

Abstract
The recent advance in technology for mass spectrometry-based targeted protein quantification has opened new avenues for a broad range of proteomic applications in clinical research. The major breakthroughs are highlighted by the capability of using a “universal” approach to perform quantitative assays for a wide spectrum of proteins with minimum restrictions, and the ease of assembling multiplex detections in a single measurement. The quantitative approach relies on the use of synthetic stable isotope labeled peptides or proteins, which precisely mimic their endogenous counterparts and act as internal standards to quantify the corresponding candidate proteins. This report reviews recently developed platform technologies for emerging applications of clinical proteomics and biomarker development.
Keywords: proteomics, absolute quantification, mass spectrometry, biomarker, MRM, MALDI TOF/TOF, AQUA, SISCAPA, stable isotope dilution
Introduction
The emerging technology of mass spectrometry-based quantitative proteomics provides a powerful tool to systematically and quantitatively assess quantitative differences in protein profiles of different samples1, and is increasingly becoming an important component of biomedical and clinical research 2-36. Discovery-based quantitative proteomics compares the proteome of a diseased sample versus normal at a global scale, and has been widely applied to study various human diseases with the goal to identify biomarkers and/or reveal the pathogenesis of diseases. These efforts have led to a significant increase in identification of novel protein candidates associated with a wide assortment of diseases. However, further characterization and validation of the vast majority of these putative biomarkers is extremely challenging because of the enormous complexity of biological systems, heterogeneity of human samples, and lack of universal quantitative technology. In fact, the number of new biomarkers validated in the past 5 years has been remarkably small 37-40. One of the critical gaps in biomarker development has been in the process of evaluating a large number of biomarker candidates to select the few that merit the effort and expense of full validation and pre-clinical testing 40. To relieve the bottleneck to preclinical biomarker assessment it will require a breakthrough in technology to provide a more universal “bridging” methodology -- a way of assessing biomarker performance in a large number of suitably selected samples -- which would be applicable for most of given protein candidates.
Traditionally, the enzyme-linked immunosorbent assay (ELISA) has been the major method used for targeted quantification of a protein, providing good sensitivity and throughput. In the cases where ELISA assays or high quality antibodies already exist, the process of validating a biomarker candidate can be relatively straightforward. Indeed, ELISA remains the “gold standard” for targeted protein quantification to date. However, for many or most novel protein candidates discovered in recent proteomics studies, the ELISA approach is limited by the lack of availability of antibodies with high specificity. The development of a high quality ELISA assay requires a significant investment in time and resources; and the lack of alternative methods for targeted protein quantification has been recognized by the community 41. Recently, the concepts of targeted quantitative proteomics 42,43 and mass spectrometry-based absolute quantification strategies 44-53 have been actively developed and implemented for clinical proteomics applications, and should help circumventing these limitations.
Mass spectrometry-based approaches for targeted protein quantification are based on the concept of isotope dilution mass spectrometry techniques commonly used for the detection of small molecules 54. The approach is comparable to modern advanced antibody-based methods, such as the multi-analyte profiling platform (MAP) 55-57, offering multiplexing detection capability with an excellent dynamic range. While the antibody development is central to the success of an ELISA based platform, mass spectrometry provides alternative and complementary approach, which can precisely detect a wide variety peptides/proteins via high mass accuracy and/or peptide sequencing. This approach provides high selectivity and specificity, and technically avoids most of the problems associated with optimization of multiple assays in a single measurement. In addition, the mass spectrometry based technique has a unique capability to perform candidate-based proteome browsing 42,43,51 and measure absolute levels of post-translational modifications (PTMs) 46,48,51,53. Figure 1 demonstrates the general components of mass spectrometry-based approaches for targeted protein quantification.
Figure 1.
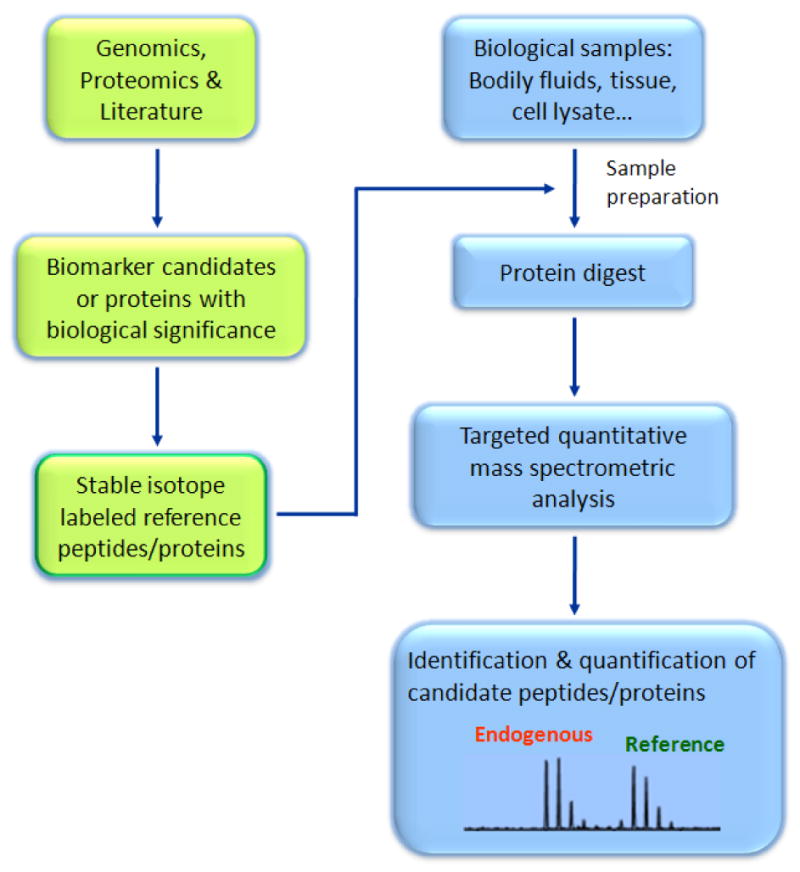
General description of mass spectrometry based targeted quantitative strategy. The approach consists of three major components: generation of stable isotope labeled peptides/proteins as references for the corresponding targeted protein; an effective sample preparation protocol to reduce sample complexity and enrich the targeted surrogates; a data-driven highly specific mass spectrometry platform for candidate-based analysis. More details of each aspect will be discussed in the following sections.
Absolute quantification strategy
The first attempts to determine the absolute amount of a specific protein using stable isotope dilution theory and mass spectrometry were reported more than two decades ago using fast atom bombardment mass spectrometry and deuterium-labeled synthetic peptides as internal standards 58,59. However, it was not until recently, that this approach benefitted from the rapid advances in mass spectrometry instrumentation and proteomics technologies; with these improvements, the mass spectrometry-based absolute quantification techniques are becoming practically available to detect candidate proteins in a complex biological sample with high sensitivity 44-51,53,60. The studies may vary according to the mass spectrometric platforms and sample preparation strategies used, but all rely on the use of synthetic reference peptides, which precisely mimic their endogenous counterparts. The synthetic reference peptides are used as internal standards which are introduced into the test sample at a known concentration in order to quantify the corresponding targeted endogenous peptides. Typically, the synthetic reference peptides are synthesized with an incorporated stable isotope labeling of 13C and 15N on one selected amino acid in the peptide sequence -- so that the reference peptide and corresponding endogenous peptide share the same physicochemical properties, including chromatographic co-elution, ionization efficiency, fragmentation pattern, but can be unambiguously distinguished by mass spectrometry with a defined mass difference. The quantification of an endogenous peptide is based on the intensity ratio of an endogenous peptide (light) to the reference peptide (heavy), which shares the identical sequence and physicochemical properties with its native counterpart, therefore, minimizing the systematic variations due to different sample complexity and analytical conditions.
Gerber et al presented a systematic approach using the strategy of absolute quantification (AQUA) to determine the absolute level of protein and phosphorylated protein expression in whole cell lysates separated by SDS/PAGE 46. An ion trap mass spectrometer coupling with microcapillary liquid chromatography (LC) was used in the study to selectively detect the targeted peptides using the selected reaction monitoring (SRM) mode 46,61. Low abundance yeast proteins involved in gene silencing were quantified. In addition, synthetic peptides with covalent modifications mimicking natural phosphorylation were also prepared to quantify cell cycle-dependent phosphorylation of Ser-1126 of human separase protein, and identify kinases capable of phosphorylating Ser-1501 of separase in an in vitro kinase assay. The study demonstrates the potential and flexibility of using absolute quantification strategy to determine the absolute level of protein expression, as well as quantitatively analyze post-translationally modified proteins in a complex biological system.
