

Abstract
Constitutional aneuploidies are rare syndromes associated with multiple developmental abnormalities and the alterations in the risk for specific cancers. Acquired somatic chromosomal aneuploidies are the most common genetic aberrations in sporadic cancers. Thus studies of these rare constitutional aneuploidy syndromes are important not only for patient counseling and clinical management, but also for deciphering the mechanisms by which chromosomal aneuploidy affect cancer initiation and progression. Here we review the major constitutional aneuploidy syndromes and suggest some general mechanisms for the associated cancer predisposition.
INTRODUCTION
Somatic acquired genomic instability is one of the hallmarks of cancer (1). This genomic instability is commonly manifested by structural or numerical chromosomal aberrations. Structural genomic aberrations leading to activation of oncogenes or elimination of tumor suppression genes have been studied extensively. However, very little is known about the oncogenic role, if any, of numerical chromosomal aberrations, aneuploidy, which are the most common abnormalities in cancer (2).
The association between constitutional aneuploidy and cancer supports a causative role of aneuploidy in cancer. The precise determination of the relative risk of cancer in many of these syndromes is hampered by their rarity and by the short life span of many of the patients. Notwithstanding these difficulties these syndromes provide a unique opportunity to study the neoplastic evolution during ontogeny and the interplay between the developmental abnormalities induced by specific chromosomal. Here we review the major disorders; Table 1 presents a summary and description of additional syndromes.
Table 1.
Summary of major constitutional aneuploidy syndromes and associated cancers
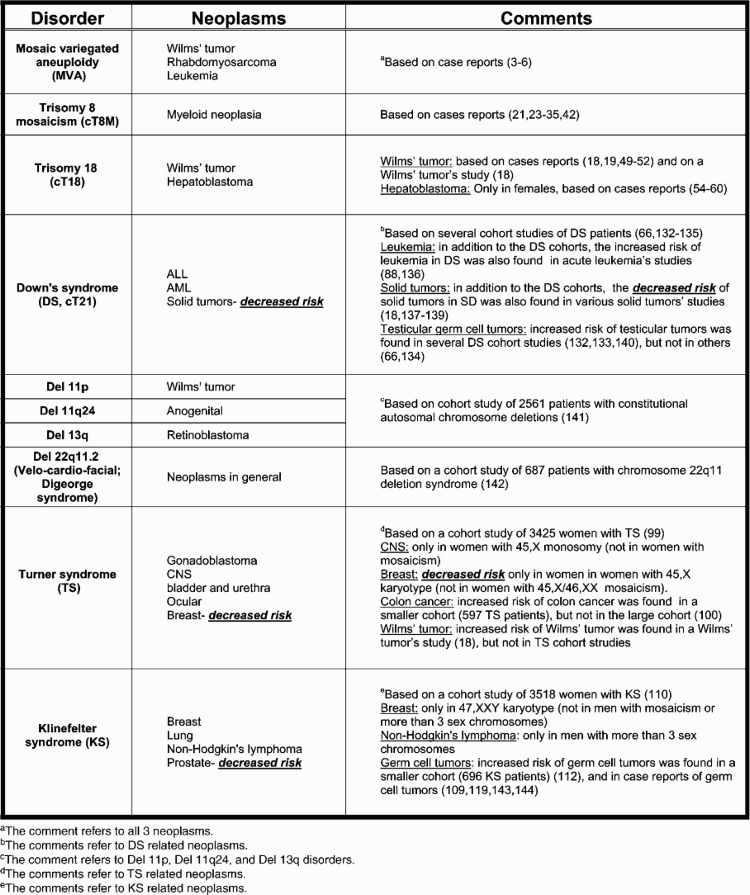 |
MOSAIC VARIEGATED ANEUPLOIDY
Unlike the rest of the constitutional aneuploidy disorders, mosaic variegated aneuploidy (MVA) is a rare autosomal recessive genomic instability syndrome characterized by multiple mosaic aneuploidies in somatic cells. Clinical manifestations include cancer predisposition, intrauterine growth retardation, microcephaly, mental retardation and CNS anomalies. Out of 35 cases of MVA syndrome that have been so far reported (3–6), 12 developed cancer: 7 had Wilms’ tumor; 4 had rhabdomyosarcoma (one patient had both tumors (7)) and 2 had leukemias.
More than half of the patients have inactivating mutations in the BUB1B gene that encodes the mitotic spindle checkpoint protein BUBR1 (4). Mitotic spindle checkpoint proteins (MAD1, MAD2, BUB1, BUB3, BUBR1, MPS1 and other proteins) arrest the metaphase by inhibiting the anaphase-promoting complex/cyclosome until the sister chromatids are correctly attached to the spindle (8,9). This is a central regulatory mechanism ensuring the maintenance of correct chromosome numbers after cell division. Not all patients with MVA have BUB1B mutations and these were reported to a have a milder phenotype without cancer (10). Bub1b+/– mice are developmentally normal but have defective spindle-checkpoint activation and develop lung and colon cancers in response to carcinogens (11). Together, the identification of BUB1B mutations as the cause of MVA and the mouse model provide a clear biological causal link between mitotic spindle dysfunction, aneuploidy and cancer development.
What is the extent of similarity between the cancers observed in patients with MVA and sporadic childhood cancers? Hanks et al. (12) reported comparative genomic hybridization analysis in an embryonal rhabdomyosarcoma (RMS) from an MVA case revealing aneuploidies typical of sporadic embryonal RMS, with gain of chromosomes 3, 8, 13 and loss of chromosomes 9, 14, X (13–15). While somatic BUB1B mutations are rare in sporadic childhood malignancies (12), it is possible that the genetic progression in RMS from MVA and non-MVA cases may be similar. Strikingly, however, the pattern of cancers in patients with MVA differs from sporadic childhood cancers. MVA seems to be specifically associated with the relatively rare embryonic myogenic and renal cancers and not with the most common sporadic childhood cancers. Abnormal mitotic checkpoint was described in sporadic neuroblastoma (16) but this tumor was not reported in patients with MVA. The specific association with RMS and Wilms’ tumors may be related to frequent predominance of trisomy 8 and trisomy 18 in MVA (17), two trisomies that are associated with RMS and Wilms’ tumors, respectively (18–20). Alternatively, the pattern of tumors in MVA possibly suggests a developmental role of BUBR1 or the mitotic checkpoint in the development of the embryonic kidney and muscle.
CONSTITUTIONAL ANEUPLOIDY OF THE AUTOSOMES AND CANCER
Trisomy 8
Full constitutional trisomy 8 (cT8) is very rare, whereas cT8 mosaicism (cT8M) is more frequent—the prevalence was estimated to be 1:25 000 (21). The full condition presents with physical stigmata, skeletal abnormalities and a mild-to-moderate mental retardation (22).
Trisomy 8 can be seen as a mosaic in the blood or in the skin or both (21). At least 23 cT8M patients were reported to have neoplasms: 18 myeloid malignancies (21,23–35) and five solid tumors (20,36–39). Since cT8M prevalence is very low, this probably represents an increased risk for myeloid neoplasms.
Acquired trisomy 8, which is restricted to the malignant cells, is the most common numeric aberration in AML and MDS (40,41). Interestingly, it is also the most common genetic aberration in the myeloid leukemias of Down’s syndrome (DS). Maserati et al. (42) suggested that some of the myeloid malignancies with ‘acquired’ trisomy 8 are actually undiagnosed cT8M—they have found 3 such cases out of 14 patients with myeloid malignancies and trisomy 8.
The association of both somatic and constitutional trisomy 8 with myeloid malignancies suggests a cell autonomous leukemogenic role for this trisomy, although the specific oncogenes are presently unknown (43). Interestingly, analysis of bone marrow from a patient with cT8M revealed increased activity of bone marrow stromal cells in supporting hematopoietic progenitor cell expansion (31). In addition, characterization of trisomy 8-positive NK cells from a cT8M patient showed an immunosenescent phenotype that may contribute to the escape and expansion of neoplastic cells as a result of altered immunosurveillance (44). Hence the micro- and macro-environment in patients with cT8M may also play an important role in the pathogenesis of MDS.
Trisomy 18
Constitutional trisomy 18 (Edwards syndrome, cT18) is the most common autosomal abnormality after trisomy 21, occurring in 1:3000 live births (45,46). Renal abnormalities, particularly horseshoe kidney, are common (47). The chances of survival over 1 year are only 10% (48). Therefore, most patients with cT18 do not survive long enough to develop cancer.
There have been nine reports of Wilms’ tumor in individuals with cT18, seven of them in females (18,19,49–52). cT18 was also found to occur at higher rates than expected based on chromosome surveys of 5854 newborns with Wilms’ tumor (18). Hepatoblastoma, another rare childhood cancer (annual incidence of 1 per million children (53)), was reported in seven patients with cT18, all females (54–60). Thus it appears that there is an increased rate of Wilms’ tumor and hepatoblastoma in female patients with cT18, especially for long-term survivors.
