoE3 or ApoE4 influence cerebral Aβ accumulation in transgenic mice (Tg-SwDI) that accumulate human Dutch/Iowa (E22Q/D23N) CAA mutant Aβ in brain, primarily in the form of fibrillar cerebral microvascular amyloid. Using Tg-SwDI mice bred onto a human APOE3/3 or human APOE4/4 background, we found that both human ApoE3 and ApoE4 proteins led to a strong reduction in the amount of cerebral microvascular amyloid with an unexpected concomitant appearance of extensive fibrillar parenchymal plaque amyloid. There was strong colocalization of all ApoE proteins with fibrillar amyloid deposits in the mice. In Tg-SwDI/hAPOE3/3 and Tg-SwDI/hAPOE4/4 mice, there was no change in the levels of total Aβ40 and Aβ42 or in the amounts of soluble and insoluble Aβ in brain compared with Tg-SwDI mice on the endogenous mouse APOE background. The shift from primarily cerebral microvascular amyloid to parenchymal plaque amyloid in Tg-SwDI/hAPOE3/3 and Tg-SwDI/hAPOE4/4 mice resulted in a parallel shift in the association of activated microglia. These findings indicate that human ApoE has a strong influence on the spatial development of human Dutch/Iowa CAA mutant amyloid accumulation in mouse brain and that microglial activation is in response to the spatial accumulation of fibrillar amyloid.
Keywords: amyloid β protein, apolipoprotein E, transgenic mice, cerebral vasculature, Alzheimer's disease, microglia
Introduction
Extracellular deposition of amyloid-β protein (Aβ) in brain is a prominent pathological feature of Alzheimer's disease (AD) and related disorders (Hardy and Selkoe, 2002). Aβ peptides are derived through sequential proteolytic processing of the Aβ precursor protein (AβPP) by β- and γ- secretase activities. Cerebral parenchymal Aβ deposition can occur as diffuse or fibrillar plaques, with the latter associated with dystrophic neurons and inflammation (Selkoe, 2001). In AD, fibrillar Aβ deposition is also commonly observed in the cerebral vasculature, a condition known as cerebral amyloid angiopathy (CAA) (Jellinger, 2002; Rensink et al., 2003, Attems et al., 2005). Additionally, several familial forms of CAA have been identified that result from specific point mutations within the Aβ peptide, including Dutch-type (E22Q) and Iowa-type (D23N), which cause early and severe cerebral vascular amyloid deposition (Levy et al., 1990; Van Broeckhoven et al., 1990; Grabowski et al., 2001).
Apolipoprotein-E (APOE) genotype has a significant effect on the development of AD. Specifically, carrying an APOE4 allele increases the incidence and decreases the age of onset of AD and CAA (Corder et al., 1993; Saunders et al., 1993). APOE4 has been associated with increased parenchymal and cerebral vascular amyloid burden in humans as well as in human AβPP transgenic mice (Schmechel et al., 1993; Olichney et al., 2000; Fryer et al., 2005). In contrast, a decrease in AD prevalence is observed with the APOE3 genotype (Mayeux et al., 1993; Rebeck et al., 1993). Although the consequences of different APOE genotypes in sporadic AD are well established, little is known about their effects in familial CAA involving mutant forms of Aβ.
Previously, we generated transgenic mice that express human CAA mutant AβPP in brain (Tg-SwDI) and develop early-onset and robust CNS deposition of Aβ, particularly in the cerebral microvasculature (Davis et al., 2004; Miao et al., 2005a). The finding that Tg-SwDI mice accumulate extensive cerebral microvascular Aβ, despite low levels of transgene-directed human AβPP expression, appears to primarily result from ineffective clearance of Dutch/Iowa mutant Aβ from brain across the blood–brain barrier into the circulation (Davis et al., 2004, 2006; Deane et al., 2004). In Tg-SwDI mice, cerebral microvascular amyloid deposition is intimately linked with a robust neuroinflammatory response, highlighted by localized strong microglial activation and deficits in spatial learning and memory (Miao et al., 2005b; Fan et al., 2007; Xu et al., 2007).
In the present study, we sought to determine the effect of human ApoE3 and ApoE4 expression on the accumulation of human CAA mutant Aβ in vivo. We show that both human ApoE3 and ApoE4 led to a dramatic reduction in the levels of microvascular fibrillar amyloid with the surprising development of abundant parenchymal fibrillar amyloid plaques. Significantly, microglial activation closely adhered to the spatial development of fibrillar amyloid. These findings show that human ApoE proteins have a dramatic impact on spatial deposition of fibrillar CAA mutant Aβ and further emphasize the role of fibrillar amyloid as a key mediator of neuroinflammation in vascular and parenchymal amyloid deposition.
Materials and Methods
Transgenic mice.
All work with animals followed National Institutes of Health guidelines and was approved by the Stony Brook University Institutional Animal Care and Use Committee (IACUC) or by the Duke University IACUC. Generation and characterization of Tg-SwDI mice were described previously (Davis et al., 2004; Miao et al., 2005). Targeted replacement human APOE3/3 or human APOE4/4 mice were generously provided by Dr. Nobuyo Maeda (University of North Carolina, Chapel Hill, NC). All mice were on a pure C57BL/6 background. Tg-SwDI/hAPOE3/3 or Tg-SwDI/hAPOE4/4 mice were generated by successive breedings of homozygous Tg-SwDI mice with the APOE3/3 or human APOE4/4 mice to yield mice that were homozygous for both human SwDI AβPP and human APOE genes. Ten to 11 Tg-SwDI/muAPOE, Tg-SwDI/hAPOE3/3, and Tg-SwDI/hAPOE4/4 mice were examined at 12 months of age for the pathological studies.
Tissue preparation.
Mice were killed with an overdose of 2.5% avertin, and the brains were immediately removed and bisected in the midsagittal plane. One hemisphere was snap frozen and used for the protein analyses. The other hemisphere was placed in 70% ethanol, followed by xylene treatment and embedding in paraffin for immunohistochemical and histological analyses.
Immunohistochemical analysis.
