

Abstract
Depth of cell invasion into the dermis is a clinical determinant for poor prognosis in cutaneous melanoma. The signaling events that promote the switch from a non-invasive to invasive tumor phenotype remain obscure. Activating mutations in the serine/threonine kinase B-RAF are prevalent in melanoma. Mutant B-RAF is required for melanoma cell invasion. The expression of Rnd3, a Rho family GTPase, is regulated by mutant B-RAF, although its role in melanoma progression is unknown. In this study, we determined the functional contribution of Rnd3 to invasive melanoma. Endogenous Rnd3 was targeted for knockdown using a doxycyclineinducible shRNA system in invasive human melanoma cells. Depletion of Rnd3 promoted prominent actin stress fibers and enlarged focal adhesions. Mechanistically, stress fiber formation induced by Rnd3 knockdown required the specific involvement of RhoA and ROCK1/2 activity but not RhoB or RhoC. Rnd3 expression in human melanoma cell lines was strongly associated with elevated ERK phosphorylation and invasive behavior in a 3-D dermal-like environment. A functional role for Rnd3 was demonstrated in the invasive outgrowth of melanoma tumor spheroids. Knockdown of Rnd3 reduced invasive outgrowth of spheroids embedded in collagen gels. Additionally, Rnd3 depletion inhibited collective and border cell movement out from spheroids in a ROCK1/2-dependent manner. Collectively, these findings implicate Rnd3 as a major suppressor of RhoA mediated actin cytoskeletal organization and in the acquisition of an invasive melanoma phenotype.
Keywords: Melanoma, Invasion, Migration, Cytoskeleton, Rnd3
Introduction
A critical event during melanoma progression is the transition from radial growth phase (RGP) to vertical growth phase (VGP). This transition is associated with movement out of the epidermis and invasion into the dermis (1). The molecular mechanisms underlying invasion in melanoma remain poorly understood. It is critical to understand these mechanisms since VGP cells acquire metastatic properties and the depth of invasion is used as a clinical determinant of a poor prognosis.
Activating mutations in the serine/threonine kinase B-RAF occur at a frequency of 50-70% in melanoma and are detected throughout various disease stages (2, 3). Mutant B-RAF has been implicated with numerous cellular functions in melanoma including invasion through matrigel (4, 5), a basement membrane-like extracellular matrix. Efficient activation of the MEK-ERK1/2 pathway downstream of mutant B-RAF mediates its effects on invasion. Non-invasive nevus cells and RGP melanoma can harbor B-RAF mutations but the MEK-ERK1/2 pathway is inefficiently activated due, in part, to negative feedback mechanisms (6, 7). When unabated, mutant B-RAF regulates melanoma cell invasion possibly via up-regulating expression of interstitial collagenase I/matrix metalloproteinase 1 (8, 9), although protease independent invasion has also been reported in mutant B-RAF melanoma cells (10). Additional micro-environmental influences and alterations in the dynamics of the actin cytoskeleton and focal adhesions (extracellular matrix interaction sites) also contribute to motility and invasive processes (11-13); however, their role in mutant B-RAF regulated melanoma invasion remain poorly described.
Rho-family GTPases (RhoA/B/C, Rac and Cdc42) are pivotal regulators of the actin cytoskeleton and cell migration (14, 15). Although rarely mutated, changes in their expression or functional activity have been associated with tumor progression (16, 17). Regulatory molecules that control Rho GTPase signaling are frequently responsible for the associated changes in actin cytoskeleton remodeling and tumor migration. Rnd3/RhoE/Rho8, a negative regulator of the Rho/ROCK pathway, has recently garnered attention as a prognostic indicator in cancer (18). Rnd3 is up-regulated in pancreatic cancer (19), melanoma (20, 21) and non-small cell lung cancer (22), whereas it is down-regulated in prostate cancer (23). Overexpression studies have implicated Rnd3 in disrupting actin stress fibers and focal adhesions in fibroblast and epithelial cells (24-27), yet conflicting roles remain for Rnd3 in transformation (28-30), tumor cell migration and invasion (24, 31). The function of endogenous Rnd3 has been less well studied. Recent literature indicate that endogenous Rnd3 participates in ROCK-mediated apoptosis (32) and myoblast alignment (33).
We recently demonstrated that mutant B-RAF expression in melanoma cells regulates actin cytoskeletal and focal adhesion organization (21). These effects were supported by mutant B-RAF control of Rnd3 expression, which acts as a regulator of cross-talk between the B-RAF/MEK/ERK and Rho/ROCK/LIM kinase/cofilin pathways (21). In the current study, we show that endogenous Rnd3 influences actin architecture through specific attenuation of RhoA stimulated actin stress fibers. Increased expression of Rnd3 correlated with the progression towards an invasive phenotype in melanoma cell lines. Notably, inducible depletion of Rnd3 by RNA interference decreased migration and invasive outgrowth on 3-D substrates. Collectively these data highlight the importance of Rnd3, a mutant B-RAF effector, to invasive melanoma behavior.
Materials and Methods
Cell culture
Human melanoma cells were provided by Dr. Meenhard Herlyn (Wistar Institute, Philadelphia, PA) and cultured as previously indicated (21). Neonatal foreskins were obtained according to Albany Medical College Institutional Review Board procedures. Normal human epidermal melanocytes (NHEM) were isolated and cultured, as previously described (21).
Antibodies and reagents
The following antibodies were used: diphosphorylated (Thr18/Ser19) myosin light chain, kindly provided by Dr. Peter Vincent (Albany Medical College, Albany, NY) (34); phospho-ERK1/2 (9106) from Cell Signaling Technology (Danvers, MA); ERK2 (sc154), RhoA (sc418), RhoB (sc180) and RhoC (sc12116) from Santa Cruz Biotech. (Santa Cruz, CA); Rnd3 (05-723) from Upstate Biotech Inc. (Lake Placid, NY); and actin (A5060) from Sigma-Aldrich (St. Louis, MO). Alexa-Fluor conjugated secondary antibodies was obtained from Molecular Probes Inc. (Eugene OR). TRITC-conjugated phalloidin and etoposide were purchased from Sigma-Aldrich.
Recombinant lentiviral infections
Inducible short hairpin RNA (shRNA) knockdowns were performed using the BLOCK-iT™ lentiviral expression system (Invitrogen, Carlsbad, CA). Oligonucleotide sequences are listed in Supplemental Methods and were based on earlier studies using short-interfering RNA (siRNA) (21). All shRNA constructs and cell lines were generated, as previously described (35). Constructs were verified by DNA sequencing. Inducible shRNA expression was achieved upon addition of 100 ng/ml doxycycline (dox) (Fisher Scientific, Fair Lawn, NJ) to cell cultures.
siRNA knockdowns
The following siRNA duplexes purchased from Dharmacon Research Inc. (Lafayette, CO) were used: RhoA (AUGGAAAGCAGGUAGAGUUUU), RhoB (ACACCGACGUCAUUCUCAUUU) and RhoC (GGAGAGAGCUGGCCAAGAUUU). The non-targeting siControl#1 was also used (Dharmacon). Melanoma cells were transfected as previously indicated (21).