Hepatoblastoma and Wilms’ tumors are embryonal tumors of the liver and kidney, respectively. Acquired trisomy 18 is frequently found in sporadic Wilms’ tumors but is rare in hepatoblastoma (http://cgap.nci.nih.gov/Chromosomes/RecurrentAberrations). Traditionally the hallmark of cancer predisposition syndromes is an earlier age of cancer occurrence. However, the age of diagnosis of Wilms’ tumor with cT18 (7 years) is double than sporadic Wilms’ tumor. This and the specific association with the female gender argue for a complex pathogenesis of cancer in patients with cT18.
Trisomy 21—Down’s syndrome
DS is reviewed by Weissman et al. in this issue. The reader is also referred to comprehensive recent reviews on the leukemias of DS (61–64).
The risk of most solid tumors is reduced in DS (65,66). A mouse model suggested that this is due to a dose-dependent tumor suppressive effect of the Hsa21 transcription factor Ets2 (67). In contrast, children (not adults) with DS have a X600 increased risk for acute myeloid (ML-DS) and X20 increased risk for B cell precursor lymphoblastic leukemias (ALL) compared with normal children (65).
The myeloid leukemias of DS (ML-DS)
These are unique to DS (68). Approximately 5% of children with DS are born with a transient clonal megakaryocytosis syndrome, named ‘transient myeloproliferative disorder’ (TMD) (69), which usually resolves spontaneously. Approximately 20% of DS patients with TMD will develop, however, a full-blown malignant acute myeloid leukemia with megakaryoblastic phenotype (AMKL) by the age of 4 years, which will not regress without chemotherapy (70). Both TMD and DS-AMKL are characterized by a mutation in the chromosome X transcription factor GATA1 that occurs in utero and invariably results in the expression of a shorter isoform GATA1s (71–73). GATA1 regulates normal development of the erythroid, megakaryocytic and basophilic/mast cell lineages. A ‘knock-in’ experiment in mice and gene expression analysis of DS-AMKL suggest that GATA1s enhances the proliferation and block the differentiation of immature embryonic megakaryoblasts (74,75).
The current model of multi-step myeloid leukemogenesis in DS (Fig. 1) suggests that trisomy 21 enhances the proliferation and self-renewal of fetal liver megakaryo-erythroid progenitors in a cell-autonomous manner (76,77). This ‘positive’ developmental pressure towards the megakaryo-erythrocytic lineage cooperates with the somatic mutation in GATA1 that further enhances the clonal proliferation of immature megakaryoblasts diagnosed at birth as TMD. GATA1 mutations are necessary but insufficient for the development of DS-AMKL. Additional genetic events such as activating mutations of JAK3 (78), or trisomy 8 (79) have been proposed to mediate the progression from TMD to DS-AMKL.
Figure 1.

Proposed model for the mechanism underlying DS predisposition to megakaryoblastic leukemias. Increased expression of chromosome Hsa21 genes in fetal liver hematopoietic stem cells leads to increased production of megakaryo-erythroid progenitors (MEPs). The somatically acquired GATA1s mutation further blocks megakaryoblastic differentiation and enhances MEPs proliferation. This can lead to transient myeloproliferative disorder (TMD) in 5% of DS newborns. Acquisition of additional somatic genetic or epigenetic events during infancy will lead to the development of full-blown megakaryoblastic leukemia (AMKL) in one-fifth of TMD patients.
There are several candidate ‘megakaryoblastic oncogenes’ on Hsa21 including RUNX1, ETS2, ERG and miRNA 125 (80–83). Trisomies result in modest elevated expression of multiple genes residing on the trisomic chromosome (84), although the expression of some genes may vary in a more drastic way (85). Thus it is possible that the tilt of normal fetal hematopoiesis towards the megakaryocytic lineage in DS results from the co-expression of several pro-megakaryopoiesis genes from the trisomic Hsa21.
This cooperation between several genes on the same chromosome may represent a general mechanism by which trisomies affect development and cancer.
Acute lymphoblastic leukemia of DS (DS-ALL)
This is similar to the ‘common’ B-cell precursor ALL affecting pre-school children. Indeed it is the most common leukemia in DS, comprising about 1–3% of total children with ALL (86–88). Acquired trisomy or tetrasomy 21 is the most common genetic aberration in sporadic ALL. Hence it is tempting to speculate that constitutional and somatic trisomy 21 may facilitate leukemogenesis in a similar fashion. Yet recent molecular and cytogenetic studies suggest that at least some of the DS-ALL have unique features.
Cytogenetically DS-ALL is characterized by reduced prevalence of the common aberrations found in childhood leukemia. Rather there are some unique features such as an additional chromosome X as single cytogenetic abnormality that suggest a yet unknown, collaborating event between genes on chromosomes 21 and X (79). More recently a mutation in the JAK2 kinase was identified in about one-fifth of the patients with DS-ALL (89–92). All mutant alleles centered around a highly conserved arginine residue (R683) within the JAK2 pseudokinase domain. The mutations immortalized primary mouse hematopoietic progenitors, and caused constitutive JAK/STAT activation and cytokine independent growth of Ba/F3 cells that was sensitive to pharmacological inhibition of JAK/STAT signaling. Mutations in JAK2 are characteristics of myeloproliferative neoplasms (MPNs). However, similar to the specificity of GATA1 mutations to DS myeloid malignancies, the novel mutations in R683 of JAK2 are unique to DS-ALL and are detected neither in MPNs nor in sporadic ALL. Modeling of JAK2 pseudokinase domain revealed that R683 is situated in an exposed conserved region separated from the one involved in MPNs. These observations not only provide the first molecular specific lesion of DS-ALL but also suggest that these leukemias are candidates for therapy with the novel JAK2 inhibitors. These findings also raise many interesting questions regarding the nature of this unique collaboration between constitutional (but not acquired) trisomy 21 and JAK2 R683 mutations.
Clinically, DS-AMKL is highly responsive to chemotherapy while the prognosis of DS-ALL is grimmer (93–95). This is in contrast to sporadic leukemias characterized by a better cure rate of ALL compared with AML. The heightened sensitivity of DS-AMKL blasts may be related to the reduced catabolism of the chemotherapeutic ARA-C caused by genes regulated by the mutated GATA1. In contrast, DS with ALL are highly sensitive to the toxic effects of Methotrexate, a drug that is not used in AML, due to the excess activity of the folate transporter coded by an Hsa21 gene (96). These examples demonstrate the pharmacogenomic effects of a trisomy on drug metabolism and response to therapy.
Aneuploidies of the sex chromosomes
Turner syndrome (45,X)
This is a relatively common (1:2000) syndrome associated with decreased adult stature, gonadal dysgenesis, reduced female sex steroids, infertility and other stigmata (97,98).
A cohort study of 3425 women with Turner syndrome (TS) (99) found an increased risk of CNS tumors, ocular cancer, gonadoblastoma and bladder and urethral cancers, while the risk for breast cancer was found to be decreased. An increased risk of colon cancer, which was seen in another study (100), was not noted.
Gonadoblastoma is a rare neoplasm that develops almost exclusively in the dysgenetic gonads of women with Y chromosome mosaicism. Malignant transformation occurs in 60% of these tumors, with 50% developing into dysgerminomas and 10% into other malignant germ cell tumors (101). The predisposition of dysgenetic gonads to develop gonadoblastoma was postulated to be associated with one or more genes on the Y chromosome, with the stimulatory effect of the gonadotropins (101), and with the presence of poorly differentiated XY gonadal tissue in an abnormal (intra-abdominal) environment (102).
The decreased risk for breast cancer was confined to women with 45,X monosomy, whereas women with 45,X/46,XX mosaicism had similar risk to the general population. This may be explained by the fact that women with 45,X/46,XX karyotype have a less severe phenotype and have a relatively high percentage of spontaneous menses and breast development (101).
An interesting finding is the lack of significant increased risk for malignant melanoma in TS, since these patients have multiple melanocytic nevi, the strongest established risk factor for melanoma (99). One possible explanation for the absence of an overt increase of incidence of melanoma is the absence of circulating sex hormones as these girls fail to undergo normal pubertal development (103). Another explanation for variation in non-hormonal sensitive tumors may involve a haploinsufficiency of autosomal X linked genes in TS (104–107).
Klinefelter syndrome (47,XXY)
Klinefelter syndrome (KS) is a group of chromosomal disorders in which there is at least one extra X chromosome to a normal male karyotype, 46,XY. XXY aneuploidy is the most common disorder of sex chromosomes in humans, with prevalence of one in 500 males (108). KS is characterized by hypergonadotrophic hypogonadism, small testes, infertility, reduced body hair, gynecomastia and tall stature (109).