Recently, full-length proteins with stable isotope labeling were used as internal standards to determine absolute quantification of biomarkers in urine 62. The study applied the methodology termed Protein Standard Absolute Quantification (PSAQ) to quantify the absolute level of staphylococcal superantigenic toxins SEA and TSST-1in urine and drinking water. A detection sensitivity of picomolar level in water samples and nanomolar level in urine samples was achieved. In a different study, stable isotope-labeled intact protein was used as an internal standard to quantify the absolute level of alcohol dehydrogenase expression in human liver specimens 63.
Mass spectrometric platforms
A variety of mass spectrometric platforms have been used for targeted quantitative proteomics analysis, including the most popular triple quadrupole (triple Q) instrument using the multiple reaction monitoring (MRM) technique 44,53,63-71, matrix assisted laser desorption/ionization time-of-flight tandem mass spectrometer (MALDI TOF/TOF) 51,72, an ion trap instrument using selective ion monitoring (SIM) mode 46,48,73 and the electrospray ionization (ESI) based QTOF mass spectrometer 62,74. The MRM technique, combined with stable isotope-labeled internal standards, has been used in the pharmaceutical industry for decades for the quantitative detection of small molecules including drug metabolites 75, protein degradation products 76, and hormones 70. The discriminating power, maturity of technology and the highly developed automation for data processing have provided great convenience in adapting the methodology for quantitative protein detection. Indeed, many of the recent reports in biomarker verification/validation using mass spectrometry based approach for absolute protein quantification utilized MRM assays. When applying quantitative protein detection with a triple Q instrument using the MRM mode, a precursor ion of interest is first selected in Q1 based on its accurate mass. The pre-selected precursor ion is transmitted and fragmented with collision excitation with a neutral gas in Q2 -- which is used as a collision cell for collision induced dissociation (CID). The resulting fragmented ions of the selected precursor ion are mass analyzed in Q3, and precursor-fragment transitions are used to quantify a peptide. It has been demonstrated that with a modern triple Q mass spectrometer, a significant number of peptide candidates (> 50) can be multiplexed and simultaneously targeted for quantitative detection in plasma in a single measurement 64.
In the past few years, significant technical advances have been made on MALDI based mass spectrometry, including the introduction of tandem instruments, coupling of off-line LC separation/spotting for sample preparation and imaging capability; these improvements greatly enhance the application and performance of MALDI based mass spectrometers in proteomics. For targeted quantitative proteome analysis, the MALDI based tandem mass spectrometer offers several unique features including: 1) the dissociation of LC separation with sample introduction for mass spectrometric analysis, allowing sample preparation and mass spectrometric analysis to be performed in parallel; 2) the decoupling of the MS and MS/MS operations enables a data-driven, selective MS/MS analysis; 3) the non-destructive sample interrogation reserves most or part of samples for repeat analysis; 4) the generation of predominant single-charge ions simplifies data analysis. These unique features allow the LC MALDI based platform to afford greater flexibility for a variety of analytical modes (e.g. discovery mode for global profiling versus browsing mode for targeted analysis), as well as providing great potential for high throughput proteome screening. In the MALDI based approach for targeted analysis, the spiked sample is first separated with LC and the fractions are spotted onto a MALDI plate with a designated format and density to form a “peptide array”. The array is then sequentially surveyed by the MALDI mass spectrometer using a high speed MS mode. The generated MS data is processed and the precursor ions with targeted masses are selectively chosen for MS/MS analysis. This data driven candidate-based process can be performed automatically using an inclusion list which is an available function for most of the modern MALDI tandem instruments. Several applications of MALDI based instruments for targeted quantitative proteomics analysis have recently reported, including targeted analysis and biomarker validation using LC MALDI TOF/TOF 51,72 and LC MALDI triple Q/MRM 77. The application of the LC MALDI based platform for targeted protein quantification is highlighted by its excellent resolution and potential for high throughput, which is particularly appealing for biomarker studies and will be further discussed below.
A different and unique application of MALDI based targeted mass spectrometric analysis is its capability to perform targeted protein or peptide imaging analysis directly on whole tissue samples 78-81. A number of biomarker and clinical studies using MALDI molecular imaging techniques have recently been reported 80,82-87. Although this technique is not directly relevant to absolute protein quantification it provides a different approach for targeted protein detection.
Design and synthesis of signature peptides
The mass spectrometric detection of a targeted peptide relies on the accurate mass and exact sequence of the peptide of interest, thus, when combined with effective sample preparation method, the technique is highly specific and capable of quantifying the corresponding protein. The selection of signature peptides to represent a candidate protein is a critical step in experimental design and assay establishment because it will affect both the specificity and accuracy of a targeted analysis. Mass spectrometric, as well as biological considerations, need to be included in determining whether a tryptic peptide can serve as a signature peptide. The factors to be considered should at least include: the mass and specific sequence of a peptide, the uniqueness of a peptide to the corresponding protein, the physicochemical properties of a peptide, mass spectrometric sensitivity for a given mass spectrometer, known PTMs of the target, and known amino acid variants. It is also important to ensure that the selected peptides do not have flanking sequence and can be robustly proteolyzed by trypsin digestion. Peptides with reactive or labile amino acid residues should be avoided as well. For instance, methionine, cysteine and tryptophan are likely to be oxidized; Asp-Pro and Asp-Gly peptide bonds are unstable; asparagine and glutamine are subjected to deamidation; and N-terminal glutamine can undergo cyclization. Some of the information can be obtained based on in silico digestion of a candidate protein, peptide and protein libraries 88 and the informatic protein databases now available. Other considerations, such as mass spectrometric sensitivity, can only be fully assessed based on empirical data and may be dependent on the specific mass spectrometry platform used. In practice, unique peptides of a targeted protein that have been observed in profiling experiments are good candidates for signature peptides. Recently, the empirical data from four different proteomics platforms has been used to construct a computational model, in which “proteotypic” peptides of a specific protein can be predicted based on their characteristic physicochemical properties for a given platform 89. While stripped peptides (peptides with no PTM) unique to a candidate protein can be used to quantify the absolute level of protein expression, peptides carrying PTMs (e.g. phosphorylation, glycosylation, methylation, acetylation, etc.) may also serve as signature peptides to probe the quantitative state of a PTM associated with a particular biological state or disease setting.
It is possible that direct measurement of target peptides could be used to identify polymorphisms and mutations in proteins of interest. For example, in our study in quantifying protein ceruloplasmin in human cerebrospinal fluid (CSF) the measurement of a polymorphic peptide with an amino acid variant (DIFT*GLIGPMK, T to I) was compared to the measurement of a non-polymorphic ceruloplasmin peptide (ALYLQYTDETFR), and showed significant difference in quantification. If validated, this phenomenon may suggest that peptides with different variants due to gene mutation or polymorphism could potentially be detected and quantified using a similar approach.
Stable isotope labeled reference peptides have traditionally been generated using stepwise chemical synthesis, purified with HPLC and quantified with amino acid analysis. Cell culture-derived stable isotopic labeled peptides have also been used as internal standards for protein quantification 90. Recently, biologically synthesized stable isotope labeled peptides using artificial QconCAT proteins have been introduced 74,91,92. A recent paper provides a comparison of chemically synthesized and biologically generated peptides for quantification and an assessment of the pros and cons of each method 93.