Western blotting
Standard methods were used for western blotting using PVDF membrane (21). Immunoreactive bands were developed using enhanced chemiluminescence kits (Pierce Chemical, Rockford IL), detected with a Fluor-S MultiImager (Bio-Rad, Hercules, CA) and band intensity quantified using Quantity-One image analysis (Bio-Rad).
Cell growth and cell cycle analysis
Experiments were performed on cells in growth medium in the absence/presence of dox. DNA synthesis was measured using Click-iT EdU Alexa Fluor 647 (Invitrogen). Briefly, knockdown cells labeled 7 hours with 10 μM EdU were resuspended, fixed and processed according to manufacturer's instruction. Propidium iodide staining was performed using 10 μg/ml propidium iodide (Invitrogen). Flow cytometer was carried out using a BD FACSCanto. Data were analyzed with FlowJo cytometry software (Tree Star, Ashland, OR).
Immunofluorescence
Cells plated on glass coverslips were treated as previously indicated (21). An Olympus BX-61 microscope equipped with a CCD sensi-camera (Cooke, Auburn Hills, MI) was used to photograph samples. Images were acquired using IPLab software (Scanalytics Inc., Fairfax VA). Quantification was performed according to the method outlined previously (36) by categorizing at least 600 cells from multiple experiments depending on their cytoskeletal organization. Images were further processed using Adobe Photoshop Software (Adobe Systems, San Jose CA).
Migration assays
Collective cell migration assays were performed on spheroids placed on top of collagen I gels and submerged in complete growth medium. Cell movement was monitored for five hours at 37°C. Images were acquired using an Olympus IX-70 inverted microscope equipped with a CCD sensi-camera (Cooke) and processed using Image-Pro Plus software (Media Cybernetics Inc., Silver Springs, MD). Collective cell migration was quantified using an integrated morphometric analysis to measure the change in spheroid surface area from identical spheroids. Migrating border cells were quantified by counting the number of individual cells separating away from the collective cell sheet during a 5 hour time period.
3-D spheroid invasive outgrowth
Melanoma spheroids were prepared and implanted into collagen gel as described (37). Spheroids, formed by growing cells in non-adherent conditions for 72 hours in the absence/presence of doxycycline, were embedded into a bovine collagen I gels and placed into a dish pre-coated with a layer of acellular collagen. After the gel solidified it was overlaid with normal growth medium, supplemented with dox, if appropriate, and incubated four days. To assess cell viability, spheroids were washed in PBS and incubated with calcein-AM and ethidium bromide, according to the manufacturer's instruction (Molecular Probes, Inc.). Photographs were taken and processed for morphometric analysis, as described above. A total of 8-20 spheroids from 2-6 experiments were analyzed per condition. Box-and-whisker plots indicate the 25th percentile (bottom line of the box), median (middle line), 75th percentile (top line of the box), 5th and 95th percentile (whiskers), and minimum and maximum measurements (•) of the entire population.
Results
Rnd3 regulates the actin cytoskeletal organization of invasive melanoma
Rnd3 is a target of oncogenic B-RAF-MEK-ERK1/2 signaling in melanoma (21). To investigate the role of endogenous Rnd3 in melanoma, we constructed a dox inducible shRNA system to knockdown Rnd3 in WM793 and WM115 human invasive melanoma cell lines. WM793 and WM115 harbor the constitutive active B-RAF mutations V600E and V600D, respectively (38). To reduce concerns about `off-target' effects, all experiments were performed using two distinct Rnd3 shRNA (Rnd3#1 or Rnd3#2) sequences and a control non-targeting shRNA. Western blot analysis of multiple experiments indicated that treatment with dox for 72 hours did not alter Rnd3 levels in control shRNA expressing WM793 cells whereas Rnd3#1 and Rnd3#2 shRNA lowered endogenous protein levels by 53% and 77%, respectively (Fig 1A and 1B).
Figure 1.
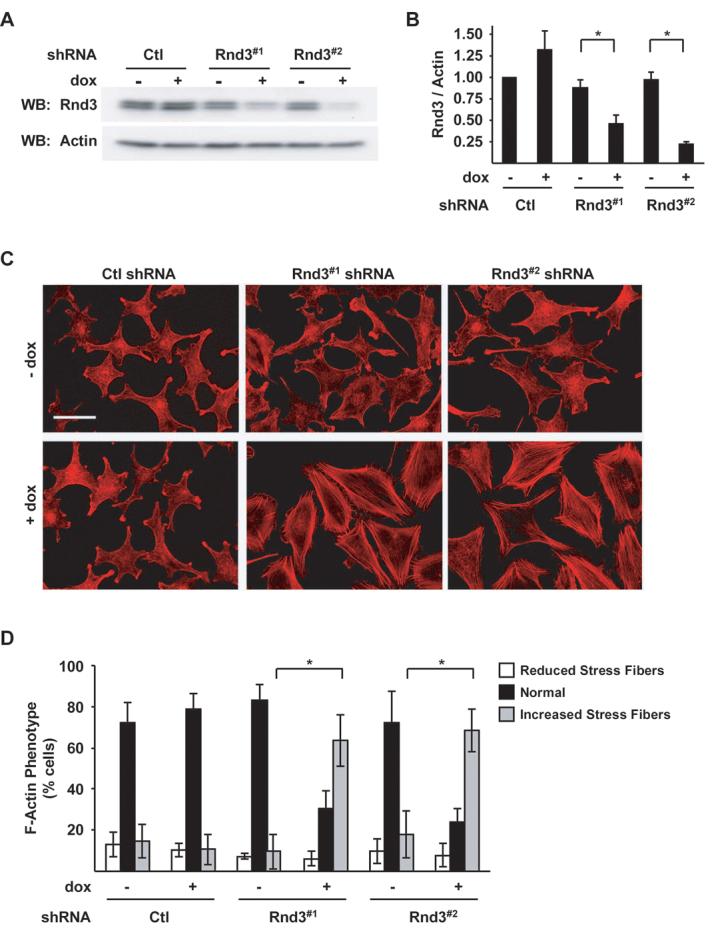
Depletion of endogenous Rnd3 induces melanoma actin cytoskeletal reorganization. Doxycycline-inducible melanoma cells expressing Ctl, Rnd3#1 or Rnd3#2 shRNA. A, Cell lysates analyzed by western blotting for Rnd3 and β-actin protein levels. B, Quantitation of endogenous Rnd3 knockdown. Graphed are the means ± SD Rnd3/actin ratios from three experiments with the Ctl shRNA condition set to one. Asterisk denotes statistical significance as determined by a two-tailed unpaired t-Test comparing Rnd3 knockdowns with Ctl knockdowns (* P<0.05). C-D, F-actin organization in inducible shRNA melanoma cells. C, Cells cultured ± dox for 72 hours were fixed and stained with TRITC-phalloidin. Bar, 50 μm. D, Quantitation of the F-actin phenotype shown in (C). The graph displays the proportion of cells with visibly altered actin stress fiber organization; means ± SD of >600 cells from multiple independent experiments; (*P<0.05).