Immunohistochemistry and histology were performed as described previously (Davis et al., 2004; Miao et al., 2005a). Briefly, sections were cut in the sagittal plane at 10 μm thickness using a microtome, deparaffinated, and rehydrated. Antigen retrieval was performed by treatment with proteinase K (0.2 mg/ml) for 10 min at 22°C for Aβ and for collagen staining and by 10 mm sodium citrate solution (pH 9.0) for 30 min at 90°C in a water bath for activated microglia staining. Primary antibodies were detected with horseradish peroxidase-conjugated or alkaline phosphatase-conjugated secondary antibodies and visualized with either a stable diaminobenzidine solution (Invitrogen, Carlsbad, CA) or the fast red substrate system (Spring Bioscience, Fremont, CA), respectively, as substrate. Sections were counterstained with hematoxylin. Thioflavin-S staining for fibrillar amyloid was performed as described previously (Dickson et al., 1990). The following antibodies were used for immunohistochemical analysis: mouse monoclonal antibody 66.1, which recognizes residues 1–5 of human Aβ (Deane et al., 2003) (1:200), rabbit polyclonal antibody to collagen type IV (1:100,; Research Diagnostics, Flanders, NJ), mouse monoclonal antibody to glial fibrillary acidic protein (GFAP) for the detection of astrocytes (1:300; Millipore Bioscience Research Reagents, Temecula, CA), mouse monoclonal anti-keratan sulfate antibody for the detection of activated microglia (clone 5D4, 1:200; Seikagaku Corporation, Tokyo, Japan), mouse monoclonal antibody to human ApoE (clone A1.4, 1:500; Santa Cruz Biotechnology, Santa Cruz, CA), and goat polyclonal antibody to mouse ApoE (M-20, 1:250; Santa Cruz Biotechnology).
Quantitative analysis of regional Aβ deposition and microvascular CAA.
Total amyloid burden in the regions of the subiculum, thalamus, and frontotemporal cortex was quantified on the same set of systematically sampled thioflavin-S-stained sections using NIH Image J 1.32 software. The percentage of thioflavin-S-labeled blood vessels in the same fields as above was determined with using stereological principles as described previously (Long et al., 1998).
Quantitative immunoblot analysis for cerebral human AβPP.
The levels of human AβPP in Tg-SwDI/muAPOE, Tg-SwDI/hAPOE3/3, and Tg-SwDI/hAPOE4/4 mouse forebrains were determined by quantitative immunoblotting using the human AβPP-specific monoclonal antibody P2-1 as described previously (Davis et al., 2004).
Immunochemical analysis of cerebral Aβ peptides.
Soluble pools of Aβ40 and Aβ42 were determined by using an specific ELISA of carbonate extracted mouse forebrain tissue, and subsequently the insoluble Aβ40 and Aβ42 levels were determined by ELISA of guanidine lysates of the insoluble pellets resulting from the carbonate extracted brain tissue (Johnson-Wood et al., 1997; DeMattos et al., 2002). Total Aβ40 and Aβ42 levels were determined by combining the soluble and insoluble levels of each form.
Quantitative analysis of reactive astrocyte and activated microglia cell densities.
Total numbers of astrocytes and activated microglia in the subiculum, thalamus, and frontotemporal cortex regions were estimated using a computerized stereology system (Stereologer; Systems Planning and Analysis, Alexandria, VA). Every 10th section was selected and generated 10–15 sections per reference space in a systematic-random manner. Immunopositive cells were counted using the optical fractionator method with the dissector principle and unbiased counting rules (Long et al., 1998).
Statistical analysis.
The data were analyzed by one-way ANOVAs for each measure at each brain region. Significant ANOVAs (p < 0.05) were followed by Fisher's post hoc tests, the results of which are reported in the corresponding figure legends.
Results
Human ApoE markedly reduces microvascular amyloid accumulation in Tg-SwDI mice
We determined whether human ApoE3 or ApoE4 influences the cerebral accumulation of Dutch/Iowa CAA mutant Aβ in Tg-SwDI mice, a model of familial cerebral microvascular amyloid (Davis et al., 2004; Miao et al., 2005a). To do this, we crossed Tg-SwDI mice onto targeted replacement human APOE3/3 or APOE4/4 backgrounds. This enabled us to evaluate the effects of the different human ApoE isoforms in the absence of endogenous mouse ApoE protein. After aging to 12 months, we initially assessed the global distribution of Aβ accumulation in the brains of mice with the different APOE backgrounds expressing mouse ApoE, human ApoE3, or human ApoE4. Quantitative immunoblotting showed that APOE background did not influence the expression of transgene encoded human AβPP in Tg-SwDI mice (data not shown). In the initial analysis of human Aβ deposition, Tg-SwDI mice on the endogenous mouse APOE background (Tg-SwDI/muAPOE) exhibited extensive accumulation of human Dutch/Iowa mutant Aβ through the cortical, hippocampal, subicular, and thalamic regions (Fig. 1 A) as reported previously (Davis et al., 2004; Miao et al., 2005a,b). We observed a similar global spatial accumulation of Dutch/Iowa mutant Aβ when Tg-SwDI mice were on a human APOE3/3 background (Tg-SwDI/hAPOE3/3) or APOE4 background (Tg-SwDI/hAPOE4/4) (Fig. 1 B,C, respectively).
Figure 1.

Gross accumulation of Aβ in Tg-SwDI mouse forebrain in the presence of different ApoE forms. A–C, Human Aβ accumulation was detected by immunostaining (brown) in the forebrain of 12-month-old Tg-SwDI/muAPOE mice (A), Tg-SwDI/hAPOE3/3 mice (B), and Tg-SwDI/hAPOE4/4 mice (C). Scale bar, 1 mm.
Closer examination of the immunostaining for Aβ accumulation in Tg-SwDI mice revealed diffuse plaque deposits in the cortical parenchyma and abundant microvascular deposits in the thalamic region that are characteristic of this model (Davis et al., 2004; Miao et al., 2005a,b) (Fig. 2 A–D). In contrast, Tg-SwDI/hAPOE3/3 and Tg-SwDI/hAPOE4/4 mice exhibited a surprising increase in the number of cortical parenchymal plaques that appeared larger and more compact (Fig. 2 E,F and I,J, respectively). In addition, there appeared a marked reduction in the extent of cerebral microvascular Aβ deposition accompanied by the presence of parenchymal plaques in the thalamic region of Tg-SwDI/hAPOE3/3 and Tg-SwDI/hAPOE4/4 mice (Fig. 2 G,H and K,L, respectively).