A cohort study of 3518 men with KS revealed increased risk of lung cancer, breast cancer and non-Hodgkin lymphoma, while a decreased risk of prostate cancer (110). Contrary to an anecdotal report (111), there was no statistically significant increase in the incidence of leukemia. The lower risk for prostate cancer is in accordance with a Danish cohort (112). The reduced prostate cancer and the increase in breast cancer likely represent the hormonal milieu of these patients characterized by high estradiol and low androgen levels.
There are contradictory reports regarding the frequency of extragonadal (mediastinal or pineal) germ cell tumors (EGGCT) in KS. No such tumors were reported in the British cohort study (110), while increased prevalence was reported in several previous studies (112–115). Studies suggested that 15% of patients with intracranial germ cell tumors and 8% of male patients with primary malignant mediastinal germ cell tumors have KS (109,113,116–119). The increased prevalence of EGGCT in KS may reflect either the role of extra chromosome X in these tumors (120) or a developmental defect in the migration of germ cells in KS (109,115,121,122).
GENERAL PRINCIPLES OF THE PATHOGENESIS OF CANCER BY CONSTITUTIONAL CHROMOSOMAL ANEUPLOIDIES
Cancer as a developmental disease
A fundamental difference between constitutional and acquired aneuploidy is that the former exists in many tissues from the earliest stage of embryonic development, whereas the latter is acquired and exists only in the transformed cells. Thus constitutional aneuploidy can predispose to cancer in variety of ways. It may exert a direct oncogenic activity in a cell autonomous manner. This may be the situation in cancers in which the same aneuploidy is also common in sporadic cancers such as trisomy 21 in ALL, trisomy 8 in myeloid leukemias and trisomy 18 in Wilms’ tumors. Alternatively, cancer may arise because of aberrant effects of the trisomy on immediate microenvironment, for example the enhancement of hematopoietic progenitor proliferation by constitutional trisomy 8. Cancer may also arise because of abnormalities in the ‘macro-environment’—for example the hormonal abnormalities in disorders of the sex chromosomes, hormonal replacement therapy (99,123), susceptibility of mal-developed organs to malignant transformation (e.g. horseshoe kidney in cT18) (47) or even altered immunosurveillance (e.g. NK cells in cT8M) (44) (Fig. 2).
Figure 2.

Factors affecting the cancer predisposition by constitutional aneuploidy. Imbalance of pro-malignant genes expression, genomic instability and cellular growth alterations coupled with specific acquired cooperative genetic events are the main mechanisms that underlie the neoplastic predisposition of constitutional aneuploidy. In addition, as the constitutional aneuploidy exists from a very early developmental stage, the developmental timing of the aneuploidic effect may have an influence on the end-point result of the aneuploidy. Moreover, as the constitutional aneuploidy resides in many tissues, it can affect the pre-neoplastic target cell in a cell-autonomous manner, and/or affect the micro- and macro-environment of the pre-neoplastic cell in a non-cell-autonomous manner.
What is common to all constitutional aneuploidies is their marked effect on embryonic development. Indeed the types of cancers observed reflect these abnormalities, e.g. the megakaryoblastic malignancies in DS and the gonadoblastomas in TS. Even more striking is the specific association of a general aneuploidy defect, as seen in MVA, with rather specific embryonal tumors (RMS and Wilms’ tumors). This type of association is seen also in other inherited cancer-predisposing syndromes. Thus mutations in Rb and p53 tumor suppressor genes are observed in most sporadic cancers, yet germline mutations in these genes are associated with childhood retinoblastoma and soft tissue sarcomas, respectively, two rare embryonal childhood cancers (124,125). Indeed, sporadic childhood cancer may be viewed as a developmental disorder. While this is obvious for the embryonal cancers, such as RMS, Wilms’ tumors, neuro-, medullo- and hepto-blastomas, the basis of the most common childhood cancer, acute lymphoblastic leukemia (ALL), is also developmental (126,127) and is reminiscent of the pattern of the myeloid leukemias of DS. Acquired somatic structural or numerical genetic aberration arising during embryonic lymphoid development leads to a proliferation of a pre-leukemic clone that can be identified by molecular means in 1–5% of normal newborns. One or more, less common, postnatal somatic genetic events are needed for transformation of these pre-leukemic cells similarly to the events required for the evolution of TMD to AMKL in DS.
Aneuploidy and cooperating oncogenic genetic aberrations in cancer
Aneuploidy alone is not sufficient for carcinogenesis. Most children with DS do not develop leukemia and even in the presence of widespread aneuploidies in MVA the prevalence of cancer is <50%. In most of the constitutional aneuploidies, the additional oncogenic events that cooperate with the aneuploidy have not been studied. The mutations in GATA1 and the JAK2 cooperating with cT21 in the myeloid and lymphoid leukemias of DS are remarkably specific to DS. They have not been described in sporadic leukemias with acquired trisomy 21. The cause of the specific association between cT21 and these mutations, outside Hsa21, is presently unknown.
A more general model that may explain these specific cooperative events emerges from recent experiments by Williams et al. (128). By generating a series of cell lines that carry an extra copy of one of four mouse chromosomes they showed that aneuploidy reduces cellular fitness by repressing cell proliferation, alters their metabolic properties and influences their immortalizing capabilities. This is consistent with the general growth inhibitory role of constitutional trisomies and with the potential for tumor suppression as observed in DS and in some experimental aneuploidy models in mice (129,130). Overcoming this proliferative block requires specific cooperating mutations. This phenomenon is similar to classical requirement for cooperative oncogenic activity to overcome the senescence or cell death induced by single oncogenes (131). Thus the association of a constitutional trisomy with cancer may depend on a specific ‘permissive cell type’ (e.g. an embryonic megakaryocytic erythroid progenitor in DS) and a specific cooperating mutation.
With few exceptions, the major challenge is in deciphering the actual genetic elements (including all type of genes) on the aneuplidic chromosomes involved in cancer initiation and/or progression. The difficulties stem from the general imbalance of expression of most genes from the aneuplidic chromosomes and from the lack of appropriate in vitro and in vivo models. Such models are required for clarification of the shared and unique pathways by which constitutional and acquired aneuploidies affect neoplastic transformation.
FUNDING
We acknowledge the funding to S.I. by the Israel Science Foundation and Morasha programs, Sam Waxman Cancer Research Foundation, Children with Leukaemia UK, Israel Ministry of Science, National Institute of Health, Israel-USA Binational Science Foundation and German Israel Foundation. I.G. is a recipient of the Stem Cell PhD fellowship from Tel-Aviv University and G.S. is the recipient of the Eshkol PhD scholarship from the Israeli Ministry of Science.