To investigate the consequence of reduced endogenous Rnd3 expression in invasive melanoma cells, we initially evaluated actin cytoskeletal organization. Microscopic evaluation of F-actin staining revealed that approximately 85% of control shRNA WM793 cells displayed either a disorganized or cortical F-actin phenotype (Fig. 1C, 1D and Supplementary Fig. S1A). In contrast, endogenous Rnd3 depletion produced a switch to an actin phenotype composed predominately of stress fibers traversing the cell (Fig. 1C and 1D). Similar results were obtained following inducible knockdown of Rnd3 in the WM115 melanoma cell line (Supplemental Fig. S1B and S1C). Actin stress fiber formation following Rnd3 depletion required serum/growth factors since enhanced stress fiber formation was not observed in serum-free conditions (Supplemental Fig. S1D).
To further characterize the role of endogenous Rnd3, we evaluated whether alterations in focal adhesion localization accompanied Rnd3 dependent changes in actin organization. Inducible knockdown WM793 cells were processed for immunofluorescence analysis of vinculin, a marker of focal adhesions (39). We did not observe discernable differences in overall number of focal adhesions following Rnd3 knockdown; however, the size of focal adhesions were markedly enlarged (Supplemental Fig. S2A). Additionally, no alterations in cell surface integrin expression were detected (Supplemental Fig S3). Collectively, these data suggest that endogenous Rnd3 may function to suppress serum/growth factor-induced actin stress fiber and focal adhesion formation in invasive melanoma.
RhoA is required for Rnd3 knockdown induced alterations in actin stress fibers
The appearance of actin stress fibers is a hallmark feature attributed to the activation of small GTPase Rho-family members, i.e. RhoA, RhoB and RhoC (14, 26). To establish which Rho isoform is required for stress fibers associated with Rnd3 depletion, we performed co-knockdown experiments. RhoA/B/C isoforms are all expressed in WM793 cells and their protein expression did not change following Rnd3 depletion (Fig 2A). Knockdown of individual Rho members by siRNA in the absence or presence of Rnd3 was confirmed by western blot (Fig. 2A and data not shown). In cells expressing Rnd3, RhoA knockdown and, to a lesser extent, RhoC knockdown further reduced the already low levels of actin stress fibers (Fig. 2B, left panel). No apparent differences in actin organization were seen in RhoB knockdown cells. In Rnd3 depleted cells, enhanced actin stress fiber formation was antagonized by RhoA knockdown (Fig. 2B right panel and 2C). Similar experiments using siRNAs targeting RhoB or RhoC did not produce any reversion of the Rnd3 knockdown phenotype. Rnd3 and RhoA have been implicated in regulating the activity of ROCK1. In nonmuscle cell types, ROCK1/2 phosphorylates myosin light chain (MLC) to promote the generation of cellular tension and actin stress fibers. Rnd3 depletion increased MLC phosphorylation on Thr18/Ser19 (Fig. 2D) which localized to actin stress fibers (Fig. 2D). Inhibition of ROCK1/2, using Y27632, reduced the Rnd3 knockdown increase in actin stress fibers and associated MLC phosphorylation (Fig. 2D). Together these data suggest that Rnd3 expression in invasive melanoma suppresses the RhoA-ROCK1/2 pathway to promote actin cytoskeletal reorganization.
Figure 2.
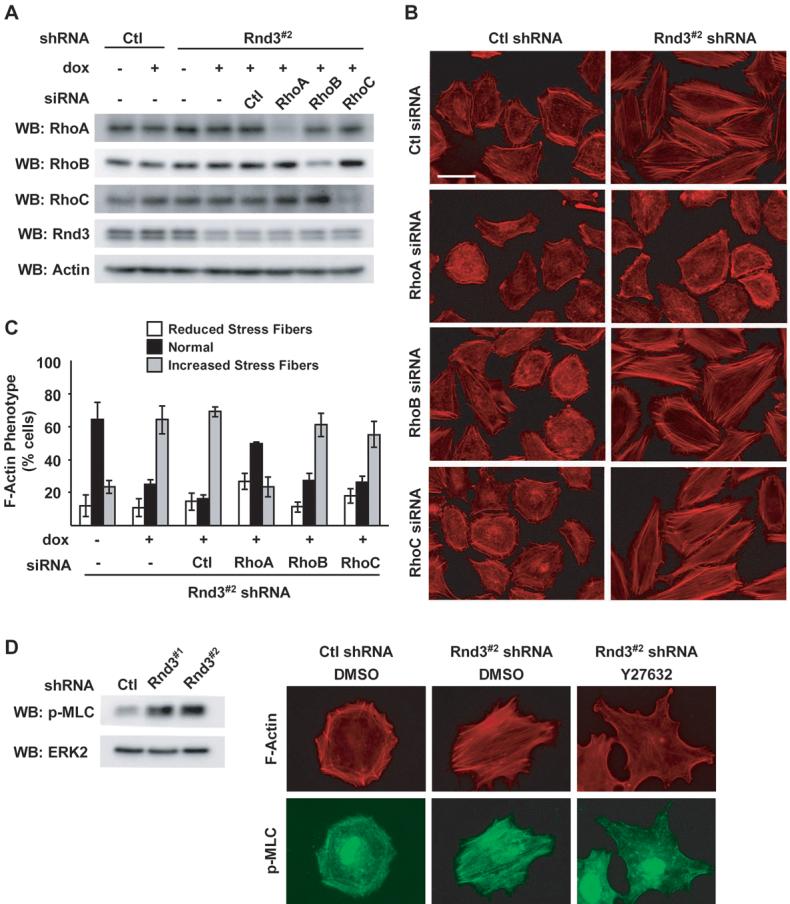
RhoA-ROCK signaling is required for Rnd3-dependent differences in actin cytoskeletal organization. A-C, Inducible WM793/TR/Rnd3#2 shRNA cells ± dox were transfected with siRNA duplexes targeting RhoA, RhoB or RhoC. A, Cell extracts subjected to western blot analysis using antibodies specific for RhoA, RhoB, RhoC, Rnd3 or β-actin. B, Cells fixed and processed to visualize F-actin organization. Bar, 50 μm. C, Quantitation of the F-actin phenotype shown in (B) (* P<0.05). D, Rnd3#2 and Ctl shRNA cells incubated with dox for 72 hours. Cells were lysed and analyzed by western blot for phospho-MLC and ERK2 or treated an additional two hours with 5 μM Y27632 or equal volume DMSO. Cells were then fixed and processed for indirect immunofluorescence using phospho-MLC (green) and TRITC-phalloidin to visualize F-actin (red). Bar, 50 μm.
Rnd3 knockdown does not alter melanoma cell cycle
In addition to F-actin organization, Rnd3 has been implicated in cell proliferation (40) and survival (32). Therefore, we determined if Rnd3 expression regulates cell cycle progression in WM793 cells. Initially, DNA synthesis was monitored by flow cytometric evaluation of 5-ethynyl-2′-deoxyuridine (EdU) incorporation (41). EdU incorporation was unaffected by Rnd3 knockdown (Figure 3A). The overall cell cycle profile of inducible knockdown cells was then measured by propidium iodide staining and under standard growth conditions. The DNA profile of Rnd3 knockdown cells was found to be similar to control cells (Fig. 3B). Analysis of signaling pathways linked to melanoma cell proliferation showed that ERK1/2 and AKT phosphorylation were similar despite Rnd3 knockdown (Supplemental Fig. S4A and S4B). The lack of effect on the cell cycle is supported by the finding that cyclin D1 expression was not decreased in Rnd3-silenced cells (Supplemental Fig. S4C). Moreover, no alteration in cell growth accompanied Rnd3 knockdown (Fig. 3C). These data indicate that Rnd3 knockdown does not alter proliferation or survival properties in melanoma cells.