Figure 2.
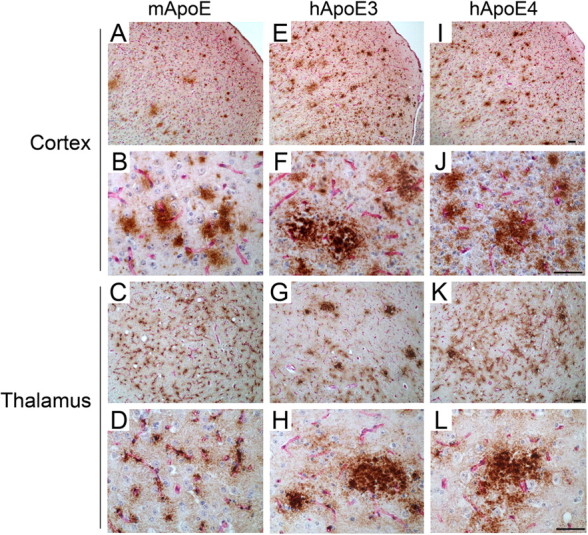
Human ApoE3 and ApoE4 alter the Aβ deposition pattern in Tg-SwDI mice. Brain sections from 12-month-old mice were double immunostained for Aβ (brown) and collagen type IV (red) to identify cerebral microvessels. A–L, Tg-SwDI/muAPOE mouse cortex (A, B) and thalamus (C, D); Tg-SwDI/hAPOE3/3 mouse cortex (E, F) and thalamus (G, H); Tg-SwDI/hAPOE4/4 mouse cortex (I, J) and thalamus (K, L). Note that the prevalent thalamic microvascular-associated Aβ deposition in Tg-SwDI/muAPOE mice (C, D) was markedly reduced in the Tg-SwDI/hAPOE3/3 mice (G, H) and Tg-SwDI/hAPOE4/4 mice (K, L). Scale bars, 50 μm.
The extent of cerebral microvascular amyloid was determined in different brain regions of Tg-SwDI mice on the different APOE backgrounds using stereological methods. As shown in Figure 3, the amount of microvascular amyloid was highest in the thalamus and subiculum with much lower levels in the frontotemporal cortex of all Tg-SwDI mice regardless of their APOE background. However, Tg-SwDI/hAPOE3/3 mice exhibited a robust 80–85% reduction in microvascular amyloid in all brain regions examined compared with Tg-SwDI/muAPOE mice (p < 0.0001). Although somewhat less severe, Tg-SwDI/hAPOE4/4 mice also showed a marked 55–70% reduction in microvascular amyloid compared with Tg-SwDI/muAPOE mice (p < 0.0001). These findings clearly demonstrate that both human ApoE3 and ApoE4 have a strong effect on reducing microvascular amyloid while increasing parenchymal Aβ deposits in Tg-SwDI/huAPOE mice.
Figure 3.

Human ApoE strongly reduces cerebral microvascular Aβ deposition in Tg-SwDI mice. Quantitative stereological estimation of cerebral microvascular amyloid load in different brain regions of Tg-SwDI/muAPOE mice (black bars), Tg-SwDI/hAPOE3/3 mice (gray bars), or Tg-SwDI/hAPOE4/4 mice (white bars). Data shown are mean ± SD (n = 8 animals per group). In all brain regions, Tg-SwDI/muAPOE mice had markedly higher amounts of microvascular CAA compared with Tg-SwDI/hAPOE3/3 mice or Tg-SwDI/hAPOE4/4 mice (*p < 0.0001). In all brain regions, Tg-SwDI/hAPOE4/4 mice exhibited higher levels of microvascular CAA compared with Tg-SwDI/hAPOE3/3 mice (**p < 0.01). F/T, Frontotemporal cortex.
Human ApoE promotes fibrillar amyloid plaque deposition in Tg-SwDI mice
We next determined the structural nature of the increased parenchymal Aβ deposits observed in Tg-SwDI mice on the human ApoE-expressing mice by performing thioflavin-S staining to detect fibrillar amyloid. At 12 months of age, Tg-SwDI/muApoE mice characteristically showed little thioflavin-S staining in the cortex with only an occasional microvascular amyloid deposit and abundant microvascular fibrillar amyloid in the thalamus (Fig. 4 A,B and C,D, respectively). Importantly, this demonstrates that the abundant parenchymal Aβ deposits in Tg-SwDI/muAPOE mice are primarily of a diffuse nature. In contrast, Tg-SwDI mice on the human APOE3/3 or APOE4/4 background exhibited the striking appearance of abundant parenchymal fibrillar amyloid plaques (Fig. 4 E,F and I,J, respectively). Complementing this finding in the thalamic region, there was an obvious loss of microvascular amyloid and, again, the appearance of parenchymal fibrillar amyloid plaques in the presence of human ApoE3 or ApoE4 proteins (Fig. 4 G,H and K,L, respectively). Specific quantitation of the regional nonvascular, parenchymal amyloid load in the mouse brains showed an ∼20-fold increase in cortical fibrillar amyloid plaques in the Tg-SwDI/hAPOE3/3 and Tg-SwDI/hAPOE4/4 mice (Fig. 5). Similarly, the nonvascular, parenchymal amyloid load in the thalamus and subiculum increased twofold to fourfold in Tg-SwDI/hAPOE3/3 and Tg-SwDI/hAPOE4/4 mice compared with Tg-SwDI/muAPOE mice. These findings convincingly show that human ApoE3 and ApoE4 strongly promote parenchymal amyloid plaque deposition and reduce microvascular amyloid deposition in Tg-SwDI transgenics.
Figure 4.