Sample preparation strategies
A robust, highly sensitive assay using mass spectrometry for multiplexing targeted protein quantification involves a variety of different technologies. Sample preparation is considered an integral part of the analytical platform and can significantly influence the overall sensitivity of the analysis for a given mass spectrometer used. A variety of sample preparation strategies have been developed for targeted protein quantification in plasma, serum, or other complex biological samples to deal with the enormous sample complexity and depth of proteome. Depletion of highly abundant proteins is one of the effective and most commonly used strategies to enhance the detection sensitivity of targeted proteins in a complex sample, such as serum or plasma 64-66,69,94-96. In a recent study by Anderson et al, with minimal sample preparation the concentrations of 47 high and medium abundance plasma proteins were quantified in a single experiment, achieving a dynamic range of 4.5 orders of magnitude 64. The depletion of 6 most abundance proteins using immuno-subtraction significantly improved the detection sensitivity and the CVs of the analysis. Notably, the depletion and tryptic digestion process are critically important in the sample processing steps, impacting the accuracy and precision of the overall quantification. Under the same experimental settings, the aforementioned study had demonstrated that depletion and tryptic digestion can be highly reproducible. A systematic comparison of the performance of a variety of different depletion columns has also been reported 97.
The depletion of highly abundant proteins in plasma or serum significantly enhances the analytical dynamic range and detection sensitivity for targeted analysis. However, the dynamic range of proteins in plasma or serum can exceed 10 orders of magnitude 38, limiting an in-depth probe of low abundance protein candidates even after the removal of abundance proteins. To address this issue, a variety of sample preparation techniques have been introduced to enrich and extract the proteins or peptides of interest from a complex background. The method of Stable Isotope Standards with Capture by Anti-Peptide Antibodies (SISCAPA) was developed by Anderson et al using peptide-based antibodies to enrich the targeted low abundance peptides 44. Rabbit anti-peptide antibodies were raised against the candidate peptide sequences and immobilized on nanoaffinity columns to enrich the specific peptides and the spiked synthetic reference peptides. The study demonstrated a 120-fold enrichment of the antigen peptides and a near 5% cycle-to-cycle coefficients of variation. A magnetic-bead based platform using SISCAPA was also developed 96 and applied to verify biomarker candidates in plasma 95. Keshishian et al took a different approach by combining immune-depletion with strong-cation-exchange chromatography (SCX) to enhance the detection sensitivity for low abundant plasma proteins 65. Using this approach they were able to reach a detection limit at high picogram/milliliter level, which is 1000-fold improvement comparing to a direct plasma analysis.
A different approach in simplifying complex biologic samples and enriching for low abundance peptides/proteins is to target a specific sub-proteome, such as glycoproteins or phosphoproteins, for targeted analysis. We introduced a platform combining solid phase glycoprotein capture and MALDI TOF/TOF mass spectrometry to selectively quantify the N-linked glycosylation of a group of candidate glycoproteins in serum 51. The de-glycosylated peptides with N-linked motifs were extracted from serum using a solid phase extraction method based on hydrazide chemistry 98,99-- the same chemistry that has been applied to quantify carbonyl motifs on proteins using spectrophotometric or ELISA assays 100. Recently Stahl-Zeng applied a similar approach to quantify N-linked glycopeptides in plasma and achieved a detection sensitivity at low nanogram/milliliter. These studies demonstrated an integrated pipeline for candidate-based glycoproteomics analysis with precise mapping of targeted N-linked motifs and absolute quantification of the glycoprotein targets. Other approaches targeting subgroup of protein or peptides for analysis include the combined fractional diagonal chromatography (COFRADIC) 101,102, which enriches N-terminal peptides for analysis. Figure 2 illustrates some of the sample preparation strategies and their overall detection limits for targeted analysis in plasma or serum. It is remarkable that for each sample preparation method the detection sensitivity for a specific targeted protein in plasma or serum also depends on the nature abundant of the protein, the proteotypic characteristics of the signature peptides and the specific analytical platform used.
Figure 2.
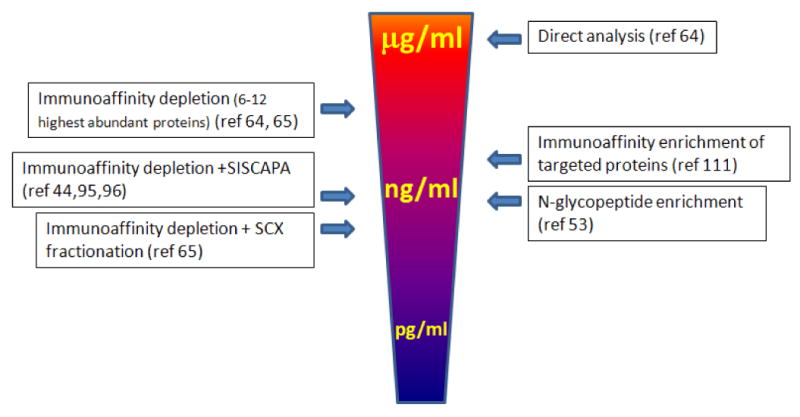
A summary of some sample preparation strategies and their overall detection sensitivity for targeted protein analysis in plasma or serum.
Potential for high throughput
For the application of biomarker verification and validation -- in which absolute quantification of a single protein or a panel of candidate proteins in a large cohort of individual samples is required -- a high throughput and multiplexing analysis is essential. While most of the recent efforts to improve targeted quantitative analysis have been focusing on the quantitative aspects of the methodology and development of sample preparation strategies, attention has also been paid to address issues related to analytical throughput. The analytical throughput can be interpreted as 1) the number of analytes that can be analyzed in a single measurement, and 2) the number of samples that can be analyzed in a defined period of time. The multiplex capability of mass spectrometry based platforms to quantitatively detect multiple candidate peptides in a complex sample is substantial and has been well demonstrated in different sample types and with different mass spectrometric protocols. It is practically feasible that more than 100 candidates could be simultaneously detected with absolute quantification in a single mass spectrometric measurement. However, the duration of each of such analysis is realistically limited by the time required for the chromatographic separation of the peptides prior to mass spectrometric analysis. For the ESI based MRM platform, a short LC gradient (75 minute total time) has been successfully used to analyze more than 50 candidate peptides in depleted plasma without sacrificing the required multiple measurements across each peak 64. In addition, it has been demonstrated that time scheduling acquisition can significantly expand the number of MRM measurements in a single LC/MS run 53,60.
The LC MALDI based platform takes a different approach in operation for targeted analysis, in which the LC separation and the mass spectrometric analysis is dissociated; in addition, within the mass spectrometric analysis, the MS and MS/MS analysis can be operated separately, providing flexibility to perform the MS/MS selectively on targeted peptides. This in turn could save significant time for high throughput analysis. For MALDI based instruments, because all the peptides are separated and deposited on a MALDI plate prior to mass spectrometric interrogation, in-situ acquisition of eluting peptides from LC is no longer required. Thus, utilization of a high throughput LC protocol that can provide adequate power of separation for a particular sample complexity is feasible and highly desirable. One important consideration for optimizing a LC protocol for MALDI spotting is to enhance the separation and resolution, so that the majority of the peptides can be separated and eluted in a minimal number of spots on the MALDI plate. Such optimization provides advantages both qualitatively and quantitatively for MALDI mass spectrometric analysis. It can also be used as a criterion to evaluate the efficiency of a LC protocol by categorizing the precursor ions detected based on the number of spots that a precursor ion eluted across. Figure 3 shows the distribution of precursor ions based on the number of consecutive spots that a precursor ion eluted across to compare the performance of a high throughput LC protocol (28 minutes) with a conventional LC protocol (100 minutes) in separation of the same de-glycosylated N-linked peptide mixture derived from human serum. The overall performance of the two protocols was similar although the high throughput protocol showed slightly fewer peptides eluted in single spot. In both cases, about 80% of the precursor ions were eluted within 3 consecutive spots; and the frequency of precursors presented in each category was comparable, showing a similar trend as the number of eluting spots increased. The high throughput potential of the LC MALDI TOF/TOF platform is better exemplified when dealing with a large number of samples for targeted quantitative analysis, in that sample separation and mass spectrometric analysis can be performed in parallel.
Figure 3.