Figure 3.
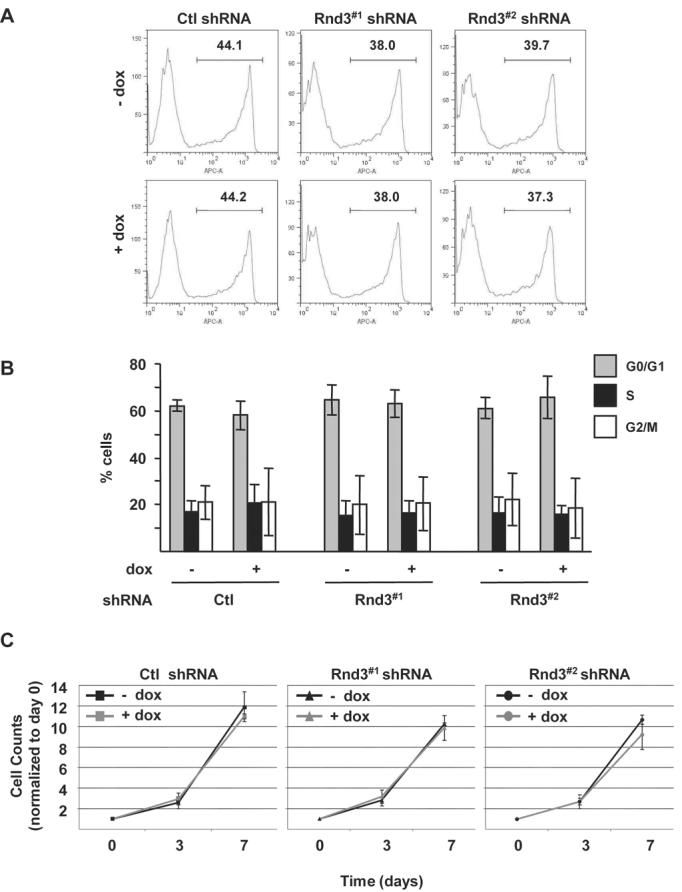
Rnd3 expression does not alter cell growth of mutant B-RAF expressing invasive melanoma cells. A, Quantitation of cell entry into S-phase. Inducible knockdown cells were labeled for 7 hours with EdU. DNA synthesis was analyzed by flow cytometry. Depicted are representative traces from one of three independent experiments. B, Melanoma cells cultured in complete medium 72 hours ± 0.1 μg/ml dox to induce expression of Ctl, Rnd3#1 or Rnd3#2 shRNA. DNA was stained with propidium iodide and analyzed by flow cytometry. The graph represents the percentage of cells in G0/G1, S, and G2/M phases of the cell cycle. Shown are means ± SD from three independent experiments. C, Cell growth of inducible knockdown cells cultured in complete growth medium ± dox. Equal numbers of cells plated at day 0 ± dox were harvested and counted at days 3 and 7. Shown are means ± SD from one experiment performed in triplicate. A total of three independent experiments were performed.
Rnd3 expression correlates with invasive properties
The functional relationship between Rnd3 expression and invasive properties of melanoma cells has not been addressed. Therefore, we next evaluated the expression of Rnd3 in normal human epidermal melanocytes (NHEM), as well as, non-invasive (SBcl2 and WM35) and invasive (WM115 and WM793) human melanoma cell lines. NHEMs harbor wild-type N-RAS and B-RAF; Sbcl2 express active N-RAS; WM35, WM793 and WM115 express mutant forms of B-RAF. Notably, only the invasive cell lines showed elevated ERK1/2 phosphorylation (Fig. 4A). These findings are consistent with previous reports demonstrating the presence of negative feedback loops in non-invasive melanoma cells to suppress ERK activation (6, 7). Rnd3 expression levels were also elevated in the invasive WM793 and WM115 cell lines (Fig. 4A).
Figure 4.
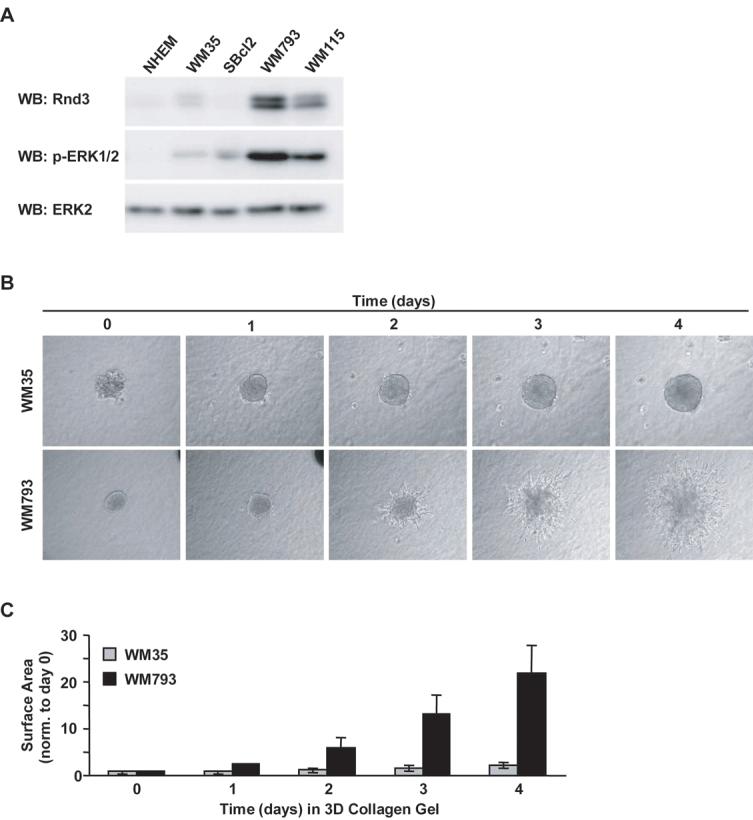
Increased Rnd3 expression in invasive human melanoma cells. A, Western blot of normal human epidermal melanocyte (NHEM), noninvasive (SBcl2 and WM35) and invasive (WM793 and WM115) human melanoma cell lines for Rnd3, phospho-ERK1/2 and ERK2. B, Micrographs depicting time-course of non-invasive (WM35) and invasive (WM793) melanoma spheroids embedded in 3-D collagen gel for the indicated times. C, Quantitation of invasive outgrowth depicted in (B). The graph reflects total spheroid surface area measured from the same spheroids monitored over time and normalized to its size at day 0. Spheroid areas at day 0 were set to 1.