Human ApoE3 and ApoE4 shift fibrillar Aβ deposition from the cerebral microvasculature to the parenchyma in Tg-SwDI mice. Brain sections from 12-month- old mice were labeled for fibrillar Aβ using thioflavin-S (green) and collagen type IV (red) to identify cerebral microvessels. A–L, Tg-SwDI/muAPOE mouse cortex (A, B) and thalamus (C, D); Tg-SwDI/hAPOE3/3 mouse cortex (E, F) and thalamus (G, H); Tg-SwDI/hAPOE4/4 mouse cortex (I, J) and thalamus (K, L). Note the prevalent fibrillar microvascular amyloid and absence of fibrillar amyloid parenchymal plaques in Tg-SwDI/muAPOE mice (A–D) and the marked reduction of fibrillar microvascular amyloid and the emergence of abundant fibrillar amyloid parenchymal plaques in the Tg-SwDI/hAPOE3/3 mice (E–H) and Tg-SwDI/hAPOE4/4 mice (I–L). Scale bars, 50 μm.
Figure 5.

Human ApoE promotes parenchymal amyloid plaque load in Tg-SwDI mice. Quantitation of thioflavin-S-positive nonvascular, parenchymal amyloid load in different brain regions of Tg-SwDI/muAPOE mice (black bars), Tg-SwDI/hAPOE3/3 mice (gray bars), or Tg-SwDI/hAPOE4/4 mice (white bars). Data shown are mean ± SD (n = 8 animals per group). In all brain regions, Tg-SwDI/muAPOE mice had markedly lower amounts of parenchymal amyloid compared with Tg-SwDI/hAPOE3/3 mice or Tg-SwDI/hAPOE4/4 mice (*p < 0.01). Specifically in the thalamus, Tg-SwDI/hAPOE3/3 mice had higher amounts of parenchymal amyloid compared with Tg-SwDI/hAPOE4/4 mice (**p < 0.01). F/T, Frontotemporal cortex.
Human ApoE3 and ApoE4 are associated with fibrillar amyloid deposits but do not affect the overall levels of cerebral Aβ peptides in Tg-SwDI mice
To determine whether human ApoE proteins exhibit a different distribution than mouse ApoE with respect to amyloid deposits, we performed double-labeling studies using thioflavin-S staining to identify fibrillar amyloid and antibodies to detect ApoE proteins in the different mice. Figure 6 shows that there is strong colocalization of ApoE with fibrillar amyloid deposits regardless of the type of ApoE. This finding suggests that the dramatic redistribution of CAA mutant fibrillar amyloid in Tg-SwDI mice in the presence of human ApoE3 or ApoE4 is not attributable to differential interaction of the mouse and human ApoE proteins with amyloid deposits.
Figure 6.

Colocalization of ApoE proteins and fibrillar amyloid deposits in Tg-SwDI mouse brain. Brain sections from 12-month-old mice were labeled for fibrillar Aβ using thioflavin-S (green) and ApoE (red). A–F, Tg-SwDI/muAPOE mouse cortex (A) and thalamus (B); Tg-SwDI/hAPOE3/3 mouse cortex (C) and thalamus (D); Tg-SwDI/hAPOE4/4 mouse cortex (E) and thalamus (F). Note the strong colocalization of mouse ApoE with microvascular amyloid deposits in Tg-SwDI/muAPOE mice and human ApoE with parenchymal amyloid plaques in Tg-SwDI/hAPOE3/3 and Tg-SwDI/hAPOE4/4 mice. Scale bar, 50 μm. F/T, Frontotemporal cortex.
To investigate whether the APOE genotype background affected the amounts of cerebral Aβ accumulation in Tg-SwDI mice, we performed quantitative ELISA measurements on brain hemispheres of the different mice. Figure 7 A shows that human ApoE3 or ApoE4 had no appreciable effect on the levels of total cerebral Aβ40 and Aβ42 compared with endogenous mouse ApoE protein. In each case, the ratios of Aβ40/Aβ42 remained ∼10:1. Similarly, the levels of soluble Aβ and insoluble Aβ (Fig. 7 B,C, respectively) in Tg-SwDI mice were unaffected by the presence of human ApoE3 or ApoE4. Although no differences were found in the amounts of Aβ in nondissected forebrain hemispheres, differences may exist in Aβ levels in specific regions, reflecting the shift from microvascular to parenchymal amyloid. ELISA analysis showed no human Aβ peptide in plasma samples of any of the Tg-SwDI mice regardless of APOE genotype (data not shown), consistent with our previous studies indicating ineffective efflux of CNS Dutch/Iowa CAA mutant Aβ into the circulation (Davis et al., 2004, 2006; Deane et al., 2004). Together, these findings indicate that the type of ApoE protein expressed had no significant quantitative effect on the total accumulation of CNS Aβ peptides in Tg-SwDI mice.
Figure 7.

Quantitation of Aβ levels in Tg-SwDI mouse forebrain. ELISA measurements for Aβ peptides were performed on nondissected brain hemispheres from the different Tg-SwDI mice. A–C, These results showed that the levels of Aβ40 and Aβ42 (A), soluble Aβ (B), and insoluble Aβ (C) were unaffected by APOE background in the total mouse forebrain tissue. ELISA data shown are mean ± SD (n = 10 or 11 animals per group). There was no significant difference between any of the groups.
Increased numbers of neuroinflammatory cells correlate with the spatial deposition of amyloid in Tg-SwDI mice of different APOE backgrounds
Previously, we showed that elevated numbers of neuroinflammatory cells were present in close association with microvascular fibrillar amyloid, but not diffuse Aβ plaque deposits, in Tg-SwDI/muAPOE mice (Miao et al., 2005a,b; Fan et al., 2007). Because the current results clearly show that Tg-SwDI mice on human APOE backgrounds shift the distribution of fibrillar amyloid from the microvascular deposits to parenchymal plaques, we investigated whether this altered the spatial accumulation of neuroinflammatory reactive astrocytes and activated microglia.
In Tg-SwDI/muAPOE mice, there were more than twice the number of reactive astrocytes in the thalamic region, which contains high levels of fibrillar microvascular amyloid, compared with the cortex, which exhibits much less fibrillar amyloid (Fig. 8). When Tg-SwDI mice are on the human APOE3/3 or APOE4/4 background, however, the numbers of reactive astrocytes in the cortex increased, whereas the numbers in the thalamic region decreased, reflecting the regional shift from microvascular amyloid to parenchymal plaque fibrillar amyloid.
Figure 8.

