

Abstract
Despite rapid advances in many fronts, pancreatic cancer (PC) remains one of the most difficult human malignancies to treat, in part due to de novo and acquired chemo- and radio-resistance. Gemcitabine alone or in combination with other conventional therapeutics is the standard of care for the treatment of advanced PC without any significant improvement in the overall survival of patients diagnosed with this deadly disease. Previous studies have shown that PC cells that are gemcitabine-resistant (GR) acquired epithelial-mesenchymal transition (EMT) phenotype which is reminiscent of “cancer stem-like cells (CSC)”; however the molecular mechanism that led to EMT phenotype has not been fully investigated. The present study demonstrates that Notch-2 and its ligand Jagged-1 are highly up-regulated in GR cells, which is consistent with the role of Notch signaling pathway in the acquisition of EMT and CSC phenotype. We also found that the down-regulation of Notch signaling was associated with decreased invasive behavior of GR cells. Moreover, down-regulation of Notch signaling by siRNA approach led to partial reversal of the EMT phenotype, resulting in the mesenchymal-epithelial transition (MET), which was associated with decreased expression of vimentin, ZEB1, Slug, Snail and NF-κB. These results provide molecular evidence showing that the activation of Notch signaling is mechanistically linked with chemo-resistance phenotype (EMT phenotype) of PC cells, suggesting that the inactivation of Notch signaling by novel strategies could be a potential targeted therapeutic approach for overcoming chemo-resistance toward the prevention of tumor progression and/or treatment of metastatic PC.
Introduction
Pancreatic cancer (PC) is a highly aggressive malignant disease, which is ranked as the fourth leading cause of cancer-related death in the United States with about 37,000 newly diagnosed cases and an approximately 34,000 deaths per year in the United States (1). In recent years, novel treatment strategies included diverse cytotoxic therapeutics such as chemotherapeutic drugs, suicide genes, γ-irradiation induced cytotoxicity or immunotherapy, have been initially encouraging, but prolonged drug exposure results in the development of acquired drug resistance impeding successful treatment (1). Thus, the aggressive behavior of PC is believed to be due to both de novo (intrinsic) and acquired (extrinsic) resistance to conventional therapeutics, and could also be due to the lack of delivery of effective doses of the drugs in the tumor because of the existence of extensive desmoplastic stroma and lack of adequate vasculature in most PCs. Although gemcitabine monotherapy (2′,2′-difluorodeoxycytidine), a deoxycytidine analogue, or its combination with other agents has become standard chemotherapy for the treatment of advanced PC, gemcitabine imparts a progression-free survival interval ranging from 0.9 to 4.2 months only (2). In summary, the effect of gemcitabine on survival has been disappointing, which could be due to many factors including intrinsic (de novo) drug resistance. This disappointing outcome strongly suggests that a better understanding of the mechanism by which chemo-resistance arises is likely to lead to novel therapeutic strategies for the successful treatment of patients diagnosed with PC.
Emerging evidence suggest molecular and phenotypic association between chemo-resistance and the acquisition of epithelial-mesenchymal (EMT)-like phenotype of cancer cells (3–6). This process is also believed to be reminiscent of “cancer stem-like cells” characteristics in many cancer systems (7–10). EMT has been classified as an unique process by which epithelial cells undergo remarkable morphological changes characterized by a transition from epithelial cobblestone phenotype to elongated fibroblastic phenotype (mesenchymal phenotype) leading to increased motility and invasion (11, 12). The processes of EMT involves loss of epithelial cell-cell junction, actin cytoskeleton reorganization, and up-regulation of mesenchymal molecular markers, such as fibronectin, α-smooth muscle actin (SMA), vimentin, and N-cadherin (12–14). A disassembly of cell-cell junction, including down-regulation and relocation of E-cadherin and β-catenin from cell membrane to nucleus, results in the induction of EMT. A number of factors which transcriptionally repress E-cadherin have emerged as potent EMT drivers during normal development and cancer. These include the zinc finger Snail homologs (Snail1, Snail2/Slug, Snail3), and several basic helix-loop-helix- factors such as Twist, ZEB1, ZEB2/SIP1 and TCF3/E47/E12 (15). EMT is a dynamic process and is triggered by the interplay of extracellular signals (such as collagen) and many secreted soluble factors, such as Wnt, transforming growth factor-β (TGF-β), fibroblast growth factor (FGF), epidermal growth factor (EGF), hepatocyte growth factor (HGF), and platelet-derived growth factors (PDGF). Among many of these signaling pathways, Wnt, TGF-β, Hedgehog, Notch, and nuclear factor-κB (NF-κB) signaling pathways are critical for EMT induction (12, 16).
Emerging evidence also suggest that there is a molecular link between EMT phenotype with chemo-or radio-resistance (3–5, 9, 10). Yang et al. reported that oxaliplatin-resistant colorectal cancer cells underwent EMT phenotype (4). Paclitaxel-resistant ovarian cancer cells showed phenotypic changes consistent with EMT with decreased expression of the epithelial adhesion molecule, E-cadherin and an increase in mesenchymal markers such as vimentin, α-SMA and fibronectin (3). Moreover, tamoxifen-resistant breast cancer cells demonstrated altered morphological characteristic of cells similar to EMT with altered β-catenin phosphorylation (5). Recently, Rho et al. reported that phenotypic changes such as a spindle-cell shape and increased pseudopodia formation suggesting EMT was present in the gefitinib-resistant lung cancer cells with a decrease in the expression of E-cadherin and an increase in the expression of vimentin, which is a mesenchymal marker (17). These studies clearly provide strong evidence linking chemo-resistance to EMT.