Following invasion through the basement membrane, melanoma tumors encounter the type I collagen-rich microenvironment of the dermis. An assay used to test for invasive behavior in a dermal-like microenvironment is the non-fibrillar 3-D collagen spheroid outgrowth assay (37, 42). Melanoma spheroids formed from both WM35 (low Rnd3 expression) and WM793 (high Rnd3 expression) cells increased size over 4 days (Fig. 4B), again suggesting that Rnd3 is not required for melanoma growth. However, WM793 and WM115 spheroids exhibited cells that progressively infiltrated into the surrounding collagen gel (Fig. 4B and data not shown). In contrast, WM35 spheroids did not display individual cells that invaded into the collagen gel (Fig. 4B). SBcl2 cells did not form compact spheroids (data not shown). Quantitation of overall invasive outgrowth after four days indicated that total surface area of WM793 spheroids increased approximately 20-fold. By contrast, WM35 spheroids increased their area of outgrowth only approximately 2-fold (Fig. 4C). Collectively, these data show that expression of Rnd3 correlates with melanoma invasive behavior.
Rnd3 knockdown disrupts melanoma invasive outgrowth
The correlation between Rnd3 expression and melanoma invasive behavior led us to examine if Rnd3 was required for invasive activity. As shown in Fig. 5A, spheroids cultured in the absence of dox or following the induction of control shRNA displayed invasive outgrowth behavior similar to parental cells. In contrast, Rnd3 knockdown spheroids showed restricted invasive movement of cells away from the spheroid edge while the area of the core spheroid remained similar to control knockdown spheroids (Fig. 5A and 5B). Notably, spheroid outgrowth area decreased approximately 10-fold in Rnd3 knockdown conditions (Fig. 5B). The spheroids were predominately composed of live cells with no noticeable accumulation of dead cells (Fig. 5C). These data demonstrate a requirement for Rnd3 in the invasive outgrowth behavior of melanoma cells.
Figure 5.
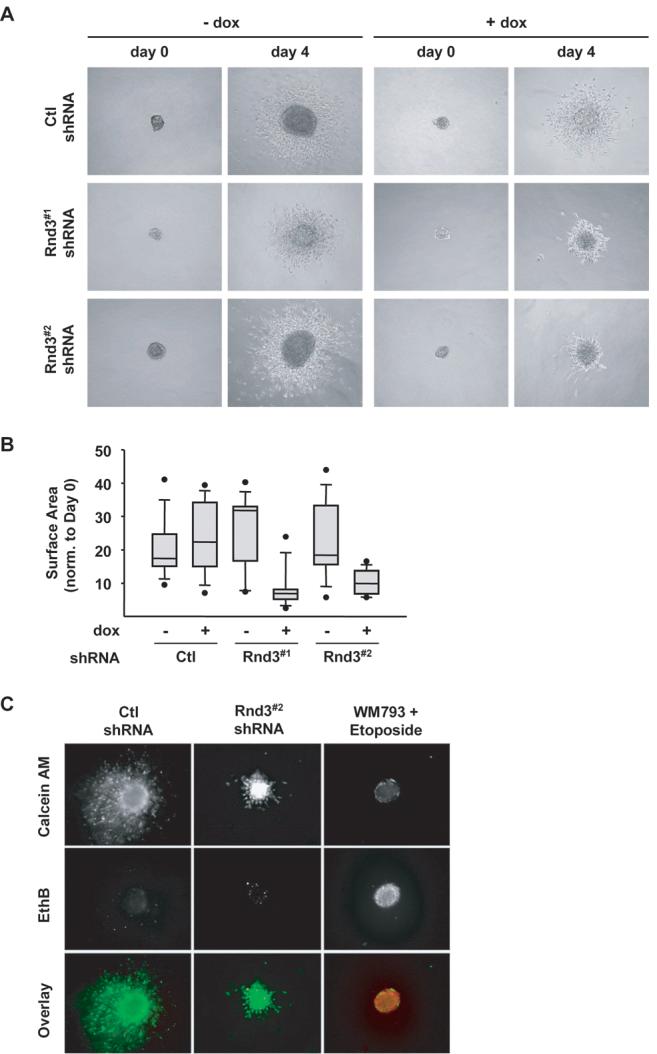
Melanoma invasive outgrowth is inhibited following Rnd3 knockdown. Spheroids formed from Ctl, Rnd3#1 and Rnd3#2 shRNA WM793TR cells embedded in a 3-D collagen gel for 4 days. A, Phase contrast pictures displaying spheroid morphology at day 0 and day 4 in ± dox. B, Quantitation of spheroid outgrowth shown in (A). Spheroid surface area measured at day 4 normalized to its area at day 0. C, Cell viability within spheroids after 4 days embedded in a collagen gel, live cells stain green (calcein-AM) and dead cells stain red (ethidium bromide).
Rnd3 regulates melanoma cell motility on 3-D matrices
The reduced invasive capacity accompanying Rnd3 depletion could result from defects in cell migration and/or extracellular matrix remodeling. Since Rnd3 knockdown affected actin cytoskeletal organization, which is known to impact cell motility, we next determined whether Rnd3 depletion altered cell migration by video microscopy. Initially, we evaluated Rnd3 depletion on the migration of cells at the edge of multi-cellular spheroids overlaid onto 3-D collagen gels (Fig. 6A). Control spheroids migrated outward as a collective cell unit, increasing spheroid surface area approximately 3-fold after five hours (Fig. 6A). In contrast, the movement of cells out from Rnd3-depleted spheroids was attenuated (Fig. 6A). Overall, collective cell migration was reduced by an average of 45% in Rnd3#1 and 60% in Rnd3#2 silenced cells compared to non-dox treated cells (Fig. 6B). Additionally, the number of individual border cells migrating away from the expanding collective cell sheet exiting the spheroid over the five hour time period was consistently reduced in the absence of Rnd3. Quantitation of the number of migrating border cells revealed a 58% reduction in Rnd3#1 shRNA expressing cells and a significant 67% decrease in Rnd3#2 shRNA expressing cells (Fig. 6C). Knockdown cells displayed no differences in cell surface expression of collagen binding integrins or cell adhesion to collagen gels (Supplemental Fig. S3A and S3B). Results from F-actin organization indicated that Rnd3 knockdown-induced changes in cytoskeletal organization could be reversed upon ROCK1/2 inhibition. Therefore, Rnd3 depleted cells were treated with Y27632 to determine if ROCK1/2 inhibition would prevent the reduction in cell migration on collagen gels. Pretreatment of Rnd3 knockdown spheroids with Y27632 one hour prior to plating on top collagen gel rescued collective cell migration after five hours (Fig. 6D). Collectively, these data highlight the function of endogenous Rnd3 to inhibit the Rho-ROCK signaling pathway to promote the migration of invasive melanoma cells on 3-D collagen matrices.
Figure 6.