Human ApoE3 and ApoE4 increase cortical and decrease thalamic reactive astrocyte numbers in Tg-SwDI mice. Brain sections from 12-month-old mice were double immunostained with GFAP to reveal astrocytes (brown) and collagen type IV (red) to identify cerebral microvessels. A–F, Tg-SwDI/muAPOE mouse cortex (A) and thalamus (B); Tg-SwDI/hAPOE3/3 mouse cortex (C) and thalamus (D); Tg-SwDI/hAPOE4/4 mouse cortex (E) and thalamus (F). Scale bar, 50 μm. G, Quantitative stereological estimation of astrocyte densities showed increased numbers in the frontotemporal cortex and decreased numbers in the thalamus of Tg-SwDI/APOE3/3 mice (white bars) and Tg-SwDI/APOE4/4 mice (gray bars) compared with Tg-SwDI/muAPOE mice (black bars). The data presented are the mean ± SD (n = 8 animals per group). In the cortex, Tg-SwDI/muAPOE mice had lower numbers of astrocytes compared with Tg-SwDI/hAPOE3/3 mice or Tg-SwDI/hAPOE4/4 mice (*p < 0.02). In the thalamus, Tg-SwDI/muAPOE mice had higher numbers of astrocytes compared with Tg-SwDI/hAPOE3/3 mice (**p < 0.01) and Tg-SwDI/hAPOE4/4 mice (p < 0.06, not significant). F/T, Frontotemporal cortex.
Similarly, in Tg-SwDI/muAPOE mice, there were greater than sixfold the number of activated microglia in the thalamic region, which is rich in microvascular amyloid, compared with the cortex, which has little microvascular amyloid (Fig. 9). In contrast, when Tg-SwDI mice are on the human APOE3/3 or APOE4/4 background, the numbers of activated microglia in the cortex increased, whereas the numbers in the thalamic region decreased, again reflecting the regional shift from microvascular amyloid to parenchymal plaque fibrillar amyloid. It was observed that, in the Tg-SwDI/muAPOE mice, the activated microglia, in either the cortex or the thalamus (Fig. 9 A,B, respectively), tightly track along the cerebral microvessels in which fibrillar amyloid deposits. However, this close association with the cerebral microvessels uncoupled when the Tg-SwDI mice were on the human APOE3/3 or APOE4/4 backgrounds (Fig. 9 C,D and E,F, respectively). To further quantitate this observation, we performed double labeling for fibrillar amyloid and activated microglia, followed by stereological counting to determine the number of activated microglia that were associated with either cerebral microvascular amyloid or parenchymal plaque amyloid. As shown in Figure 10, Tg-SwDI/muAPOE mice show a tight association between activated microglia with cerebral microvascular amyloid in the cortex and the thalamus. Although the overall numbers of activated microglia are lower in the cortex compared with the thalamus (Fig. 9), in both regions, the vast majority of these neuroinflammatory cells are associated with microvascular amyloid (65 and 90%, respectively). Conversely, when the Tg-SwDI mice are on either the human APOE3/3 or APOE4/4 background, conditions that markedly reduce fibrillar cerebral microvascular amyloid and promote fibrillar parenchymal plaque amyloid, there is a dramatic shift in which now 60–80% of the activated microglia are tightly associated with fibrillar parenchymal plaque amyloid (Fig. 10). Together, these findings show that neuroinflammatory cells, in particular activated microglia, accumulate in direct response to the spatial location of fibrillar amyloid in the Tg-SwDI mice.
Figure 9.

Human ApoE3 and ApoE4 increase cortical and decrease thalamic activated microglia numbers in Tg-SwDI mice. Brain sections from 12-month-old mice were double immunostained with monoclonal antibody 5D4 to keratan sulfate to reveal activated microglia (brown) and collagen type IV (red) to identify cerebral microvessels. A–F, Tg-SwDI/muAPOE mouse cortex (A) and thalamus (B); Tg-SwDI/hAPOE3/3 mouse cortex (C) and thalamus (D); Tg-SwDI/hAPOE4/4 mouse cortex (E) and thalamus (F). Scale bar, 50 μm. G, Quantitative stereological estimation of activated microglial densities showed increased numbers in the frontotemporal cortex (F/T cortex) and decreased numbers in the thalamus of Tg-SwDI/hAPOE3/3 mice (white bars) and Tg-SwDI/hAPOE4/4 mice (gray bars) compared with Tg-SwDI/muAPOE mice (black bars). The data presented are the mean ± SD (n = 8 animals per group). In the cortex, Tg-SwDI/muAPOE mice had lower numbers of microglia compared with Tg-SwDI/hAPOE3/3 mice or Tg-SwDI/hAPOE4/4 mice (*p < 0.05). In the thalamus, Tg-SwDI/muAPOE mice had higher numbers of microglia compared with Tg-SwDI/hAPOE3/3 mice (**p < 0.01) and Tg-SwDI/hAPOE4/4 mice (p < 0.06, not significant).
Figure 10.

Human ApoE3 and ApoE4 shift activated microglia from the cerebral microvasculature to parenchymal fibrillar plaques. Brain sections from 12-month-old mice were labeled with thioflavin-S to identify fibrillar amyloid (green) and monoclonal antibody 5D4 to keratan sulfate to reveal activated microglia (red). A–F, Tg-SwDI/muAPOE mouse cortex (A) and thalamus (B); Tg-SwDI/hAPOE3/3 mouse cortex (C) and thalamus (D); Tg-SwDI/hAPOE4/4 mouse cortex (E) and thalamus (F). Scale bar, 50 μm. G, H, Quantitative stereological estimation of the percentage of microvascular fibrillar amyloid-associated (G) or parenchymal plaque fibrillar amyloid-associated (H) activated microglia. Tg-SwDI/muAPOE mice (black bars) show a strong microvascular amyloid association, whereas Tg-SwDI/hAPOE3/3 mice (white bars) and Tg-SwDI/hAPOE4/4 mice (gray bars) exhibit a robust parenchymal amyloid association. The data presented are the mean ± SD (n = 8 animals per group). In the cortex and thalamus, Tg-SwDI/muAPOE mice had higher numbers of vascular-associated microglia and lower numbers of parenchymal-associated microglia compared with Tg-SwDI/hAPOE3/3 mice or Tg-SwDI/hAPOE4/4 mice (*p < 0.01). F/T, Frontotemporal cortex.