ACKNOWLEDGEMENTS
We apologize to many authors whose work could not be cited due to space limitations and thank physicians and scientists in Israel, Europe, USA and Japan collaborating with us on the research of Down Syndrome leukemias.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Hanahan D., Weinberg R.A. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Teixeira M.R., Heim S. Multiple numerical chromosome aberrations in cancer: what are their causes and what are their consequences? Semin. Cancer. Biol. 2005;15:3–12. doi: 10.1016/j.semcancer.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 3.Callier P., Faivre L., Cusin V., Marle N., Thauvin-Robinet C., Sandre D., Rousseau T., Sagot P., Lacombe E., Faber V., et al. Microcephaly is not mandatory for the diagnosis of mosaic variegated aneuploidy syndrome. Am. J. Med. Genet. A. 2005;137:204–207. doi: 10.1002/ajmg.a.30783. [DOI] [PubMed] [Google Scholar]
- 4.Hanks S., Coleman K., Reid S., Plaja A., Firth H., Fitzpatrick D., Kidd A., Mehes K., Nash R., Robin N., et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat. Genet. 2004;36:1159–1161. doi: 10.1038/ng1449. [DOI] [PubMed] [Google Scholar]
- 5.Matsuura S., Matsumoto Y., Morishima K., Izumi H., Matsumoto H., Ito E., Tsutsui K., Kobayashi J., Tauchi H., Kajiwara Y., et al. Monoallelic BUB1B mutations and defective mitotic-spindle checkpoint in seven families with premature chromatid separation (PCS) syndrome. Am. J. Med. Genet. A. 2006;140:358–367. doi: 10.1002/ajmg.a.31069. [DOI] [PubMed] [Google Scholar]
- 6.Micale M.A., Schran D., Emch S., Kurczynski T.W., Rahman N., Van Dyke D.L. Mosaic variegated aneuploidy without microcephaly: implications for cytogenetic diagnosis. Am. J. Med. Genet. A. 2007;143A:1890–1893. doi: 10.1002/ajmg.a.31848. [DOI] [PubMed] [Google Scholar]
- 7.Kajii T., Ikeuchi T., Yang Z.Q., Nakamura Y., Tsuji Y., Yokomori K., Kawamura M., Fukuda S., Horita S., Asamoto A. Cancer-prone syndrome of mosaic variegated aneuploidy and total premature chromatid separation: report of five infants. Am. J. Med. Genet. 2001;104:57–64. doi: 10.1002/ajmg.1580. [DOI] [PubMed] [Google Scholar]
- 8.Cahill D.P., da Costa L.T., Carson-Walter E.B., Kinzler K.W., Vogelstein B., Lengauer C. Characterization of MAD2B and other mitotic spindle checkpoint genes. Genomics. 1999;58:181–187. doi: 10.1006/geno.1999.5831. [DOI] [PubMed] [Google Scholar]
- 9.Kops G.J., Weaver B.A., Cleveland D.W. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer. 2005;5:773–785. doi: 10.1038/nrc1714. [DOI] [PubMed] [Google Scholar]
- 10.Garcia-Castillo H., Vasquez-Velasquez A.I., Rivera H., Barros-Nunez P. Clinical and genetic heterogeneity in patients with mosaic variegated aneuploidy: delineation of clinical subtypes. Am. J. Med. Genet. A. 2008;146A:1687–1695. doi: 10.1002/ajmg.a.32315. [DOI] [PubMed] [Google Scholar]
- 11.Dai W., Wang Q., Liu T., Swamy M., Fang Y., Xie S., Mahmood R., Yang Y.M., Xu M., Rao C.V. Slippage of mitotic arrest and enhanced tumor development in mice with BubR1 haploinsufficiency. Cancer Res. 2004;64:440–445. doi: 10.1158/0008-5472.can-03-3119. [DOI] [PubMed] [Google Scholar]
- 12.Hanks S., Coleman K., Summersgill B., Messahel B., Williamson D., Pritchard-Jones K., Strefford J., Swansbury J., Plaja A., Shipley J., et al. Comparative genomic hybridization and BUB1B mutation analyses in childhood cancers associated with mosaic variegated aneuploidy syndrome. Cancer Lett. 2006;239:234–238. doi: 10.1016/j.canlet.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 13.Weber-Hall S., Anderson J., McManus A., Abe S., Nojima T., Pinkerton R., Pritchard-Jones K., Shipley J. Gains, losses, and amplification of genomic material in rhabdomyosarcoma analyzed by comparative genomic hybridization. Cancer Res. 1996;56:3220–3224. [PubMed] [Google Scholar]
- 14.Pandita A., Zielenska M., Thorner P., Bayani J., Godbout R., Greenberg M., Squire J.A. Application of comparative genomic hybridization, spectral karyotyping, and microarray analysis in the identification of subtype-specific patterns of genomic changes in rhabdomyosarcoma. Neoplasia. 1999;1:262–275. doi: 10.1038/sj.neo.7900036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bridge J.A., Liu J., Qualman S.J., Suijkerbuijk R., Wenger G., Zhang J., Wan X., Baker K.S., Sorensen P., Barr F.G. Genomic gains and losses are similar in genetic and histologic subsets of rhabdomyosarcoma, whereas amplification predominates in embryonal with anaplasia and alveolar subtypes. Genes Chromosomes Cancer. 2002;33:310–321. doi: 10.1002/gcc.10026. [DOI] [PubMed] [Google Scholar]
- 16.Hernando E., Nahle Z., Juan G., Diaz-Rodriguez E., Alaminos M., Hemann M., Michel L., Mittal V., Gerald W., Benezra R., et al. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature. 2004;430:797–802. doi: 10.1038/nature02820. [DOI] [PubMed] [Google Scholar]
- 17.Plaja A., Vendrell T., Smeets D., Sarret E., Gili T., Catala V., Mediano C., Scheres J.M. Variegated aneuploidy related to premature centromere division (PCD) is expressed in vivo and is a cancer-prone disease. Am. J. Med. Genet. 2001;98:216–223. doi: 10.1002/1096-8628(20010122)98:3<216::aid-ajmg1091>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 18.Olson J.M., Hamilton A., Breslow N.E. Non-11p constitutional chromosome abnormalities in Wilms’ tumor patients. Med. Pediatr. Oncol. 1995;24:305–309. doi: 10.1002/mpo.2950240507. [DOI] [PubMed] [Google Scholar]
- 19.Anderson C.E., Punnett H.H., Huff V., de Chadarevian J.P. Characterization of a Wilms tumor in a 9-year-old girl with trisomy 18. Am. J. Med. Genet. A. 2003;121A:52–55. doi: 10.1002/ajmg.a.20141. [DOI] [PubMed] [Google Scholar]
- 20.Riccardi V.M., Crandall B.F. Karyotype-phenotype correlation: mosaic trisomy 8 and partial trisomies of different segments of chromosome 8. Hum. Genet. 1978;41:363–367. doi: 10.1007/BF00284772. [DOI] [PubMed] [Google Scholar]
- 21.Hasle H., Clausen N., Pedersen B., Bendix-Hansen K. Myelodysplastic syndrome in a child with constitutional trisomy 8 mosaicism and normal phenotype. Cancer Genet. Cytogenet. 1995;79:79–81. doi: 10.1016/0165-4608(94)00099-w. [DOI] [PubMed] [Google Scholar]
- 22.Secker-Walker L.M., Fitchett M. Constitutional and acquired trisomy 8. Leuk. Res. 1995;19:737–740. doi: 10.1016/0145-2126(95)00051-o. [DOI] [PubMed] [Google Scholar]
- 23.Gafter U., Shabtal F., Kahn Y., Halbrecht I., Djaldetti M. Aplastic anemia followed by leukemia in congenital trisomy 8 mosaicism. Ultrastructural studies of polymorphonuclear cells in peripheral blood. Clin. Genet. 1976;9:134–142. doi: 10.1111/j.1399-0004.1976.tb01559.x. [DOI] [PubMed] [Google Scholar]
- 24.Cornaglia-Ferraris P., Ghio R., Barabino A., Perlino G.F., Maggio A., Parodi M.T., Massimo L. Diminished in vitro colony forming capacity of bone marrow cells in a case of chromosome 8 trisomy (mosaicism): criteria for ‘high risk’ pre-leukemia syndrome. Boll Ist Sieroter Milan. 1981;60:69–73. [PubMed] [Google Scholar]
- 25.Palmer C.G., Provisor A.J., Weaver D.D., Hodes M.E., Heerema N. Juvenile chronic granulocytic leukemia in a patient with trisomy 8, neurofibromatosis, and prolonged Epstein-Barr virus infection. J Pediatr. 1983;102:888–892. doi: 10.1016/s0022-3476(83)80020-1. [DOI] [PubMed] [Google Scholar]
- 26.Kapaun P., Kabisch H., Held K.R., Walter T.A., Hegewisch S., Zander A.R. Atypical chronic myelogenous leukemia in a patient with trisomy 8 mosaicism syndrome. Ann. Hematol. 1993;66:57–58. doi: 10.1007/BF01737691. [DOI] [PubMed] [Google Scholar]