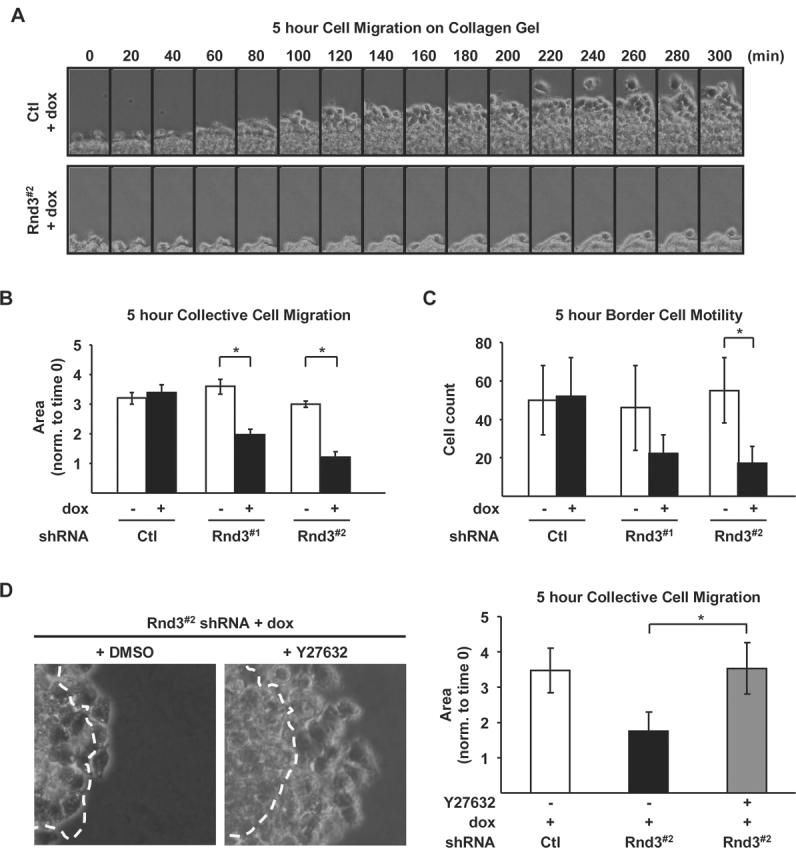
Rnd3 expression is required for directed melanoma cell migration on 3-D collagen gels. Spheroids from Ctl, Rnd3#1 and Rnd3#2 shRNA cells cultured in complete medium ± dox on top of a collagen gel. A, Micrographs of spheroids acquired from time lapse microscopy over a five hour imaging period. B-C, Quantitation of spheroid migration depicted in (A). B, Collective cell movement, measured as the increase in spheroid surface area over time (* P<0.05). C, The number of border cells, determined by counting individual cells separated from the cell sheet (* P<0.05). D, Collective cell movement of Ctl and Rnd3#2 shRNA cells treated ± 5 μM Y27632 one hour prior to plating on collagen gels. Images show an overlay of the spheroid surface area at time 0 (outlined by white line) superimposed onto its area five hours later. Graph depicting quantitation of the collective cell movement in spheroids treated ± Y27632 (* P<0.05).
Discussion
The frequent mutational activation of B-RAF in melanoma coupled with the growing number of studies demonstrating its significant role in melanoma initiation and progression has validated B-RAF as an important therapeutic target. Although clinical grade RAF inhibitors have been developed (43), their effective use to date has been diminished due to pharmacokinetic and specificity concerns (44). In addition, recent evidence suggests that increased C-RAF expression may provide for a mechanism of acquired drug resistance (45). Our studies have focused on delineating mutant B-RAF regulated targets involved in malignant traits. Complicating the analysis of these targets is the varying cellular contexts used. In this study, we analyzed the functional role of the B-RAF regulated GTPase Rnd3 in invasive melanoma cells. For the first time, our study addresses three critical issues pertaining to the role of Rnd3 in melanoma. Initially, we show that inducible knockdown of endogenous Rnd3 regulates actin cytoskeletal organization in a manner dependent on RhoA but not RhoB or RhoC. Next, we show that Rnd3 expression is elevated in melanoma cells that display invasive outgrowth from tumor spheroids. Finally, silencing of Rnd3 profoundly reduced the invasive component of spheroid outgrowth in 3-D collagen gels and effectively blocked collective and border cell movements on 3-D. Thus, the present work advances our knowledge of the signaling pathways influenced by Rnd3 and establishes its expression as a crucial regulator of invasive melanoma cytoskeletal organization and cell migration (Supplemental Fig. S5).
Oncogene-induced alterations in the actin cytoskeleton are observed in a variety of cancerous cell types (21, 46). Here, we show that depletion of endogenous Rnd3 promotes an increase in actin stress fibers, consistent with findings in osteosarcoma cells (32). Actin stress fiber formation results from activation of Rho GTPases and their subsequent effects on ROCK1/2 signaling (14, 26). Rnd3 was previously shown to inhibit Rho/ROCK signaling through its interaction with p190RhoGAP (47) and ROCK1 (30, 48). However, Rnd3 was recently shown to disassemble stress fibers independent of ROCK1 binding (49), which suggests that the Rnd3-p190RhoGAP interaction facilitates cytoskeletal disassembly. The RhoGAP domain of p190 RhoGAP can catalyze GTP hydrolysis of RhoA, RhoB and RhoC (50). Currently, there is limited information addressing a potential isoform-specific role(s) for Rnd3 regulation of RhoA/B/C. Moreover, RhoA/B/C can all mediate stress fiber formation (26) and all are expressed in melanoma. The current manuscript is the first to directly examine the contribution of RhoA/B/C isoforms to Rnd3-mediated cytoskeletal effects at the molecular level. Our results demonstrate that Rnd3 preferentially restricts RhoA signaling to regulate actin organization. This is consistent with a previous report in breast carcinoma demonstrating that the roles of RhoA/B/C are not redundant (17). While our studies in VGP melanoma cells implicate inhibition of RhoA signaling in the control of actin stress fibers and cell migration, we cannot rule out a requirement for additional Rho isoforms during later stages of melanoma progression.
In non-melanoma cell types, Rnd3 expression has been linked to either increased or decreased cell proliferation (29, 40) and pro-survival effects (32). In invasive melanoma which express mutant B-RAF, we did not observe changes in the cell cycle profile or growth following Rnd3 knockdown. Furthermore, the levels of ERK1/2 and AKT phosphorylation that regulate melanoma proliferation and survival, as well as cyclin D1 expression were similar between the control and Rnd3 depleted melanoma cell lines. Thus, it seems that the relationship between Rnd3 expression and cell proliferation varies between different types of cells. One explanation for these discrepancies may be that mutant B-RAF signaling in melanoma overrides any change in cell growth imposed by Rnd3.
The seemingly contradictory studies on Rnd3 expression in cancer (19, 22, 23) as well as our previous results showing induction of Rnd3 by mutant B-RAF (21), prompted us to examine the expression of Rnd3 in cells characteristic of non-invasive and invasive stages of melanoma. Data presented here show that increased Rnd3 expression correlated with the transition from non-invasive to invasive melanoma. Interestingly SBcl2 and WM35 cells, which harbor mutant N-RAS and mutant B-RAF, respectively, displayed low Rnd3 expression. This is likely due to the reduced phosphorylation of active ERK1/2 in SBcl2 and WM35. Differences in ERK1/2 activation in melanoma cells which harbor mutant B-RAF may be due to amplification of mutant B-RAF alleles in invasive cells or the presence of a negative feedback loop in non-invasive cells (6, 7). The mechanism of ERK1/2 regulation of Rnd3 in melanoma is largely unknown. Rnd3 has recently been reported to be a p53 transcriptional target gene (32); however, knockdown of wild-type p53 in WM793 cells did not disrupt Rnd3 expression (Supplemental Fig. S6). Now that a role for Rnd3 in invasive melanoma has been established, additional studies are warranted to identify the mechanism(s) responsible for ERK1/2 control of Rnd3 expression. In sum, it appears that Rnd3 expression in melanoma is associated with elevated B-RAF/MEK/ERK signaling, as well as a pro-invasive phenotype.