Discussion
The present findings show that human ApoE3 and ApoE4 proteins have a dramatic effect on the spatial accumulation of Dutch/Iowa CAA mutant fibrillar amyloid in Tg-SwDI transgenic mice, a model of cerebral microvascular amyloid deposition. Both human ApoE isoforms strongly reduce cerebral microvascular fibrillar amyloid while concomitantly promoting abundant parenchymal fibrillar amyloid plaques, pathological features not normally seen in Tg-SwDI mice on the endogenous mouse APOE background.
There is a strong association of APOE genotype with the risk for development of AD and CAA (Schmechel et al., 1993; Holtzman, 2001; Chalmers et al., 2003; Ashford, 2004; Holtzman, 2004). However, the precise mechanism as to how ApoE3 or ApoE4 influences this risk for the development of sporadic AD and CAA is not well understood. Nevertheless, a number of in vitro studies have suggested that it may result from differential effects of ApoE3 and ApoE4 on Aβ binding, perhaps altering fibrillogenesis, cytotoxicity, and/or clearance of the peptide from the CNS (Strittmatter et al., 1993; Wisniewski et al., 1994; Ji et al., 2001; Wilhelmus et al., 2005). For example, ApoE4 was found to bind normal, unmutated Aβ more tightly compared with ApoE3 (Strittmatter et al., 1993). Also, ApoE4 was reported to promote normal, unmutated Aβ fibrillogenesis (Wisniewski et al., 1994).
Nevertheless, how these in vitro effects of ApoE proteins translate to their actions regarding Aβ deposition in vivo is unclear. For example, previous work in human AβPP transgenic mice showed that the absence of endogenous mouse ApoE strongly reduced the deposition of normal human Aβ in the CNS (Bales et al., 1999; Holtzman, 2001, 2004; Fryer et al., 2003). Similarly, we found that the lack of endogenous mouse ApoE prevented Dutch/Iowa CAA mutant cerebral microvascular amyloid accumulation and primarily reduced parenchymal diffuse Aβ deposits in Tg-SwDI mice (Miao et al., 2005b). Furthermore, in this study, we showed that the strong reduction of fibrillar cerebral microvascular Aβ deposition in Tg-SwDI mice decreases microvascular amyloid-induced neuroinflammation. These combined findings indicate that endogenous mouse ApoE is an essential component to promote amyloid formation and deposition in transgenic mice producing either human normal Aβ or CAA mutant Aβ.
Subsequently, Fryer et al. (2005) showed that, when transgenic mice producing normal, unmutated Aβ were bred onto a human APOE3/3 background, this resulted in delayed parenchymal amyloid deposition and the absence of CAA. However, the same AβPP transgenic mice bred onto a human APOE4/4 background had delayed amyloid deposition but developed more extensive CAA. These authors also demonstrated that ApoE4 elevated the Aβ40/Aβ42 ratio and suggested that this alteration promotes CAA formation. This latter concept is similar to the findings of Herzig et al. (2004) who showed that selectively increasing the amount of Aβ42 can reduce CAA and promote parenchymal amyloid formation in a transgenic model of Dutch CAA mutant Aβ. Together, these reports suggest that altering the levels of Aβ40 or Aβ42 can drive cerebral vascular or parenchymal amyloid formation in transgenic mice.
Conversely, here we show that human ApoE3 and ApoE4 both markedly inhibit cerebral microvascular amyloid formation in Tg-SwDI transgenics producing Dutch/Iowa CAA mutant Aβ. However, the surprising finding was the extensive accumulation of parenchymal fibrillar amyloid deposits in Tg-SwDI/hAPOE3/3 and Tg-SwDI/hAPOE4/4 mice. This dramatic redistribution of fibrillar amyloid deposition occurred in the absence of any significant change in the overall levels of Aβ40 and Aβ42 or in the pools of soluble and insoluble Aβ, suggesting that some other mechanism is responsible for this robust alteration. There are several possibilities that may account for this effect of human ApoE in Tg-SwDI mice. For example, human ApoE proteins may exhibit different binding to Dutch/Iowa CAA mutant Aβ than endogenous mouse ApoE in the Tg-SwDI mouse brain or human ApoE proteins may interact with other binding proteins and/or receptors in a manner that alters their interactions with Dutch/Iowa CAA mutant Aβ. These possibilities seem less likely because we found robust colocalization of ApoE, regardless of whether it was mouse or human ApoE, with fibrillar amyloid deposits in the Tg-SwDI mice. Furthermore, the lack of detectable Dutch/Iowa CAA mutant Aβ in plasma of the Tg-SwDI mice, regardless of APOE background, suggests that all ApoE proteins may similarly be unable to influence its clearance into plasma. Alternatively, the observed preponderance of parenchymal fibrillar amyloid in Tg-SwDI mice on the human APOE3/3 or APOE4/4 backgrounds may result not from a direct effect of ApoE on Aβ but rather may change the landscape of the mouse brain in a way that promotes parenchymal amyloid formation.
The effects of human ApoE3 and ApoE4 on CAA mutant amyloid deposition in Tg-SwDI mice are similar in certain respects to those observed in Dutch familial CAA patients. For example, in a limited histopathological study, Bornebroek et al. (1997) reported that APOE genotype had no effect on amyloid-related structural lesions in Dutch CAA patients with the E22Q Aβ mutation. This is consistent with our results showing that both human ApoE3 and ApoE4 exert similar effects on amyloid lesions in Tg-SwDI mice. There have been too few Iowa familial CAA patients studied to determine whether APOE genotype influences amyloid lesions associated with the D23N Aβ mutation. However, the human ApoE effects on amyloid pathology in Tg-SwDI mice differ from Dutch CAA patients in that the mice exhibit a loss of fibrillar CAA and gain of extensive parenchymal fibrillar amyloid plaque pathology. APOE genotype appeared to have no effect on the characteristically extensive CAA distribution and no gain of parenchymal fibrillar plaques in Dutch CAA patients (Bornebroek et al., 1997).