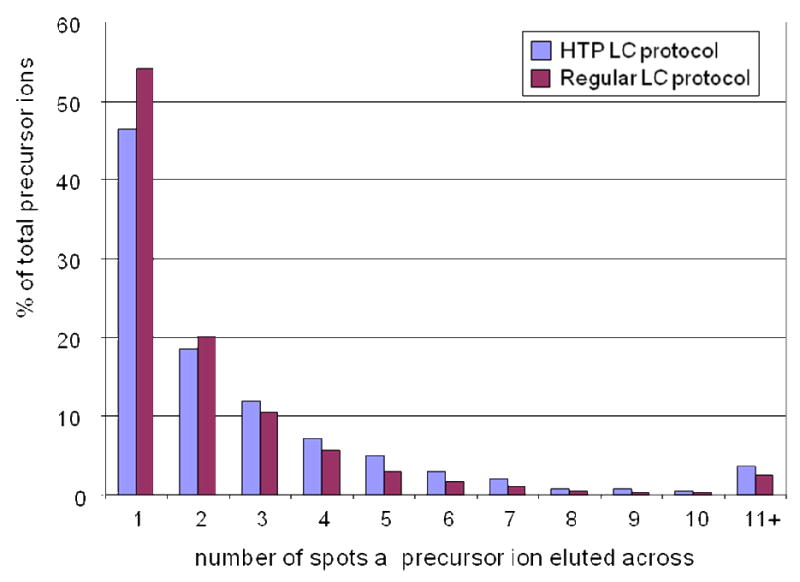
The evaluation of the performance of a high throughput LC protocol for a LC MADLI TOF/TOF based platform. The precursor ions detected were categorized based on the number of consecutive spots that a precursor ion was eluted across. The high throughput LC protocol showed similar behavior with the conventional protocol in terms of the distribution of the precursor ions.
Quantification of PTMs
PTMs play pivotal roles in many of biological processes because it is the chemical modifications of key regulatory or structural proteins that dictates the activation state for most cellular physiological events 103. Many protein activities are modulated by PTMs. Quantification of PTMs status can provide better understanding of disease mechanisms and facilitate the discovery of molecular markers that are invaluable for their potential discriminatory power in molecular classification of disease, which in turn can predict clinical outcome and response to drugs.
Phosphorylation plays an important role in normal physiological states as well as aberrant signaling pathways in cancer and other diseases. For example, the phosphorylation status of protein kinase B/Akt is an important biomarker in predicting the response of gliomas to tyrosine kinase inhibitors 103. With the recent development of mass spectrometry based quantitative proteomics technology, precise and absolute quantification of a particular protein phosphorylation and related kinases is becoming available. Gerber et al reported the first AQUA study of phosphorylation, in which reference peptides were synthesized to mimic natural tryptic peptides and phosphopeptides containing a targeted phosphorylation site and then used as internal standards for absolute phosphorylation quantification 46. The study quantitatively determined the dynamic changes of the cell cycle-dependent phosphorylation of Ser-1126 of the human separase protein and identified site-specific phosphorylation of separase at Ser-1501 in response to an in vitro kinase assay. In a different study by Mayya et al, the same AQUA strategy was applied to quantify multi-site phosphorylation status of the cyclin-dependent kinases (CDKs), which are important regulators of cell cycle transitions and apoptosis 48. The quantification of the four possible phosphorylated and non-phosphorylated versions of CDKs (T14p-Y15p, T14p-Y15, T14-Y15p, and T14-Y15) revealed the quantitative dynamics of the status of the CDK phosphorylation during cell transition and apoptosis. The same approach could potentially be extended to quantify the status of other PTMs, such as methylation and acetylation, using synthetic peptides with covalent modifications mimicking natural PTMs.
Glycosylation is another important common form of PTMs. Most secreted and membrane-bound proteins produced by mammalian cells contain covalently linked sugar chains. Abnormal glycosylation of proteins is often involved in tumor progression and metastasis 104-106. This could explain why many of the blood-based tumor markers are glycoproteins. Thus, the development of a strategy that allows multiplexing absolute quantification of targeted glycoproteins may be especially useful for biomarker verification and validation. However, the idea of using synthetic peptides to mimic natural peptides with a specific glycan to quantify the status of a glycosylation site may not be technically practical because of the structure and complexity of the sugar chain. A different approach combining the glycoprotein capturing technique and a targeted mass spectrometric platform was reported to quantify selected N-linked glycosylation peptides derived from a group of glycoproteins in serum 51 and plasma 53. In the first study, the N-linked glycopeptides were extracted from serum using a solid phase extraction method based on hydrazide chemistry 98 and de-glycosylated with PNGase F enzyme. The de-glycosylation resulted in a conversion of asparagine to aspartic acid in the peptide sequence, introducing a mass difference of 0.984 Da. This phenomenon was utilized to design a synthetic peptide to mimic the endogenous N-linked glycopeptide in its de-glycosylation form. The study demonstrated an integrated, highly specific platform for quantitative analysis of specific N-linked glycopeptides derived from a selected group of serum glycoproteins using LC MALDI TOF/TOF. Figure 4 shows the detection of 6 reference peptides as well as the corresponding endogenous de-glycosylated N-linked peptides derived from serum glycoproteins, demonstrating the effectiveness of detecting selected N-linked glycoproteins in human serum using the high throughput platform. Notably, a protein candidate, lumican, which was up-regulated in pancreatic cancer tissue 107, was found detectable in serum using the platform. Recently, Stahl-Zeng et al applied a similar approach to quantify a selected group of N-linked glycopeptides in plasma using Triple Q/linear ion trap instrument with MRM technique 53. The study demonstrated a nanogram/milliliter detection sensitivity and a dynamic range of accurate quantification over 5 orders of magnitude in detection of targeted glycoproteins in plasma. It is worthy to note that glycan alternations of glycoproteins have long been associated with cancer and been widely studied. The study of glycans itself is a very important field, and technically beyond the coverage and discussion of this report.
Figure 4.
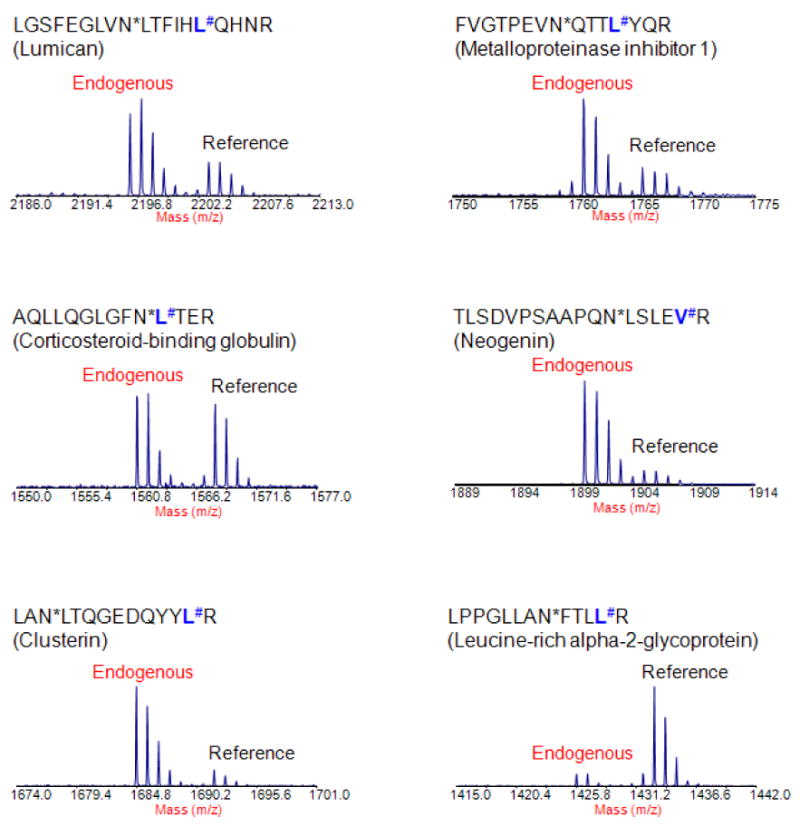
The detection of a subset of candidate N-linked glycopeptides in serum using an LC MALDI TOF/TOF based platform for targeted quantitative analysis. The targeted endogenous peptides were unambiguously identified within a complex background and can be quantified using the corresponding synthetic reference peptides. (Note: # indicates the amino acid that was stable isotope labeled (13C and 15N) in reference peptides; * indicates enzyme-catalyzed conversion of asparagine to aspartic acid at the site of carbohydrate attachment.)