- 27.Mastrangelo R., Tornesello A., Mastrangelo S., Zollino M., Neri G. Constitutional trisomy 8 mosaicism evolving to primary myelodysplastic syndrome: a new subset of biologically related patients? Am. J. Hematol. 1995;48:67–68. doi: 10.1002/ajh.2830480122. [DOI] [PubMed] [Google Scholar]
- 28.Zollino M., Genuardi M., Bajer J., Tornesello A., Mastrangelo S., Zampino G., Mastrangelo R., Neri G. Constitutional trisomy 8 and myelodysplasia: report of a case and review of the literature. Leuk. Res. 1995;19:733–736. doi: 10.1016/0145-2126(95)00050-x. [DOI] [PubMed] [Google Scholar]
- 29.Seghezzi L., Maserati E., Minelli A., Dellavecchia C., Addis P., Locatelli F., Angioni A., Balloni P., Miano C., Cavalli P., et al. Constitutional trisomy 8 as first mutation in multistep carcinogenesis: clinical, cytogenetic, and molecular data on three cases. Genes Chromosomes Cancer. 1996;17:94–101. doi: 10.1002/(SICI)1098-2264(199610)17:2<94::AID-GCC4>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 30.Maserati E., Aprili F., Vinante F., Locatelli F., Amendola G., Zatterale A., Milone G., Minelli A., Bernardi F., Lo Curto F., et al. Trisomy 8 in myelodysplasia and acute leukemia is constitutional in 15-20% of cases. Genes Chromosomes Cancer. 2002;33:93–97. doi: 10.1002/gcc.1214. [DOI] [PubMed] [Google Scholar]
- 31.Narendran A., Hawkins L.M., Ganjavi H., Vanek W., Gee M.F., Barlow J.W., Johnson G., Malkin D., Freedman M.H. Characterization of bone marrow stromal abnormalities in a patient with constitutional trisomy 8 mosaicism and myelodysplastic syndrome. Pediatr. Hematol. Oncol. 2004;21:209–221. doi: 10.1080/08880010490276917. [DOI] [PubMed] [Google Scholar]
- 32.Ando S., Maemori M., Sakai H., Ando S., Shiraishi H., Sakai K., Ruhnke G.W. Constitutional trisomy 8 mosaicism with myelodysplastic syndrome complicated by intestinal Behcet disease and antithrombin III deficiency. Cancer Genet. Cytogenet. 2005;162:172–175. doi: 10.1016/j.cancergencyto.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 33.Riccardi V.M., Humbert J.R., Peakman D. Acute leukemia associated with trisomy 8 mosaicism and a familial translocation 46,XY,t(7;20)(p13;p12) Am. J Med. Genet. 1978;2:15–21. doi: 10.1002/ajmg.1320020104. [DOI] [PubMed] [Google Scholar]
- 34.Brady A.F., Waters C.S., Pocha M.J., Brueton L.A. Chronic myelomonocytic leukaemia in a child with constitutional partial trisomy 8 mosaicism. Clin. Genet. 2000;58:142–146. doi: 10.1034/j.1399-0004.2000.580209.x. [DOI] [PubMed] [Google Scholar]
- 35.Yamamoto K., Okamura A., Kawano H., Katayama Y., Shimoyama M., Matsui T. A novel t(8;18)(q13;q21) in acute monocytic leukemia evolving from constitutional trisomy 8 mosaicism. Cancer Genet. Cytogenet. 2007;176:144–149. doi: 10.1016/j.cancergencyto.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 36.Niss R., Passarge E. Trisomy 8 restricted to cultured fibroblasts. J Med. Genet. 1976;13:229–234. doi: 10.1136/jmg.13.3.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakamura Y., Nakashima H., Fukuda S., Hashimoto T., Maruyama M. Bilateral cystic nephroblastomas and multiple malformations with trisomy 8 mosaicism. Hum. Pathol. 1985;16:754–756. doi: 10.1016/s0046-8177(85)80166-0. [DOI] [PubMed] [Google Scholar]
- 38.Lessick M., Israel J., Szego K., Wong P. Leiomyosarcoma in a patient with trisomy 8 mosaicism. J. Med. Genet. 1990;27:643–644. doi: 10.1136/jmg.27.10.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mark F.L., Ahearn J., Lathrop J.C. Constitutional trisomy 8 mosaicism and gestational trophoblastic disease. Cancer Genet. Cytogenet. 1995;80:150–154. doi: 10.1016/0165-4608(94)00188-h. [DOI] [PubMed] [Google Scholar]
- 40.Hasle H. Myelodysplastic syndromes in childhood–classification, epidemiology, and treatment. Leuk. Lymphoma. 1994;13:11–26. doi: 10.3109/10428199409051647. [DOI] [PubMed] [Google Scholar]
- 41.Heim S., Mitelman F. Cytogenetic analysis in the diagnosis of acute leukemia. Cancer. 1992;70:1701–1709. doi: 10.1002/1097-0142(19920915)70:4+<1701::aid-cncr2820701609>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 42.Maserati E., Pressato B., Valli R., Patitucci F., Lo Curto F., Pasquali F., Minelli A., Danesino C., Marchetti M., Barosi G. Constitutional trisomy 8 mosaicism in primary myelofibrosis: relevance to clinical practice and warning for trisomy 8 studies. Cancer Genet. Cytogenet. 2007;179:79–81. doi: 10.1016/j.cancergencyto.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 43.Virtaneva K., Wright F.A., Tanner S.M., Yuan B., Lemon W.J., Caligiuri M.A., Bloomfield C.D., de La Chapelle A., Krahe R. Expression profiling reveals fundamental biological differences in acute myeloid leukemia with isolated trisomy 8 and normal cytogenetics. Proc. Natl Acad. Sci. USA. 2001;98:1124–1129. doi: 10.1073/pnas.98.3.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marti S., Galan F.M., Casero J.M., Merino J., Rubio G. Characterization of trisomic natural killer cell abnormalities in a patient with constitutional trisomy 8 mosaicism. Pediatr. Hematol. Oncol. 2008;25:135–146. doi: 10.1080/08880010801890135. [DOI] [PubMed] [Google Scholar]
- 45.Nielsen J., Wohlert M. Chromosome abnormalities found among 34,910 newborn children: results from a 13-year incidence study in Arhus, Denmark. Hum. Genet. 1991;87:81–83. doi: 10.1007/BF01213097. [DOI] [PubMed] [Google Scholar]
- 46.Pal S., Siti M.I., Ankathil R., Zilfalil B.A. Two cases of isochromosome 18q syndrome. Singapore. Med. J. 2007;48:e146–e150. [PubMed] [Google Scholar]
- 47.Kinoshita M., Nakamura Y., Nakano R., Morimatsu M., Fukuda S., Nishimi Y., Hashimoto T. Thirty-one autopsy cases of trisomy 18: clinical features and pathological findings. Pediatr. Pathol. 1989;9:445–457. doi: 10.3109/15513818909022365. [DOI] [PubMed] [Google Scholar]
- 48.Kelly M., Robinson B.W., Moore J.W. Trisomy 18 in a 20-year-old woman. Am. Med. Genet. 2002;112:397–399. doi: 10.1002/ajmg.10638. [DOI] [PubMed] [Google Scholar]
- 49.Miller R.W., Fraumeni J.F., Jr, Manning M.D. Association of Wilms’s Tumor with Aniridia, Hemihypertrophy and Other Congenital Malformations. N. Engl. J. Med. 1964;270:922–927. doi: 10.1056/NEJM196404302701802. [DOI] [PubMed] [Google Scholar]
- 50.Geiser C.F., Schindler A.M. Long survival in a male with 18-trisomy syndrome and Wilms’ tumor. Pediatrics. 1969;44:111–116. [PubMed] [Google Scholar]
- 51.Karayalcin G., Shanske A., Honigman R. Wilms’ tumor in a 13-year old girl with trisomy 18. Am. J Dis. Child. 1981;135:665–666. doi: 10.1001/archpedi.1981.02130310069024. [DOI] [PubMed] [Google Scholar]
- 52.Kullendorff C.M., Wiebe T. Chromosomal aberrations in Wilms’ tumour. Eur. J Pediatr. Surg. 1997;7:286–287. doi: 10.1055/s-2008-1071173. [DOI] [PubMed] [Google Scholar]
- 53.Perilongo G., Shafford E.A. Liver tumours. Eur. J. Cancer. 1999;35:953–958. doi: 10.1016/s0959-8049(99)00049-0. discussion 958-959. [DOI] [PubMed] [Google Scholar]
- 54.Bove K.E., Soukup S., Ballard E.T., Ryckman F. Hepatoblastoma in a child with trisomy 18: cytogenetics, liver anomalies, and literature review. Pediatr. Pathol. Lab. Med. 1996;16:253–262. [PubMed] [Google Scholar]
- 55.Dasouki M., Barr M., Jr Trisomy 18 and hepatic neoplasia. Am. J. Med. Genet. 1987;27:203–205. doi: 10.1002/ajmg.1320270122. [DOI] [PubMed] [Google Scholar]
- 56.Mamlok V., Nichols M., Lockhart L., Mamlok R. Trisomy 18 and hepatoblastoma. Am. J. Med. Genet. 1989;33:125–126. doi: 10.1002/ajmg.1320330119. [DOI] [PubMed] [Google Scholar]
- 57.Maruyama K., Ikeda H., Koizumi T. Hepatoblastoma associated with trisomy 18 syndrome: a case report and a review of the literature. Pediatr. Int. 2001;43:302–305. doi: 10.1046/j.1442-200x.2001.01380.x. [DOI] [PubMed] [Google Scholar]