Any differences between humans and mice concerning APOE genotype effects on CAA mutant amyloid pathology may result for several reasons. For example, the D23N Iowa Aβ mutation may exert an effect on amyloid deposition not seen with the E22Q Dutch Aβ mutation. Furthermore, the presence of both the Dutch E22Q and Iowa D23N mutations in Aβ, as produced in Tg-SwDI mice, may exert different effects than either single CAA mutation alone. Alternatively, the mouse CNS may differ from the human CNS in a manner in which human ApoE isoforms promote parenchymal fibrillar plaque formation in mice but not in humans. In any case, the effect of human ApoE3 or ApoE4 on spatial amyloid formation and deposition in Tg-SwDI mice is clear and robust.
On a final note, the redistribution of activated microglia in Tg-SwDI mice on the human APOE3/3 or APOE4/4 backgrounds further supports the relationship between fibrillar amyloid deposition and the resulting neuroinflammatory response. In Tg-SwDI/muAPOE mice, activated microglia are tightly associated with the development of cerebral microvascular fibrillar amyloid (Miao et al., 2005a; Fan et al., 2007; Xu et al., 2007). In these mice, the abundant parenchymal diffuse Aβ deposits do not appear to promote microglial activation. However, when the Tg-SwDI mice are bred onto the human APOE3/3 or APOE4/4 background, there is a dramatic shift with the activated microglia intimately associating with the now present parenchymal fibrillar amyloid plaques (Fig. 10). This strongly suggests that fibrillar amyloid, regardless of its spatial development, is the stimulus for the recruitment and activation of the microglia in the Tg-SwDI model. Approaches to suppress fibrillar amyloid stimulation of microglial activation remain a potential therapeutic avenue in amyloid-depositing diseases.
Footnotes
This work was supported in part by National Institute of Neurological Disorders and Stroke Grants RO1-NS36645, RO1-NS52533, and RO1-NS55118 and National Institute on Aging Grants RO1-AG19780 and RO1-AG23084. Antibody reagents for the Aβ ELISAs were generously provided by Lilly Research Laboratories. We thank Dr. John Robinson (Department of Psychology, Stony Brook University) for his expert assistance with the statistical analysis of the data.
References
- Ashford JW. APOE genotype effects on Alzheimer's disease onset and epidemiology. J Mol Neurosci. 2004;23:157–165. doi: 10.1385/JMN:23:3:157. [DOI] [PubMed] [Google Scholar]
- Attems J, Jellinger KA, Lintner F. Alzheimer's disease pathology influences severity and topographical distribution of cerebral amyloid angiopathy. Acta Neuropathol. 2005;110:222–231. doi: 10.1007/s00401-005-1064-y. [DOI] [PubMed] [Google Scholar]
- Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, Fishman CE, DeLong CA, Piccardo P, Petegnief V, Ghetti B, Paul SM. Apolipoprotein E is essential for amyloid deposition in the APPV717F transgenic mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 1999;96:15233–15238. doi: 10.1073/pnas.96.26.15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornebroek M, Haan J, van Duinen SG, Maat-Schieman MLC, Van Buchem MA, Bakker E, Van Broeckhoven C, Roos RAC. Dutch hereditary cerebral amyloid angiopathy: structural lesions and apolipoprotein E genotype. Ann Neurol. 1997;41:695–698. doi: 10.1002/ana.410410523. [DOI] [PubMed] [Google Scholar]
- Chalmers K, Wilcock GK, Love S. APOE e4 influences the pathological phenotype of Alzheimer's disease by favouring cerebrovascular over parenchymal accumulation of Aβ protein. Neuropathol Appl Neurobiol. 2003;29:231–238. doi: 10.1046/j.1365-2990.2003.00457.x. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Davis J, Xu F, Deane R, Romanov G, Previti ML, Zeigler K, Zlokovic BV, Van Nostrand WE. Early-onset and robust cerebral microvascular accumulation of amyloid β-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid β-protein precursor. J Biol Chem. 2004;279:20296–20306. doi: 10.1074/jbc.M312946200. [DOI] [PubMed] [Google Scholar]
- Davis J, Xu F, Miao J, Previti ML, Romanov G, Ziegler K, Van Nostrand WE. Deficient cerebral clearance of vasculotropic Dutch/Iowa double mutant Aβ in human AβPP transgenic mice. Neurobiol Aging. 2006;26:946–954. doi: 10.1016/j.neurobiolaging.2005.05.031. [DOI] [PubMed] [Google Scholar]
- Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B. RAGE mediates amyloid β-peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- Deane R, Wu Z, Sagare A, Davis J, Yan SD, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP-amyloid β-peptide (Aβ) interaction regulates differential brain efflux of Aβ isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, O'dell MA, Parsadanian M, Taylor JW, Harmony JA, Bales KR, Paul SM, Aronow BJ, Holtzman DM. Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2002;99:10843–10848. doi: 10.1073/pnas.162228299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, Wertkin A, Mattiace LA, Fier E, Kress Y, Davies P, Yen SH. Ubiquitin immunoelectron microscopy of dystrophic neurites in cerebellar senile plaques of Alzheimer's disease. Acta Neuropathol. 1990;79:486–493. doi: 10.1007/BF00296107. [DOI] [PubMed] [Google Scholar]
- Fan R, Xu F, Previti ML, Davis J, Grande AM, Robinson JK, Van Nostrand WE. Minocycline reduces microglial activation and improves behavioral deficits in a transgenic model of cerebral microvascular amyloid. J Neurosci. 2007;27:3057–3063. doi: 10.1523/JNEUROSCI.4371-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer JD, Taylor JW, DeMattos RB, Bales KR, Paul SM, Parasdanian M, Holtzman DM. Apolipoprotein E markedly facilitates age-depenent cerebral amyloid angiopathy and spontaneous hemorrhage in amyloid precursor protein transgenic mice. J Neurosci. 2003;23:7889–7896. doi: 10.1523/JNEUROSCI.23-21-07889.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer JD, Simmons K, Parsadanian M, Bales KR, Paul SM, Sullivan PM, Holtzman DM. Human apolipoprotein E4 alters the amyloid-beta 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J Neurosci. 2005;25:2803–2810. doi: 10.1523/JNEUROSCI.5170-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski TJ, Cho HS, Vonsattel JPG, Rebeck GW, Greenberg SM. Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann Neurol. 2001;49:697–705. doi: 10.1002/ana.1009. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD, Danner S, Abramowski D, Sturchler-Pierrat C, Burki K, van Duinen SG, Maat-Schieman ML, Staufenbiel M, Mathews PM, Jucker M. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci. 2004;7:954–960. doi: 10.1038/nn1302. [DOI] [PubMed] [Google Scholar]