Previous studies have shown that gemcitabine-resistant (GR) PC cells acquired EMT characteristics (18). However, the exact mechanism for the acquisition of EMT phenotype of GR cells remains to be elucidated. The present study demonstrates that Notch signaling pathway is involved in the acquisition of EMT phenotype of GR cells. We also found that down-regulation of Notch signaling was associated with decreased invasive behavior of GR cells. Moreover, down-regulation of Notch signaling led to partial reversal of the EMT phenotype, resulting in mesenchymal-epithelial transition (MET), which was associated with decreased expression of vimentin, ZEB1, Slug, Snail and NF-κB. These results suggest that the increased Notch signaling is mechanistically associated with chemo-resistance and EMT characteristics of PC cells and, as such, Notch pathways could be a novel target for the treatment of PC.
Materials and methods
Cell culture
Human pancreatic cancer (PC) gemcitabine-resistant (GR) cell line was derived as described earlier (18). Briefly, gemcitabine resistant (GR) PC cells were derived from gemcitabine-sensitive (GS) L3.6pl cells by exposing them to increasing concentrations of gemcitabine (up to 1000nM of gemcitabine). The resulting cell line was designated GR cells showing EMT characteristics (18); however 200nM of gemcitabine was used for maintaining the EMT phenotype of GR cells during multiple passaging. These two cell lines were cultured in minimal essential medium (MEM) supplemented with 10% fetal bovine serum, L-glutamine, and 1% penicillin and streptomycin in a 5% CO2 atmosphere at 37°C unless otherwise indicated.
Experimental reagents
Antibodies against β-catenin were purchased from Cell Signaling Technology (Beverly, MA). Antibodies against vimentin and nestin were purchased from Abcam (Cambridge, MA). Primary antibodies for E-cadherin, Notch-1, Notch-2, Notch-3, Notch-4, Jagged-1, Jagged-2, Dll-1, Dll-4, uPA, MMP-2, Snail, Slug, VEGF and MMP-9 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). All other secondary antibodies were obtained from Pierce (Rockford, IL).
Real-time RT-PCR analysis for gene expression
The total RNA from GS, GR, or siRNA transfected GR cells was isolated by Trizol (Invitrogen, Carlsbad, CA) and purified by RNeasy Mini Kit and RNase-free DNase Set (QIAGEN, Valencia, CA) according to the manufacturer’s protocols. The primers used in the PCR reaction are described before (19–22). The other primers used in the PCR reaction are: ZEB1, forward primer (5′-GCA CAA CCA AGT GCA GAA GA-3′) and reverse primer (5′-GCC TGG TTC AGG AGA AGA TG-3′); Vimentin, forward primer (5′-AGA TGG CCC TTG ACA TTG AG-3′) and reverse primer (5′-TGG AAG AGG CAG AGA AAT CC-3′). Real-time PCR amplifications were performed as described earlier (19).
Western blot analysis
Cells were lysed in lysis buffer by incubating for 20 minutes at 4°C. The protein concentration was determined using the Bio-Rad assay system (Bio-Rad, Hercules, CA). Total proteins were fractionated using SDS-PAGE and transferred onto nitrocellulose membrane for Western blotting as described earlier (19)
Immunofluorescence Microscopy
Cells were fixed with 4% paraformaldehyde for 10 minutes, permeabilized in 0.5% Triton X-100 for 10 minutes, and incubated in PBS and 10% goat serum blocking solution for 1 hour. The cells were incubated for 2 hours with anti-Notch-2, anti-Notch-4, anti-Jagged-1, anti-E-cadherin, anti-vimentin, anti-β-catenin, anti-F-actin, in 5% goat serum and were analyzed as described earlier (13).
Urokinase plasminogen activator (uPA) activity assay
The culture medium of the GS or GR cells grown in 6-well plates was collected. After collection, the medium was spun at 800 × g for 3 min at 4°C to remove cell debris. The supernatant was either frozen at -20°C for uPA assay later or assayed immediately using commercially available ELISA kits (American Diagnostica, Inc., Stamford, CT).
MMP-9 activity assay
The GS or GR cells were seeded in 6 well plates and incubated at 37°C. After 24 hours, the complete medium was removed and the cells were washed with serum-free medium. The cells were then incubated in serum-free medium for 48 hours. MMP-9 activity in the medium and cell lysate was detected by using Fluorokine E Human MMP-9 Activity Assay Kit (R&D Systems, Inc., Minneapolis, MN) according to the manufacturer’s protocol.
Plasmids and transfections
GS and GR cells were transfected with Notch-2 siRNA, Notch-4 siRNA, Jagged-1 siRNA and siRNA control (Santa Cruz, CA), respectively, using Lipofectamine 2000 as described earlier (19).
Cell attachment and detachment assay
Cell attachment assay was performed as follows. GS, GR and GR cells transfected with siRNA were seeded in 24-well plates at 5 × 104 cells per well. After 1 hour incubation, unattached cells were removed, and the attached cells were counted after trypsinization. The data were presented as a percentage of the cells attached to the plate compared to total cells. For cell detachment assay, the cells were seeded in 24-well plates at 5 × 104 cells per well. After 24 hours incubation, the medium was removed and the cells were incubated with 0.05% trypsin for 3 minutes to detach the cells from the culture plates. The medium containing 10% FBS was added into the cell to inactivate the trypsin and the detached cells were collected into tubes. The remaining cells were incubated with 0.25% trypsin to detach all the cells and collected into fresh tubes. The cells were counted and the data were presented as a percentage of the detached cells to total cells.