- 58.Takada K., Hamada Y., Sato M., Fujii Y., Teraguchi M., Kaneko K., Kamiyama Y. Cecal volvulus in children with mental disability. Pediatr. Surg. Int. 2007;23:1011–1014. doi: 10.1007/s00383-007-1987-6. [DOI] [PubMed] [Google Scholar]
- 59.Tanaka K., Uemoto S., Asonuma K., Katayama T., Utsunomiya H., Akiyama Y., Sasaki M.S., Ozawa K. Hepatoblastoma in a 2-year-old girl with trisomy 18. Eur. J. Pediatr. Surg. 1992;2:298–300. doi: 10.1055/s-2008-1063464. [DOI] [PubMed] [Google Scholar]
- 60.Teraguchi M., Nogi S., Ikemoto Y., Ogino H., Kohdera U., Sakaida N., Okamura A., Hamada Y., Kobayashi Y. Multiple hepatoblastomas associated with trisomy 18 in a 3-year-old girl. Pediatr. Hematol. Oncol. 1997;14:463–467. doi: 10.3109/08880019709028777. [DOI] [PubMed] [Google Scholar]
- 61.Hitzler J.K. Acute megakaryoblastic leukemia in Down syndrome. Pediatr. Blood Cancer. 2007;49:1066–1069. doi: 10.1002/pbc.21353. [DOI] [PubMed] [Google Scholar]
- 62.Izraeli S., Rainis L., Hertzberg L., Smooha G., Birger Y. Trisomy of chromosome 21 in leukemogenesis. Blood Cells Mol. Dis. 2007;39:156–159. doi: 10.1016/j.bcmd.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 63.Malinge S., Izraeli S., Crispino J.D. Insights into the manifestations, outcomes and mechanisms of leukemogenesis in Down syndrome. Blood. 2009 doi: 10.1182/blood-2008-11-163501. [Epub ahead of print] http://bloodjournal.hematologylibrary.org/cgi/reprint/blood-2008-11-163501v1 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Izraeli S. Trisomy 21 tilts the balance. Blood. 2008;112:4361–4362. doi: 10.1182/blood-2008-09-176719. [DOI] [PubMed] [Google Scholar]
- 65.Hasle H. Pattern of malignant disorders in individuals with Down’s syndrome. Lancet Oncol. 2001;2:429–436. doi: 10.1016/S1470-2045(00)00435-6. [DOI] [PubMed] [Google Scholar]
- 66.Hasle H., Clemmensen I.H., Mikkelsen M. Risks of leukaemia and solid tumours in individuals with Down’s syndrome. Lancet. 2000;355:165–169. doi: 10.1016/S0140-6736(99)05264-2. [DOI] [PubMed] [Google Scholar]
- 67.Sussan T.E., Yang A., Li F., Ostrowski M.C., Reeves R.H. Trisomy represses Apc(Min)-mediated tumours in mouse models of Down’s syndrome. Nature. 2008;451:73–75. doi: 10.1038/nature06446. [DOI] [PubMed] [Google Scholar]
- 68.Hasle H., Niemeyer C.M., Chessells J.M., Baumann I., Bennett J.M., Kerndrup G., Head D.R. A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia. 2003;17:277–282. doi: 10.1038/sj.leu.2402765. [DOI] [PubMed] [Google Scholar]
- 69.Zipursky A. Transient leukaemia–a benign form of leukaemia in newborn infants with trisomy 21. Br. J. Haematol. 2003;120:930–938. doi: 10.1046/j.1365-2141.2003.04229.x. [DOI] [PubMed] [Google Scholar]
- 70.Klusmann J.H., Creutzig U., Zimmermann M., Dworzak M., Jorch N., Langebrake C., Pekrun A., Macakova-Reinhardt K., Reinhardt D. Treatment and prognostic impact of transient leukemia in neonates with Down syndrome. Blood. 2008;111:2991–2998. doi: 10.1182/blood-2007-10-118810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wechsler J., Greene M., McDevitt M.A., Anastasi J., Karp J.E., Le Beau M.M., Crispino J.D. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat. Genet. 2002;32:148–152. doi: 10.1038/ng955. [DOI] [PubMed] [Google Scholar]
- 72.Rainis L., Bercovich D., Strehl S., Teigler-Schlegel A., Stark B., Trka J., Amariglio N., Biondi A., Muler I., Rechavi G., et al. Mutations in exon 2 of GATA1 are early events in megakaryocytic malignancies associated with trisomy 21. Blood. 2003;102:981–986. doi: 10.1182/blood-2002-11-3599. [DOI] [PubMed] [Google Scholar]
- 73.Taub J.W., Mundschau G., Ge Y., Poulik J.M., Qureshi F., Jensen T., James S.J., Matherly L.H., Wechsler J., Crispino J.D. Prenatal origin of GATA1 mutations may be an initiating step in the development of megakaryocytic leukemia in Down syndrome. Blood. 2004;104:1588–1589. doi: 10.1182/blood-2004-04-1563. [DOI] [PubMed] [Google Scholar]
- 74.Bourquin J.P., Subramanian A., Langebrake C., Reinhardt D., Bernard O., Ballerini P., Baruchel A., Cave H., Dastugue N., Hasle H., et al. Identification of distinct molecular phenotypes in acute megakaryoblastic leukemia by gene expression profiling. Proc. Natl Acad. Sci. USA. 2006;103:3339–3344. doi: 10.1073/pnas.0511150103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li Z., Godinho F.J., Klusmann J.H., Garriga-Canut M., Yu C., Orkin S.H. Developmental stage-selective effect of somatically mutated leukemogenic transcription factor GATA1. Nat. Genet. 2005;37:613–619. doi: 10.1038/ng1566. [DOI] [PubMed] [Google Scholar]
- 76.Chou S.T., Opalinska J.B., Yao Y., Fernandes M.A., Kalota A., Brooks J.S., Choi J.K., Gewirtz A.M., Danet-Desnoyers G.A., Nemiroff R.L., et al. Trisomy 21 enhances human fetal erythro-megakaryocytic development. Blood. 2008;112:4503–4506. doi: 10.1182/blood-2008-05-157859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tunstall-Pedoe O., Roy A., Karadimitris A., de la Fuente J., Fisk N.M., Bennett P., Norton A., Vyas P., Roberts I. Abnormalities in the myeloid progenitor compartment in Down syndrome fetal liver precede acquisition of GATA1 mutations. Blood. 2008;112:4507–4511. doi: 10.1182/blood-2008-04-152967. [DOI] [PubMed] [Google Scholar]
- 78.Sato T., Toki T., Kanezaki R., Xu G., Terui K., Kanegane H., Miura M., Adachi S., Migita M., Morinaga S., et al. Functional analysis of JAK3 mutations in transient myeloproliferative disorder and acute megakaryoblastic leukaemia accompanying Down syndrome. Br. J. Haematol. 2008;141:681–688. doi: 10.1111/j.1365-2141.2008.07081.x. [DOI] [PubMed] [Google Scholar]
- 79.Forestier E., Izraeli S., Beverloo B., Haas O., Pession A., Michalova K., Stark B., Harrison C.J., Teigler-Schlegel A., Johansson B. Cytogenetic features of acute lymphoblastic and myeloid leukemias in pediatric patients with Down syndrome: an iBFM-SG study. Blood. 2008;111:1575–1583. doi: 10.1182/blood-2007-09-114231. [DOI] [PubMed] [Google Scholar]
- 80.Rainis L., Toki T., Pimanda J.E., Rosenthal E., Machol K., Strehl S., Gottgens B., Ito E., Izraeli S. The proto-oncogene ERG in megakaryoblastic leukemias. Cancer Res. 2005;65:7596–7602. doi: 10.1158/0008-5472.CAN-05-0147. [DOI] [PubMed] [Google Scholar]
- 81.Xu G., Kanezaki R., Toki T., Watanabe S., Takahashi Y., Terui K., Kitabayashi I., Ito E. Physical association of the patient-specific GATA1 mutants with RUNX1 in acute megakaryoblastic leukemia accompanying Down syndrome. Leukemia. 2006;20:1002–1008. doi: 10.1038/sj.leu.2404223. [DOI] [PubMed] [Google Scholar]
- 82.Stankiewicz M.J., Crispino J.D. ETS2 and ERG1 promote megakaryopoiesis and immortalize GATA1 knockdown (neoHS) fetal liver progenitors: implications for leukemogenesis in Down Syndrome. Blood (ASH Annual Meeting Abstracts) 2007;110:824. [Google Scholar]
- 83.Ge Y., LaFiura K.M., Dombkowski A.A., Chen Q., Payton S.G., Buck S.A., Salagrama S., Diakiw A.E., Matherly L.H., Taub J.W. The role of the proto-oncogene ETS2 in acute megakaryocytic leukemia biology and therapy. Leukemia. 2008;22:521–529. doi: 10.1038/sj.leu.2405066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hertzberg L., Betts D.R., Raimondi S.C., Schafer B.W., Notterman D.A., Domany E., Izraeli S. Prediction of chromosomal aneuploidy from gene expression data. Genes Chromosomes Cancer. 2007;46:75–86. doi: 10.1002/gcc.20391. [DOI] [PubMed] [Google Scholar]