Clinical applications
One of the main goals of developing mass spectrometry based absolute quantification strategies is for its clinical applications. Targeted analysis has been used to quantify candidate proteins in plasma or serum, including major plasma proteins 64, intermediate abundance proteins in serum 94, low abundance proteins in plasma 65, N-linked glycoproteins in serum 51 and plasma 53. Biomarkers and putative marker proteins associated with a variety of diseases have also been detected in different bodily fluids using mass spectrometry based quantitative methods. A list of 177 protein candidates associated with cardiovascular diseases and stroke has been proposed for targeted analysis in plasma 108. Barnidge et al demonstrated the feasibility of absolute quantification of Prostate-Specific Antigen, a biomarker for prostate cancer, as a model biomarker in serum 109. Kuhn et al quantified C-reactive protein (CRP), a diagnostic marker of rheumatoid arthritis (RA), in serum samples taken from patients with either erosive or nonerosive RA and compared the results to healthy individuals 69. More recently, Whiteaker et al applied SISCAPA and mass spectrometry to confirm breast cancer biomarkers using a mouse model 95. The quantitative study verified osteopontin and fibulin-2 in plasma as circulating biomarkers for breast cancer, and further suggested osteopontin could be used for early disease detection in mouse model. Staphylococcus aureus superantigenic toxins are the causes of several human diseases, including the highly lethal staphylococcal toxic shock syndrome 110. Currently, there is not a specific immunological assay or diagnostic test available for staphylococcal toxic shock syndrome because of the structure and sequence similarity of a variety of staphylococcal enterotoxins 62. Brun et al presented a highly specific, mass spectrometry based methodology to quantitatively detect the staphylococcal superantigenic toxins SEA and TSST-1 at a picomolar level in water and nanomolar level in urine for absolute quantification 62. Kirsch et al reported a study, in which two biomarkers (IGF-1 and IGFBP-3) of growth hormone abuse were simultaneously detected in serum 68. In oncology, there is considerable inter-patient variability in toxicity and response to chemotherapy and this can significantly impact the outcomes of treatment and patient management. Currently, there is a lack of clinical assays that can prospectively identify the patients at risk for specific chemotherapy treatments. McKay et al presented an empirically driven mass spectrometry method to simultaneously monitor a panel of 18 liver-derived proteins in plasma samples from common colorectal cancer (CRC) patients who were undergoing chemotherapy 66. Their study suggested that a targeted quantitative mass spectrometry method can be optimized to produce assays for diagnosis or prognosis, especially when multiplexing detection is required.
More recently, we evaluated a subset of proteomics identified biomarker candidates of neurodegenerative diseases, including Alzheimer's disease and Parkinson's disease, in human CSF using an LC MALDI TOF/TOF platform 72. Lange et al investigated the quantitative behavior of low abundant virulence factors from cultures of the human pathogen Streptococcus pyogenes exposed to increasing amounts of plasma 60. Nicol et al quantitatively assessed the level of a known biomarker -- carcinoembryonic antigen, and a subset of putative biomarker candidates in sera samples from lung cancer patients 111. In addition to the studies on biomarker detection in bodily fluids, applications have been reported using other clinical samples, such as liver tissue specimens 63. Using a stable isotope-labeled intact protein as an internal standard, Janecki et al precisely quantified the absolute level of an ADH isoenzyme, ADH1C1, in human liver tissue 63. Some of the recent studies using mass spectrometry based targeted quantification strategies are summarized in Table 1.
Table 1.
Summary of some recent studies using mass spectrometry-based targeted protein quantification
| Specimen | Quantitative study | Sample preparation | MS platform | Reference |
|---|---|---|---|---|
| plasma | Major human plasma proteins | Immunodepletion | LC ESI Triple Q / MRM | 64 |
| Low abundance human plasma proteins | Immunodepletion+SCX | LC ESI Triple Q / MRM | 65 | |
| Selected N-glycoproteins in human plasma | N-Glycopeptide enrichment | LC ESI Triple Q / MRM | 53 | |
| Liver-derived proteins in human plasma | Immunodepletion | LC ESI Triple Q / MRM | 66 | |
| Biomarker candidates in mouse model of breast cancer | Immunodepletion+SISCAPA | LC ESI Triple Q / MRM | 95 | |
| Serum | Selected N-glycoproteins in human serum | N-Glycopeptide enrichment | LC MALDI TOF/TOF | 51 |
| Protein markers of lung cancer in human serum | Immunoprecipitation | LC ESI Triple Q / MRM | 111 | |
| Growth hormone biomarkers in human serum | LC ESI Triple Q / MRM | 68 | ||
| C-reactive protein for rheumatoid arthritis in human serum | Immunodepletion+size exclusion chromatography (SEC) | LC ESI Triple Q / MRM | 69 | |
| Intermediate abundance proteins in human serum | Immunodepletion | LC ESI single Q | 94 | |
| Prostate-Specific Antigen in human serum | SCX | LC ESI Triple Q / SRM | 109 | |
| CSF | Biomarker candidates of Alzheimer's disease and Parkinson's disease | SCX | LC MALDI TOF/TOF | 72 |
| Urine | Staphylo coccus aureus superantigenic toxins | 1D gel | LC ESI Q/TOF | 62 |
| Tissue | Biomarker candidates in mouse model of breast cancer | LC ESI Triple Q / MRM | 95 | |
| Human liver alcohol dehydrogenase ADH1C1 isoenzyme | LC ESI Triple Q / MRM | 63 | ||
| Selected proteins of Postsynaptic Density from rat brain | 1D gel | LC ESI ion trap / SRM | 73 | |
| Cell lysate | Yeast proteins involved in gene silencing, cell cycle-dependent phosphorylation of Ser-1126 of human separase protein | 1D gel | LC ESI ion trap / SRM | 46 |
| Cyclin-dependent kinases | Immunoprecipitation | LC ESI ion trap / SRM | 48 | |
| virulence factors of the gram-positive bacterium Streptococcus pyogenes | Isoelectric focusing | LC ESI Triple Q / MRM | 60 | |
| Biomarker of Candida albicans | SCX | LCMALDI Triple Q / MRM | 77 |
Concluding remarks
The recent development of mass spectrometry based strategies for absolute protein quantification opens new avenues for a broad range of applications from clinical diagnosis and prognosis to drug development and personalized medicine. Complementary to the other existing techniques, the major breakthrough of this technology is highlighted in its capability of providing a universal approach for developing assays of quantification for a wide spectrum of proteins with minimum restrictions, and the ease of assembling multiplex detection in a single measurement. It is conceptually feasible that these new strategies could be applied to design personalized medicine and treatments in the future. Biomarkers from single or multiple diseases can also be assembled into a panel providing multiplex diagnosis in a single assay. The development of absolute quantification techniques should also help researchers with data standardization and validation of biomarker candidates across different proteomics platforms and laboratories, a major challenge for current proteomics studies.
While the prospects of this technology are exciting and promising, it is a long journey to develop a robust, high throughput multi-functional system that can provide sophisticated function that we desperately need in clinical diagnosis and therapy. Most of the current mass spectrometry based platform technologies require a combination of multiple sample preparations, such as immuno-subtraction, multidimensional LC separation, immunoaffinity and/or solid phase extraction to enhance the analytical dynamic range and detection sensitivity. These requirements limit the capability for high throughput. Thus, the major caveat of the current mass spectrometry based approaches for targeted analysis is the lack of a high throughput pipeline integrating a highly specific and, ideally, single-step sample preparation strategy. Nevertheless, the development of mass spectrometry based techniques has already provided a complementary platform, in which a surging number of novel protein biomarker candidates for a variety of different diseases can potentially be verified and validated before a significant effort of time and resources is invested for assay development and pre-clinical testing. As a technology that is quickly evolving it is anticipated that many of the current technical limitations may be transient. The development of a quantitative detection technology that provides high specificity and sensitivity, multiplexing, absolute quantification and PTM monitoring should provide us a powerful tool for a broad range of applications and greatly enhance our effort in developing better medicine.
Acknowledgments
This work was supported in part with federal funds from National Institutes of Health (R21NS060252, R01CA107209, K07CA116296, R01AG025327, R01ES012703, NO1-HV-28179), a grant from Canary Foundation, American Association for Cancer Research (AACR) / PanCAN career development award to RC, and a grant from the Swiss national Science Foundation to RA (Grant No. 31000-10767). The authors thank the proteomics center at Institute for Systems Biology for the supports.