Cell migration and invasion assay
Cell migration was assessed using 24-well inserts (BD Biosciences, Bedford, MA) with 8μm pores according to the manufacturer’s protocol. The invasive activity of the GR siRNA transfected cells was tested by using the BD BioCoat Tumor Invasion Assay System (BD Biosciences, Bedford, MA) as described earlier (19).
Data Analyses
Experiments presented in the figures are representative of three or more different repetitions. The data are presented as the mean values ± SE. Comparisons between groups were evaluated by a two-tailed Student’s t test. Values of p < .05 were considered to be statistically significant.
Results
GR cells have the morphologic changes consistent with EMT
As shown in Fig. 1A, the GR cells displayed elongated, irregular fibroblastoid morphology. In contrast, GS cells had a rounded shape, typical of an epithelial cobblestone appearance and these cells grow in clusters. These changes in phenotype suggested that GR cells have undergone the EMT as reported previously (18). To further confirm whether GR cells underwent EMT, we determined the expression of markers of epithelial and mesenchymal phenotypes. We found that E-cadherin, β-catenin, and Twist expression was greatly reduced in GR cells (Fig. 1C). However, elevated levels of nestin, ZEB1, alpha-SMA, vimentin and fibronectin were observed in GR cells, suggesting that the expression of these factors may be important to gemcitabine-induced EMT of PC cells.
Figure 1.

L3.6pl Gemcitabine-resistant (GR) cells undergo an epithelial-mesenchymal transition (EMT) phenotype compared to the epithelial phenotype of L3.6pl gemcitabine-sensitive (GS) cells.
A: Morphology of GS and GR cells.
B: Real-time reverse transcription-polymerase chain reaction (RT-PCR) was used to quantify E-cadherin, vimentin and ZEB1 mRNA expression in GS and GR cells. **, P<0.01 compared with GS.
C: Western blot analysis showing the expression of markers of epithelial and mesenchymal phenotypes of cells.
To further confirm whether GR cells are truly EMT-like cells, we assessed the expression and localization of E-cadherin and β-catenin using immunofluorescence staining. GR cells acquired their elongated shape, which was consistent with β-catenin nuclear translocation and relocation of E-cadherin from cell plasma membrane to the nuclear compartment (Fig. 2). The results from immunostaining also indicate that GR cells had increased levels of expression of vimentin. F-actin reorganization and a diffuse pattern were also observed in GR cells, which were correlated with EMT phenotype. Since we confirmed that the GR cells are EMT-like cells, we investigated the role of Notch signaling especially because Notch signaling is known to play important roles during normal development as well as in the processes of EMT that is reminiscent of “cancer stem-like cells” (23, 24).
Figure 2.
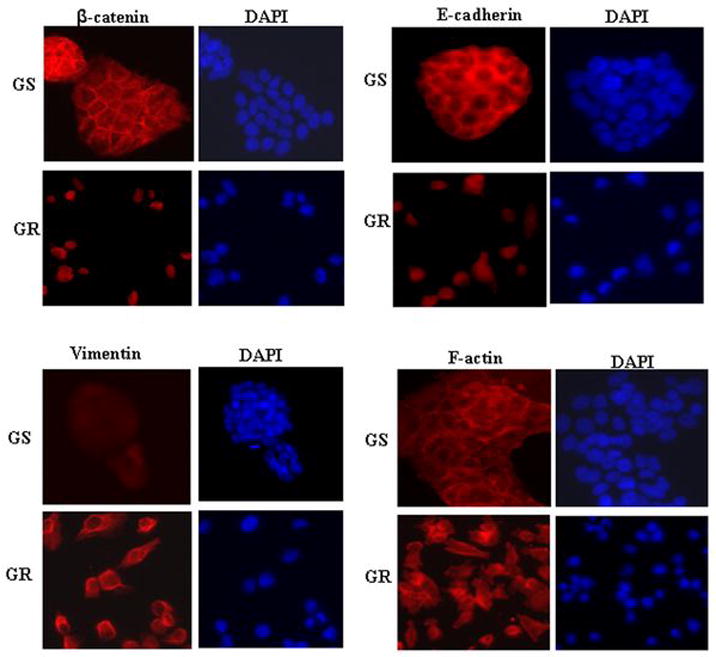
Immunofluorescence staining for the expression and cellular localization of β-catenin, E-cadherin, vimentin and F-actin.
Activation of Notch pathway is involved in EMT in GR cells
Notch pathway is involved in the EMT induction during tumor progression and converts polarized epithelial cells into motile, invasive cells (23). To determine whether Notch pathway is involved in GR cells, we assessed the levels of Notch pathway at mRNA and protein levels by Real-time RT-PCR and Western blotting, respectively. GR cells displayed an increased activation of Notch-2 and Jagged-1 at mRNA and protein levels. Notch-4 expression was also increased in GR cells. However, other Notch receptors and ligands were not significantly changed (Fig. 3A, 3B). To further confirm whether Notch pathway is involved in EMT, we determined the expression and localization of Notch-2, Notch-4, and Jagged-1 using immunofluorescence staining. We found that Notch-2 and Notch-4 were up-regulated in nuclear compartment in GR cells. We also found that the expression of Jagged-1 was increased in GR cells compared with GS cells (Fig. 3C).
Figure 3.

Notch pathway is up-regualted in GR cells.
A: Real-time RT-PCR was used to quantify Notch pathway genes at the mRNA levels in GS and GR cells. *, P<0.05, **, P<0.01 compared with GS.