VGP melanoma is distinguished from RGP melanoma by its invasive movement into the dermis (1). In a 3-D model system, we demonstrate that knockdown of endogenous Rnd3 dramatically attenuates the ability of spheroid cells to invade out into the surrounding collagen gel. Notably, the spheroid core increased in size and was composed of live cells, suggesting that overall growth was maintained. Analysis of spheroids placed on top of collagen gels revealed that migration of collective cell layers, as well as individual border cells was reduced in Rnd3-depleted spheroids. Although a precise mechanism for reduced migration in 3-D matrices is currently under investigation, the results from 2-D assays implicate deregulated RhoA signaling. This would be consistent with current reports implicating Rho/ROCK signaling in the regulation of tissue compliance and tumor cell migration (10, 13, 36). Future studies will examine whether Rnd3 regulation of RhoA facilitates melanoma migration in 3-D matrices through its control of focal adhesion dynamics, extracellular matrix protease activity and/or cellular tension.
In conclusion, the current data advance our knowledge of Rnd3 and Rho GTPases in melanoma. Increased expression of Rnd3 attenuates RhoA-ROCK signaling in invasive melanoma to regulate 3-D migration. Our results potentially serve as the framework for new therapeutic strategies that target oncogene-regulated effector pathways to reduce invasive behavior and restrict malignancy in human cancer.
Supplementary Material
Acknowledgements
Grant Support: American Cancer Society (PF-08-032-01-CSM; to R.M. Klein), American Cancer Society (RSG-08-033-01-CSM; to A.E. Aplin), and National Institutes of Health (R01-GM067893; to A.E. Aplin).
We thank Drs. C. Michael DiPersio, Peter Vincent and Jihe Zhao for critically reviewing this manuscript and Dr. G. Liu for technical assistance.
Footnotes
Disclosure of Potential Conflicts of Interest No potential conflicts of interest were disclosed.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
References
- 1.Miller AJ, Mihm MC., Jr Melanoma. N Engl J Med. 2006;355:51–65. doi: 10.1056/NEJMra052166. [DOI] [PubMed] [Google Scholar]
- 2.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 3.DeLuca AM, Srinivas A, Alani RM. BRAF kinase in melanoma development and progression. Expert Rev Mol Med. 2008;10:e6. doi: 10.1017/S1462399408000604. [DOI] [PubMed] [Google Scholar]
- 4.Sumimoto H, Miyagishi M, Miyoshi H, et al. Inhibition of growth and invasive ability of melanoma by inactivation of mutated BRAF with lentivirus-mediated RNA interference. Oncogene. 2004;23:6031–9. doi: 10.1038/sj.onc.1207812. [DOI] [PubMed] [Google Scholar]
- 5.Tsai J, Lee JT, Wang W, et al. From the Cover: Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci. 2008;105:3041–6. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park S, Yeung ML, Beach S, Shields JM, Yeung KC. RKIP downregulates B-Raf kinase activity in melanoma cancer cells. Oncogene. 2005;24:3535–40. doi: 10.1038/sj.onc.1208435. [DOI] [PubMed] [Google Scholar]
- 7.Schuierer MM, Bataille F, Hagan S, Kolch W, Bosserhoff A-K. Reduction in Raf kinase inhibitor protein expression is associated with increased Ras-extracellular signal-regulated kinase signaling in melanoma cell lines. Cancer Res. 2004;64:5186–92. doi: 10.1158/0008-5472.CAN-03-3861. [DOI] [PubMed] [Google Scholar]
- 8.Blackburn JS, Rhodes CH, Coon CI, Brinckerhoff CE. RNA interference inhibition of matrix metalloproteinase-1 prevents melanoma metastasis by reducing tumor collagenase activity and angiogenesis. Cancer Res. 2007;67:10849–58. doi: 10.1158/0008-5472.CAN-07-1791. [DOI] [PubMed] [Google Scholar]
- 9.Huntington JT, Shields JM, Der CJ, et al. Overexpression of collagenase 1 (MMP-1) is mediated by the ERK pathway in invasive melanoma cells: role of BRAF mutation and fibroblast growth factor signaling. J Biol Chem. 2004;279:33168–76. doi: 10.1074/jbc.M405102200. [DOI] [PubMed] [Google Scholar]
- 10.Sahai E, Marshall CJ. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol. 2003;5:711–9. doi: 10.1038/ncb1019. [DOI] [PubMed] [Google Scholar]
- 11.Hoek KS, Eichhoff OM, Schlegel NC, et al. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008;68:650–6. doi: 10.1158/0008-5472.CAN-07-2491. [DOI] [PubMed] [Google Scholar]
- 12.Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3:362–74. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- 13.Paszek MJ, Zahir N, Johnson KR, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–54. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 14.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–14. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 15.Ridley AJ. Rho GTPases and cell migration. J Cell Sci. 2001;114:2713–22. doi: 10.1242/jcs.114.15.2713. [DOI] [PubMed] [Google Scholar]
- 16.Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406:532–5. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]
- 17.Bellovin DI, Simpson KJ, Danilov T, et al. Reciprocal regulation of RhoA and RhoC characterizes the EMT and identifies RhoC as a prognostic marker of colon carcinoma. Oncogene. 2006;25:6959–67. doi: 10.1038/sj.onc.1209682. [DOI] [PubMed] [Google Scholar]
- 18.Zhang C, Zhou F, Li N, et al. Overexpression of RhoE has a prognostic value in non-small cell lung cancer. Ann Surg Oncol. 2007;14:2628–35. doi: 10.1245/s10434-007-9457-x. [DOI] [PubMed] [Google Scholar]
- 19.Gress TM, Muller-Pillasch F, Geng M, et al. A pancreatic cancer-specific expression profile. Oncogene. 1996;13:1819–30. [PubMed] [Google Scholar]
- 20.Shields JM, Thomas NE, Cregger M, et al. Lack of extracellular signal-regulated kinase mitogen-activated protein kinase signaling shows a new type of melanoma. Cancer Res. 2007;67:1502–12. doi: 10.1158/0008-5472.CAN-06-3311. [DOI] [PubMed] [Google Scholar]
- 21.Klein RM, Spofford LS, Abel EV, Ortiz A, Aplin AE. B-RAF regulation of Rnd3 participates in actin cytoskeletal and focal adhesion organization. Mol Biol Cell. 2008;19:498–508. doi: 10.1091/mbc.E07-09-0895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cuiyan Z, Jie H, Fang Z, et al. Overexpression of RhoE in non-small cell lung cancer (NSCLC) is associated with smoking and correlates with DNA copy number changes. Cancer Biol Ther. 2007;6:335–42. doi: 10.4161/cbt.6.3.3663. [DOI] [PubMed] [Google Scholar]