- 85.Taub J.W., Huang X., Matherly L.H., Stout M.L., Buck S.A., Massey G.V., Becton D.L., Chang M.N., Weinstein H.J., Ravindranath Y. Expression of chromosome 21-localized genes in acute myeloid leukemia: differences between Down syndrome and non-Down syndrome blast cells and relationship to in vitro sensitivity to cytosine arabinoside and daunorubicin. Blood. 1999;94:1393–1400. [PubMed] [Google Scholar]
- 86.Ravindranath Y. Down syndrome and leukemia: new insights into the epidemiology, pathogenesis, and treatment. Pediatr. Blood Cancer. 2005;44:1–7. doi: 10.1002/pbc.20242. [DOI] [PubMed] [Google Scholar]
- 87.Ross J.A., Spector L.G., Robison L.L., Olshan A.F. Epidemiology of leukemia in children with Down syndrome. Pediatr. Blood Cancer. 2005;44:8–12. doi: 10.1002/pbc.20165. [DOI] [PubMed] [Google Scholar]
- 88.Zeller B., Gustafsson G., Forestier E., Abrahamsson J., Clausen N., Heldrup J., Hovi L., Jonmundsson G., Lie S.O., Glomstein A., et al. Acute leukaemia in children with Down syndrome: a population-based Nordic study. Br. J. Haematol. 2005;128:797–804. doi: 10.1111/j.1365-2141.2005.05398.x. [DOI] [PubMed] [Google Scholar]
- 89.Bercovich D., Ganmore I., Scott L.M., Wainreb G., Birger Y., Elimelech A., Shochat C., Cazzaniga G., Biondi A., Basso G., et al. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down’s syndrome. Lancet. 2008;372:1484–1492. doi: 10.1016/S0140-6736(08)61341-0. [DOI] [PubMed] [Google Scholar]
- 90.Malinge S., Ben-Abdelali R., Settegrana C., Radford-Weiss I., Debre M., Beldjord K., Macintyre E.A., Villeval J.L., Vainchenker W., Berger R., et al. Novel activating JAK2 mutation in a patient with Down syndrome and B-cell precursor acute lymphoblastic leukemia. Blood. 2007;109:2202–2204. doi: 10.1182/blood-2006-09-045963. [DOI] [PubMed] [Google Scholar]
- 91.Gaikwad A., Rye C.L., Devidas M., Heerema N.A., Carroll A.J., Izraeli S., Plon S.E., Basso G., Pession A., Rabin K.R. Prevalence and clinical correlates of JAK2 mutations in Down syndrome acute lymphoblastic leukaemia. Br. J. Haematol. 2008 doi: 10.1111/j.1365-2141.2008.07552.x. [Epub ahead of print] http://www3.interscience.wiley.com/cgi-bin/fulltext/121581546/PDFSTART . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kearney L., Gonzalez De Castro D., Yeung J., Procter J., Horsley S.W., Eguchi-Ishimae M., Bateman C.M., Anderson K., Chaplin T., Young B.D., et al. Specific JAK2 mutation (JAK2R683) and multiple gene deletions in Down syndrome acute lymphoblastic leukemia. Blood. 2009;113:646–648. doi: 10.1182/blood-2008-08-170928. [DOI] [PubMed] [Google Scholar]
- 93.Arico M., Ziino O., Valsecchi M.G., Cazzaniga G., Baronci C., Messina C., Pession A., Santoro N., Basso G., Conter V. Acute lymphoblastic leukemia and Down syndrome: presenting features and treatment outcome in the experience of the Italian Association of Pediatric Hematology and Oncology (AIEOP) Cancer. 2008;113:515–521. doi: 10.1002/cncr.23587. [DOI] [PubMed] [Google Scholar]
- 94.Creutzig U., Reinhardt D., Diekamp S., Dworzak M., Stary J., Zimmermann M. AML patients with Down syndrome have a high cure rate with AML-BFM therapy with reduced dose intensity. Leukemia. 2005;19:1355–1360. doi: 10.1038/sj.leu.2403814. [DOI] [PubMed] [Google Scholar]
- 95.Whitlock J.A., Sather H.N., Gaynon P., Robison L.L., Wells R.J., Trigg M., Heerema N.A., Bhatia S. Clinical characteristics and outcome of children with Down syndrome and acute lymphoblastic leukemia: a Children’s Cancer Group study. Blood. 2005;106:4043–4049. doi: 10.1182/blood-2003-10-3446. [DOI] [PubMed] [Google Scholar]
- 96.Matherly L.H., Taub J.W. Methotrexate pharmacology and resistance in childhood acute lymphoblastic leukemia. Leuk. Lymphoma. 1996;21:359–368. doi: 10.3109/10428199609093433. [DOI] [PubMed] [Google Scholar]
- 97.Gravholt C.H. Epidemiology of Turner syndrome. Lancet Oncol. 2008;9:193–195. doi: 10.1016/S1470-2045(08)70045-7. [DOI] [PubMed] [Google Scholar]
- 98.Stochholm K., Juul S., Juel K., Naeraa R.W., Gravholt C.H. Prevalence, incidence, diagnostic delay, and mortality in Turner syndrome. J Clin. Endocrinol. Metab. 2006;91:3897–3902. doi: 10.1210/jc.2006-0558. [DOI] [PubMed] [Google Scholar]
- 99.Schoemaker M.J., Swerdlow A.J., Higgins C.D., Wright A.F., Jacobs P.A. Cancer incidence in women with Turner syndrome in Great Britain: a national cohort study. Lancet Oncol. 2008;9:239–246. doi: 10.1016/S1470-2045(08)70033-0. [DOI] [PubMed] [Google Scholar]
- 100.Hasle H., Olsen J.H., Nielsen J., Hansen J., Friedrich U., Tommerup N. Occurrence of cancer in women with Turner syndrome. Br. J. Cancer. 1996;73:1156–1159. doi: 10.1038/bjc.1996.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Elsheikh M., Dunger D.B., Conway G.S., Wass J.A. Turner’s syndrome in adulthood. Endocr. Rev. 2002;23:120–140. doi: 10.1210/edrv.23.1.0457. [DOI] [PubMed] [Google Scholar]
- 102.Verp M.S., Simpson J.L. Abnormal sexual differentiation and neoplasia. Cancer Genet. Cytogenet. 1987;25:191–218. doi: 10.1016/0165-4608(87)90180-4. [DOI] [PubMed] [Google Scholar]
- 103.Gibbs P., Brady B.M., Gonzalez R., Robinson W.A. Nevi and melanoma: lessons from Turner’s syndrome. Dermatology. 2001;202:1–3. doi: 10.1159/000051575. [DOI] [PubMed] [Google Scholar]
- 104.Cheng L., MacLennan G.T., Pan C.X., Jones T.D., Moore C.R., Zhang S., Gu J., Patel N.B., Kao C., Gardner T.A. Allelic loss of the active X chromosome during bladder carcinogenesis. Arch. Pathol. Lab. Med. 2004;128:187–190. doi: 10.5858/2004-128-187-ALOTAX. [DOI] [PubMed] [Google Scholar]
- 105.Indsto J.O., Nassif N.T., Kefford R.F., Mann G.J. Frequent loss of heterozygosity targeting the inactive X chromosome in melanoma. Clin. Cancer Res. 2003;9:6476–6482. [PubMed] [Google Scholar]
- 106.Knuutila S., Aalto Y., Autio K., Bjorkqvist A.M., El-Rifai W., Hemmer S., Huhta T., Kettunen E., Kiuru-Kuhlefelt S., Larramendy M.L., et al. DNA copy number losses in human neoplasms. Am. J. Pathol. 1999;155:683–694. doi: 10.1016/S0002-9440(10)65166-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lekanne Deprez R.H., Riegman P.H., van Drunen E., Warringa U.L., Groen N.A., Stefanko S.Z., Koper J.W., Avezaat C.J., Mulder P.G., Zwarthoff E.C., et al. Cytogenetic, molecular genetic and pathological analyses in 126 meningiomas. J. Neuropathol. Exp. Neurol. 1995;54:224–235. doi: 10.1097/00005072-199503000-00009. [DOI] [PubMed] [Google Scholar]
- 108.Visootsak J., Graham J.M., Jr Klinefelter syndrome and other sex chromosomal aneuploidies. Orphanet J. Rare Dis. 2006;1:42. doi: 10.1186/1750-1172-1-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Aguirre D., Nieto K., Lazos M., Pena Y.R., Palma I., Kofman-Alfaro S., Queipo G. Extragonadal germ cell tumors are often associated with Klinefelter syndrome. Hum. Pathol. 2006;37:477–480. doi: 10.1016/j.humpath.2006.01.029. [DOI] [PubMed] [Google Scholar]
- 110.Swerdlow A.J., Schoemaker M.J., Higgins C.D., Wright A.F., Jacobs P.A. Cancer incidence and mortality in men with Klinefelter syndrome: a cohort study. J. Natl Cancer Inst. 2005;97:1204–1210. doi: 10.1093/jnci/dji240. [DOI] [PubMed] [Google Scholar]
- 111.Geraedts J.P., Mol A., Briet E., Hartgrink-Groeneveld C.A., den Ottolander G.J. Klinefelter syndrome: predisposition to acute non-lymphocytic leukaemia? Lancet. 1980;1:774. doi: 10.1016/s0140-6736(80)91274-x. [DOI] [PubMed] [Google Scholar]
- 112.Hasle H., Mellemgaard A., Nielsen J., Hansen J. Cancer incidence in men with Klinefelter syndrome. Br. J. Cancer. 1995;71:416–420. doi: 10.1038/bjc.1995.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hasle H., Jacobsen B.B., Asschenfeldt P., Andersen K. Mediastinal germ cell tumour associated with Klinefelter syndrome. A report of case and review of the literature. Eur. J. Pediatr. 1992;151:735–739. doi: 10.1007/BF01959079. [DOI] [PubMed] [Google Scholar]