B: Western blot analysis showing the expression of Notch pathway related proteins.
C: Immunofluorescence staining for the expression and cellular localization for Notch-2, Notch-4 and Jagged-1.
Notch downstream target, NF-κB and its downstream genes contribute to EMT characteristics
Notch pathway has been reported to strongly regulate NF-κB activity and induce expression of several NF-κB subunits (25). NF-κB has been identified as a central mediator of EMT in cancer progression (12). Therefore, we assessed whether Notch downstream target NF-κB subunit p65 expression is altered in GR cells or not. We found that p65 was up-regulated in GR cells. The expression of MMP-9, MMP-2, uPA and VEGF are known to be transcriptionally regulated by NF-κB and has been reported to play important roles in tumor migration and invasion (26). We therefore investigated whether MMP-9, MMP-2, uPA and VEGF correlate with the acquisition of EMT characteristics in GR cells. Indeed, we found that both MMP-9 mRNA and protein levels were dramatically increased in the GR cells (Fig. 4A, 4B). Next, we examined whether the GR cells have increased MMP-9 activity or not. We found a dramatic increase in the activity of MMP-9 in GR cells (Fig. 4C). The uPA and uPAR are known to regulate the MMP-9 activity in PC (27) and our data showed that both uPA mRNA and protein levels are significantly increased in the GR cells (Fig. 4A, 4B). Most importantly, GR cells showed higher uPA activity (Fig. 4B). However, we did not find any changes in the expression of MMP-2 or VEGF. Our results clearly showed up-regulation of MMP-9 and uPA in GR cells, results consistent with the up-regulation of NF-κB induced by the activation of Notch signaling pathway. To obtain direct proof in support of the role of Notch in EMT of GR cells, we used Notch inactivation strategies in GR cells.
Figure 4.
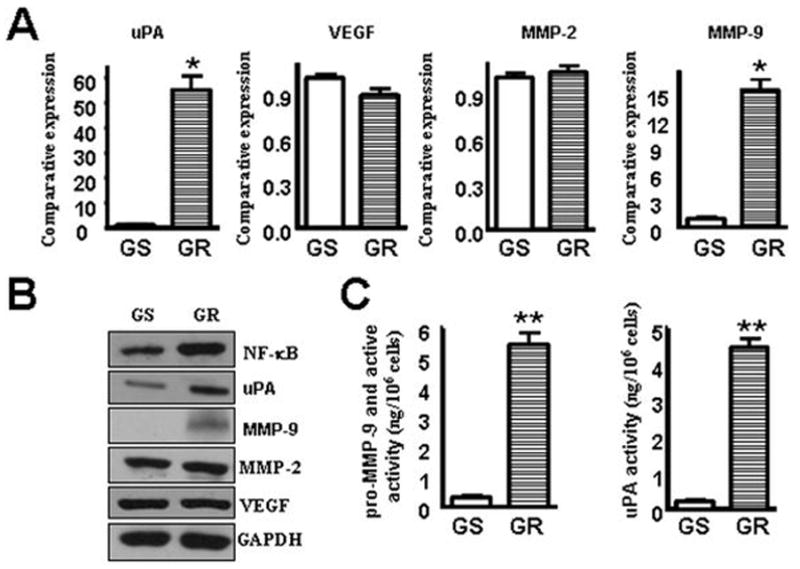
MMP-9 and uPA were up-regulated in GR cells.
A: Real-time RT-PCR reaction was used to quantify uPA, VEGF, MMP-2 and MMP-9 mRNA expression in GS and GR cells. *, P<0.05 compared with GS.
B: Western blot analysis showing MMP-9 and uPA protein levels were significantly increased in the GR cells.
C: Left, MMP-9 activity assay showing that MMP-9 was up-regulated in GR cells; right, uPA assay showing that uPA level in culture medium was up-regulated in GR cells.
Down-regulation of Notch signaling induces reversal of EMT to mesenchymal-epithelial transition (MET) in GR cells
We have shown that GR cells have a fibroblast-like morphology that is typical of mesenchymal phenotype of cells associated with the loss of epithelial markers. Therefore, to further confirm a direct mechanistic role of Notch in GR cells containing EMT characteristics, we decreased the expression of Notch-2 and Jagged-1 using specific siRNAs. As shown in Figure 5A and 5D, specific siRNAs for Notch-2 or Jagged-1 were effective in reducing the expression of these proteins compared to GAPDH protein (used as protein loading control). Subsequently, we assessed whether EMT phenotype was reversed in GR cells transfected with Notch-2 and Jagged-1 siRNAs. We found that GR cells transfected with Notch-2 or Jagged-1 siRNA displayed round cell-like morphology three days after transfection (Fig. 5B). The results from Real-time RT-PCR showed that the expression of E-cadherin was significantly increased, which clearly suggests that the down-regulation of Notch-2 or Jagged-1 expression in GR cells results in the reversal of EMT phenotype to MET phenotype (Fig. 5C). However, down-regulation of Notch-4 by siRNA transfection did not show any sign of the reversal of EMT (data not shown). We also found that the pattern of expression of mesenchymal markers was changed in GR cells transfected with Notch-2 and Jagged-1 siRNA, showing a dramatic reduction in the expression of vimentin, which is consistent with the acquisition of MET phenotype of GR cells (Fig. 5C, 5D).
Figure 5.