- 23.Bektic J, Pfeil K, Berger AP, et al. Small G-protein RhoE is underexpressed in prostate cancer and induces cell cycle arrest and apoptosis. Prostate. 2005;64:332–40. doi: 10.1002/pros.20243. [DOI] [PubMed] [Google Scholar]
- 24.Guasch RM, Scambler P, Jones GE, Ridley AJ. RhoE regulates actin cytoskeleton organization and cell migration. Mol Cell Biol. 1998;18:4761–71. doi: 10.1128/mcb.18.8.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nobes CD, Lauritzen I, Mattei MG, Paris S, Hall A, Chardin P. A new member of the Rho family, Rnd1, promotes disassembly of actin filament structures and loss of cell adhesion. J Cell Biol. 1998;141:187–97. doi: 10.1083/jcb.141.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aspenstrom P, Fransson A, Saras J. Rho GTPases have diverse effects on the organization of the actin filament system. Biochem J. 2004;377:327–37. doi: 10.1042/BJ20031041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chardin P. Function and regulation of Rnd proteins. Nat Rev Mol Cell Biol. 2006;7:54–62. doi: 10.1038/nrm1788. [DOI] [PubMed] [Google Scholar]
- 28.Hansen SH, Zegers MMP, Woodrow M, et al. Induced expression of Rnd3 is associated with transformation of polarized epithelial cells by the Raf-MEK-Extracellular signal-regulated kinase pathway. Mol Cell Biol. 2000;20:9364–75. doi: 10.1128/mcb.20.24.9364-9375.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Villalonga P, Guasch RM, Riento K, Ridley AJ. RhoE inhibits cell cycle progression and Ras-induced transformation. Mol Cell Biol. 2004;24:7829–40. doi: 10.1128/MCB.24.18.7829-7840.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Riento K, Totty N, Villalonga P, Garg R, Guasch R, Ridley AJ. RhoE function is regulated by ROCK I-mediated phosphorylation. EMBO J. 2005;24:1170–80. doi: 10.1038/sj.emboj.7600612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gadea G, de Toledo M, Anguille C, Roux P. Loss of p53 promotes RhoA-ROCK-dependent cell migration and invasion in 3D matrices. J Cell Biol. 2007;178:23–30. doi: 10.1083/jcb.200701120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ongusaha PP, Kim H-G, Boswell SA, et al. RhoE Is a pro-survival p53 target gene that inhibits ROCK I-mediated apoptosis in response to genotoxic stress. Cur Biol. 2006;16:2466–72. doi: 10.1016/j.cub.2006.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Fortier M, Comunale F, Kucharczak J, Blangy A, Charrasse S, Gauthier-Rouviere C. RhoE controls myoblast alignment prior fusion through RhoA and ROCK. Cell Death Differ. 2008 doi: 10.1038/cdd.2008.34. [DOI] [PubMed] [Google Scholar]
- 34.Clements RT, Minnear FL, Singer HA, Keller RS, Vincent PA. RhoA and Rho-kinase dependent and independent signals mediate TGF-{beta}-induced pulmonary endothelial cytoskeletal reorganization and permeability. Am J Physiol Lung Cell Mol Physiol. 2005;288:L294–306. doi: 10.1152/ajplung.00213.2004. [DOI] [PubMed] [Google Scholar]
- 35.Boisvert-Adamo K, Aplin AE. Mutant B-RAF mediates resistance to anoikis via Bad and Bim. Oncogene. 2008;27:3301–12. doi: 10.1038/sj.onc.1211003. [DOI] [PubMed] [Google Scholar]
- 36.Pinner S, Sahai E. PDK1 regulates cancer cell motility by antagonising inhibition of ROCK1 by RhoE. Nat Cell Biol. 2008;10:127–37. doi: 10.1038/ncb1675. [DOI] [PubMed] [Google Scholar]
- 37.Smalley KSM, Haass NK, Brafford PA, Lioni M, Flaherty KT, Herlyn M. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol Cancer Ther. 2006;5:1136–44. doi: 10.1158/1535-7163.MCT-06-0084. [DOI] [PubMed] [Google Scholar]
- 38.Satyamoorthy K, Li G, Gerrero MR, et al. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res. 2003;63:756–9. [PubMed] [Google Scholar]
- 39.Zamir E, Geiger B. Molecular complexity and dynamics of cell-matrix adhesions. J Cell Sci. 2001;114:3583–90. doi: 10.1242/jcs.114.20.3583. [DOI] [PubMed] [Google Scholar]
- 40.Poch E, Minambres R, Mocholi E, et al. RhoE interferes with Rb inactivation and regulates the proliferation and survival of the U87 human glioblastoma cell line. Exp Cell Res. 2007;313:719–31. doi: 10.1016/j.yexcr.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 41.Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci. 2008;105:2415–20. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haass NK, Sproesser K, Nguyen TK, et al. The mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in melanoma cells and tumor regression when combined with Docetaxel. Clin Cancer Res. 2008;14:230–9. doi: 10.1158/1078-0432.CCR-07-1440. [DOI] [PubMed] [Google Scholar]
- 43.Fecher LA, Amaravadi RK, Flaherty KT. The MAPK pathway in melanoma. Curr Opin Oncol. 2008;20:183–9. doi: 10.1097/CCO.0b013e3282f5271c. [DOI] [PubMed] [Google Scholar]
- 44.Li N, Batt D, Warmuth M. B-Raf kinase inhibitors for cancer treatment. Curr Opin Investig Drugs. 2007;8:452–6. [PubMed] [Google Scholar]
- 45.Montagut C, Sharma SV, Shioda T, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68:4853–61. doi: 10.1158/0008-5472.CAN-07-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vial E, Sahai E, Marshall CJ. ERK-MAPK signaling coordinately regulates activity of Rac1 and RhoA for tumor cell motility. Cancer Cell. 2003;4:67–79. doi: 10.1016/s1535-6108(03)00162-4. [DOI] [PubMed] [Google Scholar]
- 47.Wennerberg K, Forget M-A, Ellerbroek SM, et al. Rnd proteins function as RhoA antagonists by activating p190 RhoGAP. Curr Biol. 2003;13:1106–15. doi: 10.1016/s0960-9822(03)00418-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Riento K, Guasch RM, Garg R, Jin B, Ridley AJ. RhoE binds to ROCK I and inhibits downstream signaling. Mol Cell Biol. 2003;23:4219–29. doi: 10.1128/MCB.23.12.4219-4229.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Komander D, Garg R, Wan PT, Ridley AJ, Barford D. Mechanism of multi-site phosphorylation from a ROCK-I:RhoE complex structure. EMBO J. 2008 doi: 10.1038/emboj.2008.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang L, Yang L, Luo Y, Zheng Y. A novel strategy for specifically down-regulating individual Rho GTPase activity in tumor cells. J Biol Chem. 2003;278:44617–25. doi: 10.1074/jbc.M308929200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.