- Holtzman DM. Role of apoE/Aβ interactions in the pathogenesis of Alzheimer's disease and cerebral amyloid angiopathy. J Mol Neurosci. 2001;17:147–155. doi: 10.1385/JMN:17:2:147. [DOI] [PubMed] [Google Scholar]
- Holtzman DM. In vivo effects of ApoE and clusterin on amyloid-β metabolism and neuropathology. J Mol Neurosci. 2004;23:247–254. doi: 10.1385/JMN:23:3:247. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Alzheimer's disease and cerebrovascular pathology: an update. J Neural Trasm. 2002;109:813–836. doi: 10.1007/s007020200068. [DOI] [PubMed] [Google Scholar]
- Ji Y, Permanne B, Sigurdsson EM, Holtzman DM, Wisniewski T. Amyloid beta 40/42 clearance across the blood-brain barrier following intra-ventricular injections in wild-type, apoE knock-out and human apoE3 or E4 expressing transgenic mice. J Alzheimers Dis. 2001;3:23–30. doi: 10.3233/jad-2001-3105. [DOI] [PubMed] [Google Scholar]
- Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L. Amyloid precursor protein processing and Aβ42 deposition in a transgenic mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberbur I, van Duinen SG, Bots GTAM, Luyendijk W, Frangione B. Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990;248:1124–1126. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- Long JM, Kalehua AN, Muth NJ, Calhoun ME, Jucker M, Hengemihle JM, Ingram DK, Mouton PR. Stereological estimation of total microglia number in mouse hippocampus. J Neurosci Methods. 1998;84:101–108. doi: 10.1016/s0165-0270(98)00100-9. [DOI] [PubMed] [Google Scholar]
- Mayeux R, Stern Y, Ottman R, Tatemichi TK, Tang MX, Maestre G, Ngai C, Tycko B, Ginsberg H. The apolipoprotein epsilon 4 allele in patients with Alzheimer's disease. Ann Neurol. 1993;34:752–754. doi: 10.1002/ana.410340527. [DOI] [PubMed] [Google Scholar]
- Miao J, Xu F, Davis J, Otte-Holler I, Verbeek MM, Van Nostrand WE. Cerebral microvascular amyloid β-protein deposition induces vascular degeneration and neuroinflammation in transgenic mice expressing human vasculotropic mutant amyloid β-protein precursor. Am J Pathol. 2005a;167:505–515. doi: 10.1016/s0002-9440(10)62993-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao J, Vitek MP, Xu F, Previti ML, Davis J, Van Nostrand WE. Reducing cerebral microvascular amyloid-β protein deposition diminishes regional neuroinflammation in vasculotropic mutant amyloid precursor protein transgenic mice. J Neurosci. 2005b;25:6271–6277. doi: 10.1523/JNEUROSCI.1306-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olichney JM, Hansen LA, Lee JH, Hofstetter CR, Katzman R, Thal LJ. Relationship between severe amyloid angiopathy, apolipoprotein E genotype, and vascular lesions in Alzheimer's disease. Ann NY Acad Sci. 2000;903:138–143. doi: 10.1111/j.1749-6632.2000.tb06360.x. [DOI] [PubMed] [Google Scholar]
- Rebeck GW, Reiter JS, Strickland DK, Hyman BT. Apolipoprotein E in sporadic Alzheimer's disease: allelic variation and receptor interactions. Neuron. 1993;11:575–580. doi: 10.1016/0896-6273(93)90070-8. [DOI] [PubMed] [Google Scholar]
- Rensink AA, de Waal RM, Kremer B, Verbeek MM. Pathogenesis of cerebral amyloid angiopathy. Brain Res Brain Res Rev. 2003;43:207–223. doi: 10.1016/j.brainresrev.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Saunders AM, Schmader K, Breitner JC, Benson MD, Brown WT, Goldfarb L, Goldgaber D, Manwaring MG, Szymanski MH, McCown N, Pericak-Vance MA, Roses AD. Apolipoprotein E epsilon 4 allele distributions in late-onset Alzheimer's disease and in other amyloid-forming dis-eases. Lancet. 1993;342:710–711. doi: 10.1016/0140-6736(93)91709-u. [DOI] [PubMed] [Google Scholar]
- Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, Pericak-Vance MA, Goldgaber D, Roses AD. Increased amyloid β-peptide deposition in cerebral cortex as a consequence of apolipoprotein genotype in late-onset Alzheimer's disease. Proc Natl Acad Sci USA. 1993;90:9649–9653. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;8:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, Schemchel D, Saunders AM, Goldgaber D, Roses AD. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:8098–8102. doi: 10.1073/pnas.90.17.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Broeckhoven C, Haan J, Bakker E, Hardy JA, Van Hul W, Wehnert A, Vegter Van der Vlis M, Roos RA. Amyloid beta protein precursor gene and hereditary cerebral hemorrhage with amyloidosis (Dutch) Science. 1990;248:1120–1122. doi: 10.1126/science.1971458. [DOI] [PubMed] [Google Scholar]
- Wilhelmus MM, Otte-Höller I, Davis J, Van Nostrand WE, de Waal RMW, Verbeek MM. Apolipoprotein E (ApoE) genotype regulates Aβ cytotoxicity. J Neurosci. 2005;25:3621–3627. doi: 10.1523/JNEUROSCI.4213-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski T, Castano EM, Golabek A, Vogel T, Frangione B. Acceleration of Alzheimer's fibril formation by apolipoprotein-E in vitro. Am J Pathol. 1994;145:1030–1035. [PMC free article] [PubMed] [Google Scholar]
- Xu F, Grande AM, Robinson JK, Previti ML, Vasek M, Davis J, Van Nostrand WE. Early-onset subicular microvascular amyloid and neuroinflammation correlate with behavioral deficits in vasculotropic mutant amyloid beta-protein precursor transgenic mice. Neuroscience. 2007;146:98–107. doi: 10.1016/j.neuroscience.2007.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]