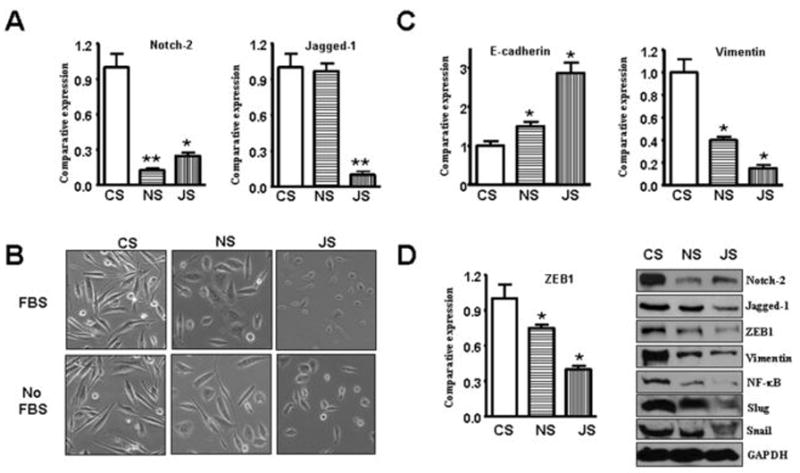
Notch pathway contributes to the regulation of molecular markers of epithelial-mesenchymal transition (EMT) in GR cells. Abbreviations: CS, control siRNA; NS, Notch-2 siRNA; JS, Jagged-1 siRNA.
A: Real-time RT-PCR was used to quantify Notch-2 and Jagged-1 mRNA expression in GR cells transfected with specific siRNA. *, P<0.05, **, P<0.01 compared with CS.
B: Down-regulation of Notch-2 and Jagged-1 could cause reversal of EMT phenotype of GR cells.
C-D: GR cells transfected with control siRNA or Notch-2 siRNA or Jagged-1 siRNA were assessing the expression of markers of epithelial and mesenchymal phenotypes using Real-time RT-PCR and Western blot analysis, respectively.
It is known that E-cadherin is a downstream target of ZEB1, and that ZEB1 is a transcriptional repressor to the expression of which down-regulates E-cadherin, resulting in the induction of the EMT phenotype. Therefore, we sought to assess the expression levels of ZEB1 in GR cells. As documented in figure-1 that the expression of ZEB1 was increased in GR cells, consistent with up-regulation of E-cadherin and the acquisition of EMT phenotype. We subsequently observed that ZEB1 expression was down-regulated in GR cells transfected with Notch-2 and Jagged-1 siRNA (Fig. 5D). Recently, Notch pathway was found to up-regulate Snail and Slug expression (24, 28). Slug is essential for Notch-mediated EMT by repressing E-cadherin expression, which results in β-catenin activation (24, 28). Therefore, we assessed the expression of Snail and Slug in GR cells transfected with Notch-2 and Jagged-1 siRNA. As we expected, the expressions of Snail and Slug were down-regulated in Notch-2 siRNA- and Jagged-1 siRNA- transfected cells (Fig. 5D).
Next, we assessed whether the increased levels in the expression of NF-κB as seen in Figure. 4 in GR cells could be down-regulated in GR cells transfected with Notch-2 and Jagged-1 siRNA. Moreover, since it is known that NF-κB (p65) could up-regulate ZEB1 expression, we also assessed the inter-relationships among Notch-2, Jagged-1, NF-κB and ZEB1 in GR cells transfected with Notch-2 and Jagged-1 siRNAs. Our results showed that the expression of NF-κB (p65 protein) was down-regulated in GR cells transfected with Notch-2 and Jagged-1 siRNAs (Fig. 5D). These results strongly suggest that down-regulation of Notch pathway contributes to the down-regulation of NF-κB and ZEB1, resulting in the up-regulation of E-cadherin and the reversal of EMT phenotype to MET phenotype.
Down-regulation of Notch signaling reduced detachment and attachment and inhibits the migration and invasion of GR cells
It is well known that cell detachment from the matrix where the tumor grows in the micro-environment and attachment to the secondary site is the “hallmark” of cell migration and invasion during metastatic process. We found that GR cells have increased capacity for attachment and detachment (Fig. 6A). Since the activation of Notch can lead to increased migration and invasion, we sought to assess the ability of GR cells for attachment, detachment and for the ability of GR cells for its migration and invasion when transfected with Notch-2 and Jagged-1 siRNA. As expected, we found that GR cells transfected with Notch-2 and Jagged-1 siRNA displayed decreased detachment and attachment (Fig. 6B). More importantly, down-regulation of Notch-2 and Jagged-1 by siRNA markedly reduced the migratory and invasive ability of GR cells (Fig. 6C), which is consistent with the cell attachment and detachment findings. These results clearly suggest that down-regulation of Notch pathway down-regulates NF-κB and ZEB1 and consequently up-regulates E-cadherin, resulting in the reversal of the EMT phenotype to a MET phenotype with less cell migration and invasion characteristics.
Figure 6.

Down-regulation of Notch-2 and Jagged-1 inhibited cell migration and invasion, and reduce attachment and detachment of GR cells. Abbreviations: GS, Gemcitabine-sensitive; GR, gemcitabine-resistant. CS, control siRNA; NS, Notch-2 siRNA; JS, Jagged-1 siRNA. *, P<0.05, **, P<0.01 compared with control.
A: GR cells showed increased cell attachment and detached.
B: Down-regulation of Notch-2 or Jagged-1 in GR cells dramatically decreased the attachment and detachment of cells.
C: Transfection of GR cells with Notch-2 siRNA or Jagged-1 siRNA inhibited the migration and invasion.