References
- 1.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422(6928):198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 2.Ali M, Manolios N. Proteomics in rheumatology: a new direction for old diseases. Semin Arthritis Rheum. 2005;35(2):67–76. doi: 10.1016/j.semarthrit.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Alpantaki K, Tsiridis E, Pape HC, Giannoudis PV. Application of clinical proteomics in diagnosis and management of trauma patients. Injury. 2007;38(3):263–271. doi: 10.1016/j.injury.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 4.Ardekani AM, Liotta LA, Petricoin EF., III Clinical potential of proteomics in the diagnosis of ovarian cancer. Expert Rev Mol Diagn. 2002;2(4):312–320. doi: 10.1586/14737159.2.4.312. [DOI] [PubMed] [Google Scholar]
- 5.Bertucci F, Birnbaum D, Goncalves A. Proteomics of breast cancer: principles and potential clinical applications. Mol Cell Proteomics. 2006;5(10):1772–1786. doi: 10.1074/mcp.R600011-MCP200. [DOI] [PubMed] [Google Scholar]
- 6.Bowler RP, Ellison MC, Reisdorph N. Proteomics in pulmonary medicine. Chest. 2006;130(2):567–574. doi: 10.1378/chest.130.2.567. [DOI] [PubMed] [Google Scholar]
- 7.Caron M, Joubert-Caron R. Proteomics in hematologic malignancies. Expert Rev Proteomics. 2005;2(4):567–576. doi: 10.1586/14789450.2.4.567. [DOI] [PubMed] [Google Scholar]
- 8.Cho WC. Application of proteomics in Chinese medicine research. Am J Chin Med. 2007;35(6):911–922. doi: 10.1142/S0192415X07005375. [DOI] [PubMed] [Google Scholar]
- 9.Conrad DH, Goyette J, Thomas PS. Proteomics as a method for early detection of cancer: a review of proteomics, exhaled breath condensate, and lung cancer screening. J Gen Intern Med. 2008;23 Suppl 1:78–84. doi: 10.1007/s11606-007-0411-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Czibere A, Grall F, Aivado M. Perspectives of proteomics in acute myeloid leukemia. Expert Rev Anticancer Ther. 2006;6(11):1663–1675. doi: 10.1586/14737140.6.11.1663. [DOI] [PubMed] [Google Scholar]
- 11.de Noo ME, Tollenaar RA, Deelder AM, Bouwman LH. Current status and prospects of clinical proteomics studies on detection of colorectal cancer: hopes and fears. World J Gastroenterol. 2006;12(41):6594–6601. doi: 10.3748/wjg.v12.i41.6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Devarajan P. Proteomics for biomarker discovery in acute kidney injury. Semin Nephrol. 2007;27(6):637–651. doi: 10.1016/j.semnephrol.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dhamoon AS, Kohn EC, Azad NS. The ongoing evolution of proteomics in malignancy. Drug Discov Today. 2007;12(1718):700–708. doi: 10.1016/j.drudis.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 14.Drake RR, Cazare LH, Semmes OJ, Wadsworth JT. Serum, salivary and tissue proteomics for discovery of biomarkers for head and neck cancers. Expert Rev Mol Diagn. 2005;5(1):93–100. doi: 10.1586/14737159.5.1.93. [DOI] [PubMed] [Google Scholar]
- 15.Feng JT, Shang S, Beretta L. Proteomics for the early detection and treatment of hepatocellular carcinoma. Oncogene. 2006;25(27):3810–3817. doi: 10.1038/sj.onc.1209551. [DOI] [PubMed] [Google Scholar]
- 16.Gonzalez-Buitrago JM, Ferreira L, Lorenzo I. Urinary proteomics. Clin Chim Acta. 2007;375(12):49–56. doi: 10.1016/j.cca.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 17.Hanash S, Brichory F, Beer D. A proteomic approach to the identification of lung cancer markers. Dis Markers. 2001;17(4):295–300. doi: 10.1155/2001/657605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hunnerkopf R, Grassl J, Thome J. Proteomics: biomarker research in psychiatry. Fortschr Neurol Psychiatr. 2007;75(10):579–586. doi: 10.1055/s-2007-959249. [DOI] [PubMed] [Google Scholar]
- 19.Hutter G, Sinha P. Proteomics for studying cancer cells and the development of chemoresistance. Proteomics. 2001;1(10):1233–1248. doi: 10.1002/1615-9861(200110)1:10<1233::AID-PROT1233>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 20.Johnson MD, Floyd JL, Caprioli RM. Proteomics in diagnostic neuropathology. J Neuropathol Exp Neurol. 2006;65(9):837–845. doi: 10.1097/01.jnen.0000235116.67558.24. [DOI] [PubMed] [Google Scholar]
- 21.Liu BC, Ehrlich JR. Proteomics approaches to urologic diseases. Expert Rev Proteomics. 2006;3(3):283–296. doi: 10.1586/14789450.3.3.283. [DOI] [PubMed] [Google Scholar]
- 22.Magi B, Bargagli E, Bini L, Rottoli P. Proteome analysis of bronchoalveolar lavage in lung diseases. Proteomics. 2006;6(23):6354–6369. doi: 10.1002/pmic.200600303. [DOI] [PubMed] [Google Scholar]
- 23.Matharoo-Ball B, Ball G, Rees R. Clinical proteomics: discovery of cancer biomarkers using mass spectrometry and bioinformatics approaches--a prostate cancer perspective. Vaccine. 2007;25 Suppl 2:B110–B121. doi: 10.1016/j.vaccine.2007.06.040. [DOI] [PubMed] [Google Scholar]
- 24.Matt P, Carrel T, White M, Lefkovits I, Van Eyk J. Proteomics in cardiovascular surgery. J Thorac Cardiovasc Surg. 2007;133(1):210–214. doi: 10.1016/j.jtcvs.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 25.Mayr M, Zhang J, Greene AS, Gutterman D, Perloff J, Ping P. Proteomics-based development of biomarkers in cardiovascular disease: mechanistic, clinical, and therapeutic insights. Mol Cell Proteomics. 2006;5(10):1853–1864. doi: 10.1074/mcp.R600007-MCP200. [DOI] [PubMed] [Google Scholar]
- 26.Shoshan SH, Admon A. Proteomics in cancer vaccine development. Expert Rev Proteomics. 2005;2(2):229–241. doi: 10.1586/14789450.2.2.229. [DOI] [PubMed] [Google Scholar]
- 27.Shoshan SH, Admon A. Proteomics in clinical laboratory diagnosis. Adv Clin Chem. 2005;39:159–184. doi: 10.1016/s0065-2423(04)39006-2. [DOI] [PubMed] [Google Scholar]
- 28.Unwin RD, Whetton AD. How will haematologists use proteomics? Blood Rev. 2007;21(6):315–326. doi: 10.1016/j.blre.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 29.Vidal BC, Bonventre JV, Hong Hsu S. Towards the application of proteomics in renal disease diagnosis. Clin Sci (Lond) 2005;109(5):421–430. doi: 10.1042/CS20050085. [DOI] [PubMed] [Google Scholar]
- 30.Wang KK, Ottens AK, Liu MC, Lewis SB, Meegan C, Oli MW, Tortella FC, Hayes RL. Proteomic identification of biomarkers of traumatic brain injury. Expert Rev Proteomics. 2005;2(4):603–614. doi: 10.1586/14789450.2.4.603. [DOI] [PubMed] [Google Scholar]
- 31.Yarbrough WG, Slebos RJ, Liebler D. Proteomics: clinical applications for head and neck squamous cell carcinoma. Head Neck. 2006;28(6):549–558. doi: 10.1002/hed.20357. [DOI] [PubMed] [Google Scholar]
- 32.Yim EK, Park JS. Role of proteomics in translational research in cervical cancer. Expert Rev Proteomics. 2006;3(1):21–36. doi: 10.1586/14789450.3.1.21. [DOI] [PubMed] [Google Scholar]
- 33.Zhang J, Goodlett DR, Montine TJ. Proteomic biomarker discovery in cerebrospinal fluid for neurodegenerative diseases. J Alzheimers Dis. 2005;8(4):377–386. doi: 10.3233/jad-2005-8407. [DOI] [PubMed] [Google Scholar]