- 114.Nichols C.R., Heerema N.A., Palmer C., Loehrer P.J., Sr, Williams S.D., Einhorn L.H. Klinefelter’s syndrome associated with mediastinal germ cell neoplasms. J. Clin. Oncol. 1987;5:1290–1294. doi: 10.1200/JCO.1987.5.8.1290. [DOI] [PubMed] [Google Scholar]
- 115.Arens R., Marcus D., Engelberg S., Findler G., Goodman R.M., Passwell J.H. Cerebral germinomas and Klinefelter syndrome. A review. Cancer. 1988;61:1228–1231. doi: 10.1002/1097-0142(19880315)61:6<1228::aid-cncr2820610628>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 116.Jennings M.T., Gelman R., Hochberg F. Intracranial germ-cell tumors: natural history and pathogenesis. J. Neurosurg. 1985;63:155–167. doi: 10.3171/jns.1985.63.2.0155. [DOI] [PubMed] [Google Scholar]
- 117.Schneider D.T., Calaminus G., Reinhard H., Gutjahr P., Kremens B., Harms D., Gobel U. Primary mediastinal germ cell tumors in children and adolescents: results of the German cooperative protocols MAKEI 83/86, 89 and 96. J. Clin. Oncol. 2000;18:832–839. doi: 10.1200/JCO.2000.18.4.832. [DOI] [PubMed] [Google Scholar]
- 118.Beasley S.W., Tiedemann K., Howat A., Werther G., Auldist A.W., Tuohy P. Precocious puberty associated with malignant thoracic teratoma and malignant histiocytosis in a child with Klinefelter’s syndrome. Med. Pediatr. Oncol. 1987;15:277–280. doi: 10.1002/mpo.2950150511. [DOI] [PubMed] [Google Scholar]
- 119.Queipo G., Aguirre D., Nieto K., Pena Y.R., Palma I., Olvera J., Chavez L., Najera N., Kofman-Alfaro S. Intracranial germ cell tumors: association with Klinefelter syndrome and sex chromosome aneuploidies. Cytogenet. Genome Res. 2008;121:211–214. doi: 10.1159/000138887. [DOI] [PubMed] [Google Scholar]
- 120.Okada Y., Nishikawa R., Matsutani M., Louis D.N. Hypomethylated X chromosome gain and rare isochromosome 12p in diverse intracranial germ cell tumors. J. Neuropathol. Exp. Neurol. 2002;61:531–538. doi: 10.1093/jnen/61.6.531. [DOI] [PubMed] [Google Scholar]
- 121.Kaido T., Sasaoka Y., Hashimoto H., Taira K. De novo germinoma in the brain in association with Klinefelter’s syndrome: case report and review of the literature. Surg. Neurol. 2003;60:553–558. doi: 10.1016/s0090-3019(03)00454-3. discussion 559. [DOI] [PubMed] [Google Scholar]
- 122.Dexeus F.H., Logothetis C.J., Chong C., Sella A., Ogden S. Genetic abnormalities in men with germ cell tumors. J. Urol. 1988;140:80–84. doi: 10.1016/s0022-5347(17)41492-3. [DOI] [PubMed] [Google Scholar]
- 123.Claus E.B., Black P.M., Bondy M.L., Calvocoressi L., Schildkraut J.M., Wiemels J.L., Wrensch M. Exogenous hormone use and meningioma risk: what do we tell our patients? Cancer. 2007;110:471–476. doi: 10.1002/cncr.22783. [DOI] [PubMed] [Google Scholar]
- 124.Knudson A.G. Two genetic hits (more or less) to cancer. Nat. Rev. Cancer. 2001;1:157–162. doi: 10.1038/35101031. [DOI] [PubMed] [Google Scholar]
- 125.Sherr C.J., McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–112. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 126.Izraeli S. Leukaemia—a developmental perspective. Br. J. Haematol. 2004;126:3–10. doi: 10.1111/j.1365-2141.2004.04986.x. [DOI] [PubMed] [Google Scholar]
- 127.Greaves M. Infection, immune responses and the aetiology of childhood leukaemia. Nat. Rev. Cancer. 2006;6:193–203. doi: 10.1038/nrc1816. [DOI] [PubMed] [Google Scholar]
- 128.Williams B.R., Prabhu V.R., Hunter K.E., Glazier C.M., Whittaker C.A., Housman D.E., Amon A. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science. 2008;322:703–709. doi: 10.1126/science.1160058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Weaver B.A., Silk A.D., Montagna C., Verdier-Pinard P., Cleveland D.W. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11:25–36. doi: 10.1016/j.ccr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 130.Baker D.J., Jeganathan K.B., Cameron J.D., Thompson M., Juneja S., Kopecka A., Kumar R., Jenkins R.B., de Groen P.C., Roche P., et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet. 2004;36:744–749. doi: 10.1038/ng1382. [DOI] [PubMed] [Google Scholar]
- 131.Land H., Parada L.F., Weinberg R.A. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature. 1983;304:596–602. doi: 10.1038/304596a0. [DOI] [PubMed] [Google Scholar]
- 132.Patja K., Pukkala E., Sund R., Iivanainen M., Kaski M. Cancer incidence of persons with Down syndrome in Finland: a population-based study. Int. J. Cancer. 2006;118:1769–1772. doi: 10.1002/ijc.21518. [DOI] [PubMed] [Google Scholar]
- 133.Hill D.A., Gridley G., Cnattingius S., Mellemkjaer L., Linet M., Adami H.O., Olsen J.H., Nyren O., Fraumeni J.F., Jr Mortality and cancer incidence among individuals with Down syndrome. Arch. Intern. Med. 2003;163:705–711. doi: 10.1001/archinte.163.6.705. [DOI] [PubMed] [Google Scholar]
- 134.Sullivan S.G., Hussain R., Glasson E.J., Bittles A.H. The profile and incidence of cancer in Down syndrome. J. Intellect. Disabil. Res. 2007;51:228–231. doi: 10.1111/j.1365-2788.2006.00862.x. [DOI] [PubMed] [Google Scholar]
- 135.Fabia J., Drolette M. Malformations and leukemia in children with Down’s syndrome. Pediatrics. 1970;45:60–70. [PubMed] [Google Scholar]
- 136.James R., Lightfoot T., Simpson J., Moorman A.V., Roman E., Kinsey S. Acute leukemia in children with Down’s syndrome: the importance of population based study. Haematologica. 2008;93:1262–1263. doi: 10.3324/haematol.12831. [DOI] [PubMed] [Google Scholar]
- 137.Narod S.A., Stiller C., Lenoir G.M. An estimate of the heritable fraction of childhood cancer. Br. J. Cancer. 1991;63:993–999. doi: 10.1038/bjc.1991.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Satge D., Sasco A.J., Carlsen N.L., Stiller C.A., Rubie H., Hero B., de Bernardi B., de Kraker J., Coze C., Kogner P., et al. A lack of neuroblastoma in Down syndrome: a study from 11 European countries. Cancer Res. 1998;58:448–452. [PubMed] [Google Scholar]
- 139.Nishi M., Miyake H., Takeda T., Hatae Y. Congenital malformations and childhood cancer. Med. Pediatr. Oncol. 2000;34:250–254. doi: 10.1002/(sici)1096-911x(200004)34:4<250::aid-mpo3>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 140.Satge D., Sasco A.J., Cure H., Leduc B., Sommelet D., Vekemans M.J. An excess of testicular germ cell tumors in Down’s syndrome: three case reports and a review of the literature. Cancer. 1997;80:929–935. [PubMed] [Google Scholar]
- 141.Swerdlow A.J., Schoemaker M.J., Higgins C.D., Wright A.F., Jacobs P.A. Cancer risk in patients with constitutional chromosome deletions: a nationwide British cohort study. Br. J. Cancer. 2008;98:1929–1933. doi: 10.1038/sj.bjc.6604391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.McDonald-McGinn D.M., Reilly A., Wallgren-Pettersson C., Hoyme H.E., Yang S.P., Adam M.P., Zackai E.H., Sullivan K.E. Malignancy in chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome) Am. J Med. Genet. A. 2006;140:906–909. doi: 10.1002/ajmg.a.31199. [DOI] [PubMed] [Google Scholar]
- 143.Schneider D.T., Schuster A.E., Fritsch M.K., Calaminus G., Gobel U., Harms D., Lauer S., Olson T., Perlman E.J. Genetic analysis of mediastinal nonseminomatous germ cell tumors in children and adolescents. Genes Chromosomes Cancer. 2002;34:115–125. doi: 10.1002/gcc.10053. [DOI] [PubMed] [Google Scholar]
- 144.Bussey K.J., Lawce H.J., Olson S.B., Arthur D.C., Kalousek D.K., Krailo M., Giller R., Heifetz S., Womer R., Magenis R.E. Chromosome abnormalities of eighty-one pediatric germ cell tumors: sex-, age-, site-, and histopathology-related differences–a Children’s Cancer Group study. Genes Chromosomes Cancer. 1999;25:134–146. [PubMed] [Google Scholar]