Discussion
Despite rapid advances in the diagnostic and surgical procedures, pancreatic cancer (PC) remains one of the most difficult human malignancies to treat. This may be due to intrinsic (de novo) and extrinsic (therapy-induced) chemo- or radio-resistant behaviors of PC and as of yet no systemic therapy has been proven effective in improving the overall survival of patients diagnosed with this deadly disease (1). It is believed that both de novo and acquired resistance to therapy could be overcome by understanding the mechanisms by which PC become chemo-resistant, which will improve the overall survival of patients. Therefore, in this study, we have used a gemcitabine-sensitive (GS) and a gemcitabine-resistant (GR) PC cell line to investigate the molecular mechanism of resistance and associated cellular behaviors. Previous studies have shown that GR cells acquired epithelial-mesenchymal transition (EMT) phenotype, which is reminiscent of “cancer stem-like cells (CSC)”, which was associated with spindle-shaped morphology and enhanced pseudopodia formation. The GR cells also showed decreased expression of the epithelial adhesion molecule, E-cadherin and an increase in the expression of mesenchymal markers such as vimentin. However, the mechanism(s) governing the acquisition of EMT phenotype by GR cells have not been fully elucidated.
Many of signaling pathways, including Notch and NF-κB signaling pathways, are found to be critical for EMT induction (12, 16, 28). The activation of Notch signaling is known to regulate the expression of its target genes, and thus plays important roles in growth and development, including regulation of proliferation and apoptosis (29–31). Therefore, not surprisingly, alterations in Notch signaling are associated with tumorigenesis (19, 29, 30). Moreover, it has been reported that Notch is involved in the EMT induction during tumor progression and converts polarized epithelial cells into motile, invasive cells (24). Over-expression of Jagged-1 and Notch-1 induces the expression of Slug and correlates with poor prognosis in various human cancers (24). Slug is also essential for Notch-mediated EMT by repressing E-cadherin expression, which results in β-catenin activation (24). In addition, Niessen et al. demonstrated that Slug is directly up-regulated by Notch in endothelial cells and that Slug expression is required for Notch-mediated repression of the vascular endothelial cadherin promoter and for promoting migration of transformed endothelial cells (32). Our results are consistent with these findings.
Recently, Notch pathway was found to up-regulate Snail expression by recruitment of the intracellular Notch (ICN) to Snail promoter (28). Moreover, Notch potentiate hypoxia-inducible factor 1α recruitment to the lysyl oxidase (LOX) promoter and elevates the hypoxia-induced up-regulation of LOX, which stabilizes the Snail protein (28). Consistent with these findings, we found that GR cells displayed an increased activation of Notch-2 and Jagged-1 both at the mRNA and protein levels, which was consistent with EMT characteristics of GR cells. To gain further insight whether Notch could directly regulate the expression of the above mentioned epithelial marker genes, we down-regulated the expression of Notch-2 and Jagged-1 by transfection of GR cells with specific siRNAs. The GR cells transfected with Notch-2 and Jagged-1 siRNA led to an increase in the expression of E-cadherin, and reduced the expression of vimentin and ZEB1. The expression of Snail and Slug were also down-regulated by Notch-2 siRNA and Jagged-1 siRNA. Taken together, these results suggest that the activation of Notch, Jagged-1 and ZEB1 could mediate the induction of EMT phenotype as observed in GR cells and that the down-regulation of Notch signaling could be useful for the reversal of the EMT phenotype. However our results raised a question with respect to how the activation of Notch-2 could regulate the expression of epithelial and mesenchymal molecular markers and whether Notch signaling could cross-talk with other signaling molecules critical to EMT. To answer this question, we determined the expression of NF-κB in GR cells. The rationale was based on the fact that Notch was reported to cross-talk with NF-κB (33). Constitutive levels of Notch activity are essential to maintain NF-κB activity in various cell types (34, 35). Recently, Fukushima et al. reported that Notch-2 could enhance NF-κB transcriptional activity (36). Indeed, we found that the expression of NF-κB protein (p65 subunit of NF-κB) was increased significantly in GR cells compared to GS cells. Moreover, the down-regulation of Notch-2 and Jagged-1 by siRNA transfection resulted in decreased NF-κB in GR cells, suggesting a molecular link or cross-talk between Notch and NF-κB, and thus the activation of these signaling pathways appears to be mechanistically associated with the acquisition of EMT phenotype of GR cells.
The activation of NF-κB is known to play critical roles in the processes of EMT and tumor cell invasion and metastasis (12). Emerging evidence also suggests that the activation of NF-κB is mechanistically linked with the processes of EMT via regulating the expression of a transcription factor, ZEB1, which negatively regulate the transcription of E-cadherin, resulting in the loss of E-cadherin in EMT-like cells (37) and our findings are consistent with this mechanism. Although the activation of NF-κB is associated with EMT, it is not known what downstream gene of NF-κB is mechanistically associated with EMT. A candidate for such a gene is MMP-9, a well-described NF-κB downstream target gene. Thus, we assessed the role of NF-κB downstream genes in the processes of EMT and found that MMP-9 expression and its activity were significantly increased in GR cells, which is consistent with the activation of Notch and NF-κB signaling. Since uPA and uPAR are known to regulate the activity of MMP-9 activity in PC cells (27) and uPA and its receptor (uPAR) are important genes in the processes of tumor cell invasion and metastasis (38, 39), we assessed the expression of uPA in our system. We found that GR cells have higher expression of uPA, which is consistent with previous findings (18). Therefore, our results suggest that GR cells could potentiate aggressive behavior in part due to the up-regulation of MMP-9 and uPA expression during the acquisition of EMT phenotype.