- 34.Chen R, Pan S, Brentnall TA, Aebersold R. Proteomic profiling of pancreatic cancer for biomarker discovery. Mol Cell Proteomics. 2005;4(4):523–533. doi: 10.1074/mcp.R500004-MCP200. [DOI] [PubMed] [Google Scholar]
- 35.Chen R, Pan S, Aebersold R, Brentnall TA. Proteomics studies of pancreatic cancer. Proteomics-Clin Appl. 2007;1:1582–1591. doi: 10.1002/prca.200700414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Faca VM, Song KS, Wang H, Zhang Q, Krasnoselsky AL, Newcomb LF, Plentz RR, Gurumurthy S, Redston MS, Pitteri SJ, Pereira-Faca SR, Ireton RC, Katayama H, Glukhova V, Phanstiel D, Brenner DE, Anderson MA, Misek D, Scholler N, Urban ND, Barnett MJ, Edelstein C, Goodman GE, Thornquist MD, McIntosh MW, DePinho RA, Bardeesy N, Hanash SM. A mouse to human search for plasma proteome changes associated with pancreatic tumor development. PLoS Med. 2008;5(6):e123. doi: 10.1371/journal.pmed.0050123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aebersold R, Anderson L, Caprioli R, Druker B, Hartwell L, Smith R. Perspective: a program to improve protein biomarker discovery for cancer. J Proteome Res. 2005;4(4):1104–1109. doi: 10.1021/pr050027n. [DOI] [PubMed] [Google Scholar]
- 38.Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics. 2002;1(11):845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 39.Gutman S, Kessler LG. The US Food and Drug Administration perspective on cancer biomarker development. Nat Rev Cancer. 2006;6(7):565–571. doi: 10.1038/nrc1911. [DOI] [PubMed] [Google Scholar]
- 40.Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol. 2006;24(8):971–983. doi: 10.1038/nbt1235. [DOI] [PubMed] [Google Scholar]
- 41.Haab BB, Paulovich AG, Anderson NL, Clark AM, Downing GJ, Hermjakob H, Labaer J, Uhlen M. A reagent resource to identify proteins and peptides of interest for the cancer community: a workshop report. Mol Cell Proteomics. 2006;5(10):1996–2007. doi: 10.1074/mcp.T600020-MCP200. [DOI] [PubMed] [Google Scholar]
- 42.Aebersold R. Constellations in a cellular universe. Nature. 2003;422(6928):115–116. doi: 10.1038/422115a. [DOI] [PubMed] [Google Scholar]
- 43.Kuster B, Schirle M, Mallick P, Aebersold R. Scoring proteomes with proteotypic peptide probes. Nat Rev Mol Cell Biol. 2005;6(7):577–583. doi: 10.1038/nrm1683. [DOI] [PubMed] [Google Scholar]
- 44.Anderson NL, Anderson NG, Haines LR, Hardie DB, Olafson RW, Pearson TW. Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA) J Proteome Res. 2004;3(2):235–244. doi: 10.1021/pr034086h. [DOI] [PubMed] [Google Scholar]
- 45.Bronstrup M. Absolute quantification strategies in proteomics based on mass spectrometry. Expert Rev Proteomics. 2004;1(4):503–512. doi: 10.1586/14789450.1.4.503. [DOI] [PubMed] [Google Scholar]
- 46.Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc Natl Acad Sci USA. 2003;100(12):6940–6945. doi: 10.1073/pnas.0832254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kirkpatrick DS, Gerber SA, Gygi SP. The absolute quantification strategy: a general procedure for the quantification of proteins and post-translational modifications. Methods. 2005;35(3):265–273. doi: 10.1016/j.ymeth.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 48.Mayya V, Rezual K, Wu L, Fong MB, Han DK. Absolute quantification of multisite phosphorylation by selective reaction monitoring mass spectrometry: determination of inhibitory phosphorylation status of cyclin-dependent kinases. Mol Cell Proteomics. 2006;5(6):1146–1157. doi: 10.1074/mcp.T500029-MCP200. [DOI] [PubMed] [Google Scholar]
- 49.Putz S, Reinders J, Reinders Y, Sickmann A. Mass spectrometry-based peptide quantification: applications and limitations. Expert Rev Proteomics. 2005;2(3):381–392. doi: 10.1586/14789450.2.3.381. [DOI] [PubMed] [Google Scholar]
- 50.Ackermann BL, Berna MJ. Coupling immunoaffinity techniques with MS for quantitative analysis of low-abundance protein biomarkers. Expert Rev Proteomics. 2007;4(2):175–186. doi: 10.1586/14789450.4.2.175. [DOI] [PubMed] [Google Scholar]
- 51.Pan S, Zhang H, Rush J, Eng J, Zhang N, Patterson D, Comb MJ, Aebersold R. High throughput proteome screening for biomarker detection. Mol Cell Proteomics. 2005;4(2):182–190. doi: 10.1074/mcp.M400161-MCP200. [DOI] [PubMed] [Google Scholar]
- 52.Domon B, Aebersold R. Mass spectrometry and protein analysis. Science. 2006;312(5771):212–217. doi: 10.1126/science.1124619. [DOI] [PubMed] [Google Scholar]
- 53.Stahl-Zeng J, Lange V, Ossola R, Eckhardt K, Krek W, Aebersold R, Domon B. High sensitivity detection of plasma proteins by multiple reaction monitoring of N-glycosites. Mol Cell Proteomics. 2007;6(10):1809–1817. doi: 10.1074/mcp.M700132-MCP200. [DOI] [PubMed] [Google Scholar]
- 54.Bowers GN, Jr, Fassett JD, White E. Isotope dilution mass spectrometry and the National Reference System. Anal Chem. 1993;65(12):475R–479R. doi: 10.1021/ac00060a620. [DOI] [PubMed] [Google Scholar]
- 55.Pang S, Smith J, Onley D, Reeve J, Walker M, Foy C. A comparability study of the emerging protein array platforms with established ELISA procedures. J Immunol Methods. 2005;302(12):1–12. doi: 10.1016/j.jim.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 56.Schwenk JM, Lindberg J, Sundberg M, Uhlen M, Nilsson P. Determination of binding specificities in highly multiplexed bead-based assays for antibody proteomics. Mol Cell Proteomics. 2007;6(1):125–132. doi: 10.1074/mcp.T600035-MCP200. [DOI] [PubMed] [Google Scholar]
- 57.Zhang J, Sokal I, Peskind ER, Quinn JF, Jankovic J, Kenney C, Chung KA, Millard SP, Nutt JG, Montine TJ. CSF multianalyte profile distinguishes Alzheimer and Parkinson diseases. Am J Clin Pathol. 2008;129(4):526–529. doi: 10.1309/W01Y0B808EMEH12L. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barr JR, Maggio VL, Patterson DG, Jr, Cooper GR, Henderson LO, Turner WE, Smith SJ, Hannon WH, Needham LL, Sampson EJ. Isotope dilution--mass spectrometric quantification of specific proteins: model application with apolipoprotein A-I. Clin Chem. 1996;42(10):1676–1682. [PubMed] [Google Scholar]
- 59.Desiderio DM, Kai M, Tanzer FS, Trimble J, Wakelyn C. Measurement of enkephalin peptides in canine brain regions, teeth, and cerebrospinal fluid with high-performance liquid chromatography and mass spectrometry. J Chromatogr. 1984;297:245–260. doi: 10.1016/s0021-9673(01)89046-4. [DOI] [PubMed] [Google Scholar]
- 60.Lange V, Malmström JA, Didion J, King NL, Johansson BP, Schäfer J, Rameseder J, Wong CH, Deutsch EW, Brusniak MY, Bühlmann P, Björck L, Domon B, Aebersold R. Targeted quantitative analysis of Streptococcus pyogenes virulence factors by multiple reaction monitoring. Mol Cell Proteomics. 2008 doi: 10.1074/mcp.M800032-MCP200. on-line published. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gerber SA, Kettenbach AN, Rush J, Gygi SP. The absolute quantification strategy: application to phosphorylation profiling of human separase serine 1126. Methods Mol Biol. 2007;359:71–86. doi: 10.1007/978-1-59745-255-7_5. [DOI] [PubMed] [Google Scholar]