Since the processes of EMT have been linked with cell migration and invasion, we hypothesized that GR cells transfected with Notch-2 and Jagged-1 siRNA could lead to decreased migration and invasion compared to cells transfected with the control siRNA. We found that the transfection of GR cells with Notch-2 and Jagged-1 siRNA significantly inhibited cell attachment and detachment, and cell migration and invasion. These results are consistent with the mechanistic role of Notch signaling in the processes of EMT and that the inactivation of Notch signaling leads to the reversal of EMT to MET phenotype with less invasive characteristics. Collectively, our results provide strong evidence suggesting that the inactivation of Notch signaling by novel therapeutic strategies could be clinically important in overcoming drug resistance and the reversal of EMT phenotype, which is likely to improve the overall survival of patients diagnosed with PC.
In conclusion, our results provided strong molecular evidence, for the first time, showing that GR cells underwent EMT partly due to the activation of Notch signaling. Therefore, we believe that novel strategies by which Notch signaling could be down-regulated may become an important approach for the prevention of tumor progression and/or treatment of invasive and metastatic PC for which there is no curative therapy.
Acknowledgments
Grant Support: This work was partly funded by grants from the National Cancer Institute, NIH (5R01CA101870-05) to F.H.S. and also partly supported by a subcontract award (F.H.S.) from the University of Texas MD Anderson Cancer Center through a SPORE grant (1P20-CA010193-01) on pancreatic cancer awarded to James Abbruzzese. We also sincerely thank the Puschelberg Foundation for their generous contribution.
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Stathopoulos GP, Androulakis N, Souglakos J, Stathopoulos J, Georgoulias V. Present treatment and future expectations in advanced pancreatic cancer. Anticancer Res. 2008;28:1303–8. [PubMed] [Google Scholar]
- 3.Kajiyama H, Shibata K, Terauchi M, et al. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int J Oncol. 2007;31:277–83. [PubMed] [Google Scholar]
- 4.Yang AD, Fan F, Camp ER, et al. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res. 2006;12:4147–53. doi: 10.1158/1078-0432.CCR-06-0038. [DOI] [PubMed] [Google Scholar]
- 5.Hiscox S, Jiang WG, Obermeier K, et al. Tamoxifen resistance in MCF7 cells promotes EMT-like behaviour and involves modulation of beta-catenin phosphorylation. Int J Cancer. 2006;118:290–301. doi: 10.1002/ijc.21355. [DOI] [PubMed] [Google Scholar]
- 6.Hiscox S, Morgan L, Barrow D, Dutkowskil C, Wakeling A, Nicholson RI. Tamoxifen resistance in breast cancer cells is accompanied by an enhanced motile and invasive phenotype: inhibition by gefitinib (‘Iressa’, ZD1839) Clin Exp Metastasis. 2004;21:201–12. doi: 10.1023/b:clin.0000037697.76011.1d. [DOI] [PubMed] [Google Scholar]
- 7.Croker AK, Allan AL. Cancer stem cells: implications for the progression and treatment of metastatic disease. J Cell Mol Med. 2008;12:374–90. doi: 10.1111/j.1582-4934.2007.00211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kondo T. Brain cancer stem-like cells. Eur J Cancer. 2006;42:1237–42. doi: 10.1016/j.ejca.2006.01.038. [DOI] [PubMed] [Google Scholar]
- 9.Ponti D, Zaffaroni N, Capelli C, Daidone MG. Breast cancer stem cells: an overview. Eur J Cancer. 2006;42:1219–24. doi: 10.1016/j.ejca.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 10.Soltysova A, Altanerova V, Altaner C. Cancer stem cells. Neoplasma. 2005;52:435–40. [PubMed] [Google Scholar]
- 11.Hugo H, Ackland ML, Blick T, et al. Epithelial--mesenchymal and mesenchymal--epithelial transitions in carcinoma progression. J Cell Physiol. 2007;213:374–83. doi: 10.1002/jcp.21223. [DOI] [PubMed] [Google Scholar]
- 12.Min C, Eddy SF, Sherr DH, Sonenshein GE. NF-kappaB and epithelial to mesenchymal transition of cancer. J Cell Biochem. 2008;104:733–44. doi: 10.1002/jcb.21695. [DOI] [PubMed] [Google Scholar]
- 13.Kong D, Wang Z, Sarkar SH, et al. Platelet-derived growth factor-D overexpression contributes to epithelial-mesenchymal transition of PC3 prostate cancer cells. Stem Cells. 2008;26:1425–35. doi: 10.1634/stemcells.2007-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Larue L, Bellacosa A. Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene. 2005;24:7443–54. doi: 10.1038/sj.onc.1209091. [DOI] [PubMed] [Google Scholar]
- 15.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–28. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 16.Wu Y, Zhou BP. New insights of epithelial-mesenchymal transition in cancer metastasis. Acta Biochim Biophys Sin (Shanghai) 2008;40:643–50. doi: 10.1111/j.1745-7270.2008.00443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rho JK, Choi YJ, Lee JK, et al. Epithelial to mesenchymal transition derived from repeated exposure to gefitinib determines the sensitivity to EGFR inhibitors in A549, a non-small cell lung cancer cell line. Lung Cancer. 2008 doi: 10.1016/j.lungcan.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 18.Shah AN, Summy JM, Zhang J, Park SI, Parikh NU, Gallick GE. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann Surg Oncol. 2007;14:3629–37. doi: 10.1245/s10434-007-9583-5. [DOI] [PubMed] [Google Scholar]
- 19.Wang Z, Banerjee S, Li Y, Rahman KM, Zhang Y, Sarkar FH. Down-regulation of Notch-1 inhibits invasion by inactivation of nuclear factor-{kappa}B, vascular endothelial growth factor, and matrix metalloproteinase-9 in pancreatic cancer cells. Cancer Res. 2006;66:2778–84. doi: 10.1158/0008-5472.CAN-05-4281. [DOI] [PubMed] [Google Scholar]
- 20.Wang Z, Banerjee S, Kong D, Li Y, Sarkar FH. Down-regulation of Forkhead Box M1 transcription factor leads to the inhibition of invasion and angiogenesis of pancreatic cancer cells. Cancer Res. 2007;67:8293–300. doi: 10.1158/0008-5472.CAN-07-1265. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, Wang Z, Ahmed F, Banerjee S, Li Y, Sarkar FH. Down-regulation of Jagged-1 induces cell growth inhibition and S phase arrest in prostate cancer cells. Int J Cancer. 2006;119:2071–7. doi: 10.1002/ijc.22077. [DOI] [PubMed] [Google Scholar]
- 22.Gregory PA, Bert AG, Paterson EL, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 23.Grego-Bessa J, Diez J, Timmerman L, de la Pompa JL. Notch and epithelial-mesenchyme transition in development and tumor progression: another turn of the screw. Cell Cycle. 2004;3:718–21. [PubMed] [Google Scholar]
- 24.Leong KG, Niessen K, Kulic I, et al. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J Exp Med. 2007;204:2935–48. doi: 10.1084/jem.20071082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Osipo C, Golde TE, Osborne BA, Miele LA. Off the beaten pathway: the complex cross talk between Notch and NF-kappaB. Lab Invest. 2008;88:11–7. doi: 10.1038/labinvest.3700700. [DOI] [PubMed] [Google Scholar]
- 26.Sarkar FH, Li Y, Wang Z, Kong D. NF-kappaB signaling pathway and its therapeutic implications in human diseases. Int Rev Immunol. 2008;27:293–319. doi: 10.1080/08830180802276179. [DOI] [PubMed] [Google Scholar]
- 27.Harvey SR, Hurd TC, Markus G, et al. Evaluation of urinary plasminogen activator, its receptor, matrix metalloproteinase-9, and von Willebrand factor in pancreatic cancer. Clin Cancer Res. 2003;9:4935–43. [PubMed] [Google Scholar]
- 28.Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci U S A. 2008;105:6392–7. doi: 10.1073/pnas.0802047105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miele L, Miao H, Nickoloff BJ. Notch signaling as a novel cancer therapeutic target. Curr Cancer Drug Targets. 2006;6:313–23. doi: 10.2174/156800906777441771. [DOI] [PubMed] [Google Scholar]
- 30.Miele L. Notch signaling. Clin Cancer Res. 2006;12:1074–9. doi: 10.1158/1078-0432.CCR-05-2570. [DOI] [PubMed] [Google Scholar]
- 31.Wang Z, Zhang Y, Li Y, Banerjee S, Liao J, Sarkar FH. Down-regulation of Notch-1 contributes to cell growth inhibition and apoptosis in pancreatic cancer cells. Mol Cancer Ther. 2006;5:483–93. doi: 10.1158/1535-7163.MCT-05-0299. [DOI] [PubMed] [Google Scholar]
- 32.Niessen K, Fu Y, Chang L, Hoodless PA, McFadden D, Karsan A. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J Cell Biol. 2008;182:315–25. doi: 10.1083/jcb.200710067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jang MS, Miao H, Carlesso N, et al. Notch-1 regulates cell death independently of differentiation in murine erythroleukemia cells through multiple apoptosis and cell cycle pathways. J Cell Physiol. 2004;199:418–33. doi: 10.1002/jcp.10467. [DOI] [PubMed] [Google Scholar]
- 34.Nickoloff BJ, Qin JZ, Chaturvedi V, Denning MF, Bonish B, Miele L. Jagged-1 mediated activation of notch signaling induces complete maturation of human keratinocytes through NF-kappaB and PPARgamma. Cell Death Differ. 2002;9:842–55. doi: 10.1038/sj.cdd.4401036. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, Chan SL, Miele L, et al. Involvement of Notch signaling in hippocampal synaptic plasticity. Proc Natl Acad Sci U S A. 2004;101:9458–62. doi: 10.1073/pnas.0308126101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fukushima H, Nakao A, Okamoto F, et al. The association of Notch2 and NF-kappaB accelerates RANKL-induced osteoclastogenesis. Mol Cell Biol. 2008;28:6402–12. doi: 10.1128/MCB.00299-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chua HL, Bhat-Nakshatri P, Clare SE, Morimiya A, Badve S, Nakshatri H. NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene. 2007;26:711–24. doi: 10.1038/sj.onc.1209808. [DOI] [PubMed] [Google Scholar]
- 38.Kong D, Li Y, Wang Z, Banerjee S, Sarkar FH. Inhibition of angiogenesis and invasion by 3,3′-diindolylmethane is mediated by the nuclear factor-kappaB downstream target genes MMP-9 and uPA that regulated bioavailability of vascular endothelial growth factor in prostate cancer. Cancer Res. 2007;67:3310–9. doi: 10.1158/0008-5472.CAN-06-4277. [DOI] [PubMed] [Google Scholar]
- 39.Dass K, Ahmad A, Azmi AS, Sarkar SH, Sarkar FH. Evolving role of uPA/uPAR system in human cancers. Cancer Treat Rev. 2008;34:122–36. doi: 10.1016/j.ctrv.2007.10.005. [DOI] [PubMed] [Google Scholar]