- 62.Brun V, Dupuis A, Adrait A, Marcellin M, Thomas D, Court M, Vandenesch F, Garin J. Isotope-labeled protein standards: toward absolute quantitative proteomics. Mol Cell Proteomics. 2007;6(12):2139–2149. doi: 10.1074/mcp.M700163-MCP200. [DOI] [PubMed] [Google Scholar]
- 63.Janecki DJ, Bemis KG, Tegeler TJ, Sanghani PC, Zhai L, Hurley TD, Bosron WF, Wang M. A multiple reaction monitoring method for absolute quantification of the human liver alcohol dehydrogenase ADH1C1 isoenzyme. Anal Biochem. 2007;369(1):18–26. doi: 10.1016/j.ab.2007.06.043. [DOI] [PubMed] [Google Scholar]
- 64.Anderson L, Hunter CL. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol Cell Proteomics. 2006;5(4):573–588. doi: 10.1074/mcp.M500331-MCP200. [DOI] [PubMed] [Google Scholar]
- 65.Keshishian H, Addona T, Burgess M, Kuhn E, Carr SA. Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol Cell Proteomics. 2007;6(12):2212–2229. doi: 10.1074/mcp.M700354-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McKay MJ, Sherman J, Laver MT, Baker MS, Clarke SJ, Molloy MP. The development of multiple reaction monitoring assays for liver-derived plasma proteins. Proteomics-Clin Appl. 2007;1:1570–1581. doi: 10.1002/prca.200700305. [DOI] [PubMed] [Google Scholar]
- 67.Unwin RD, Griffiths JR, Leverentz MK, Grallert A, Hagan IM, Whetton AD. Multiple reaction monitoring to identify sites of protein phosphorylation with high sensitivity. Mol Cell Proteomics. 2005;4(8):1134–1144. doi: 10.1074/mcp.M500113-MCP200. [DOI] [PubMed] [Google Scholar]
- 68.Kirsch S, Widart J, Louette J, Focant JF, De Pauw E. Development of an absolute quantification method targeting growth hormone biomarkers using liquid chromatography coupled to isotope dilution mass spectrometry. J Chromatogr A. 2007;1153(12):300–306. doi: 10.1016/j.chroma.2007.03.058. [DOI] [PubMed] [Google Scholar]
- 69.Kuhn E, Wu J, Karl J, Liao H, Zolg W, Guild B. Quantification of C-reactive protein in the serum of patients with rheumatoid arthritis using multiple reaction monitoring mass spectrometry and 13C-labeled peptide standards. Proteomics. 2004;4(4):1175–1186. doi: 10.1002/pmic.200300670. [DOI] [PubMed] [Google Scholar]
- 70.Tai SS, Bunk DM, White E, Welch MJ. Development and evaluation of a reference measurement procedure for the determination of total 3,3′,5-triiodothyronine in human serum using isotope-dilution liquid chromatography-tandem mass spectrometry. Anal Chem. 2004;76(17):5092–5096. doi: 10.1021/ac049516h. [DOI] [PubMed] [Google Scholar]
- 71.Wienkoop S, Weckwerth W. Relative and absolute quantitative shotgun proteomics: targeting low-abundance proteins in Arabidopsis thaliana. J Exp Bot. 2006;57(7):1529–1535. doi: 10.1093/jxb/erj157. [DOI] [PubMed] [Google Scholar]
- 72.Pan S, Rush J, Peskind ER, Galasko D, Chung K, Quinn J, Jankovic J, Leverenz JB, Zabetian C, Pan C, Wang Y, Oh JH, Gao J, Zhang J, Montine T, Zhang J. Application of targeted quantitative proteomics analysis in human cerebrospinal fluid using a liquid chromatography matrix-assisted laser desorption/ionization time-of-flight tandem mass spectrometer (LC MALDI TOF/TOF) platform. J Proteome Res. 2008;7(2):720–730. doi: 10.1021/pr700630x. [DOI] [PubMed] [Google Scholar]
- 73.Cheng D, Hoogenraad CC, Rush J, Ramm E, Schlager MA, Duong DM, Xu P, Wijayawardana SR, Hanfelt J, Nakagawa T, Sheng M, Peng J. Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol Cell Proteomics. 2006;5(6):1158–1170. doi: 10.1074/mcp.D500009-MCP200. [DOI] [PubMed] [Google Scholar]
- 74.Rivers J, Simpson DM, Robertson DH, Gaskell SJ, Beynon RJ. Absolute multiplexed quantitative analysis of protein expression during muscle development using QconCAT. Mol Cell Proteomics. 2007;6(8):1416–1427. doi: 10.1074/mcp.M600456-MCP200. [DOI] [PubMed] [Google Scholar]
- 75.Kostiainen R, Kotiaho T, Kuuranne T, Auriola S. Liquid chromatography/atmospheric pressure ionization-mass spectrometry in drug metabolism studies. J Mass Spectrom. 2003;38(4):357–372. doi: 10.1002/jms.481. [DOI] [PubMed] [Google Scholar]
- 76.Ahmed N, Thornalley PJ. Quantitative screening of protein biomarkers of early glycation, advanced glycation, oxidation and nitrosation in cellular and extracellular proteins by tandem mass spectrometry multiple reaction monitoring. Biochem Soc Trans. 2003;31(Pt 6):1417–1422. doi: 10.1042/bst0311417. [DOI] [PubMed] [Google Scholar]
- 77.Melanson JE, Chisholm KA, Pinto DM. Targeted comparative proteomics by liquid chromatography/matrix-assisted laser desorption/ionization triple-quadrupole mass spectrometry. Rapid Commun Mass Spectrom. 2006;20(5):904–910. doi: 10.1002/rcm.2391. [DOI] [PubMed] [Google Scholar]
- 78.Chaurand P, Norris JL, Cornett DS, Mobley JA, Caprioli RM. New developments in profiling and imaging of proteins from tissue sections by MALDI mass spectrometry. J Proteome Res. 2006;5(11):2889–2900. doi: 10.1021/pr060346u. [DOI] [PubMed] [Google Scholar]
- 79.Cornett DS, Reyzer ML, Chaurand P, Caprioli RM. MALDI imaging mass spectrometry: molecular snapshots of biochemical systems. Nat Methods. 2007;4(10):828–833. doi: 10.1038/nmeth1094. [DOI] [PubMed] [Google Scholar]
- 80.Reyzer ML, Caprioli RM. MALDI mass spectrometry for direct tissue analysis: a new tool for biomarker discovery. J Proteome Res. 2005;4(4):1138–1142. doi: 10.1021/pr050095+. [DOI] [PubMed] [Google Scholar]
- 81.Reyzer ML, Caprioli RM. MALDI-MS-based imaging of small molecules and proteins in tissues. Curr Opin Chem Biol. 2007;11(1):29–35. doi: 10.1016/j.cbpa.2006.11.035. [DOI] [PubMed] [Google Scholar]
- 82.Chaurand P, Sanders ME, Jensen RA, Caprioli RM. Proteomics in diagnostic pathology: profiling and imaging proteins directly in tissue sections. Am J Pathol. 2004;165(4):1057–1068. doi: 10.1016/S0002-9440(10)63367-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Herring KD, Oppenheimer SR, Caprioli RM. Direct tissue analysis by matrix-assisted laser desorption ionization mass spectrometry: application to kidney biology. Semin Nephrol. 2007;27(6):597–608. doi: 10.1016/j.semnephrol.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stauber J, Lemaire R, Franck J, Bonnel D, Croix D, Day R, Wisztorski M, Fournier I, Salzet M. MALDI imaging of formalin-fixed paraffin-embedded tissues: application to model animals of parkinson disease for biomarker hunting. J Proteome Res. 2008;7(3):969–978. doi: 10.1021/pr070464x. [DOI] [PubMed] [Google Scholar]
- 85.Wisztorski M, Lemaire R, Stauber J, Menguelet SA, Croix D, Mathe OJ, Day R, Salzet M, Fournier I. New developments in MALDI imaging for pathology proteomic studies. Curr Pharm Des. 2007;13(32):3317–3324. doi: 10.2174/138161207782360672. [DOI] [PubMed] [Google Scholar]
- 86.Stoeckli M, Knochenmuss R, McCombie G, Mueller D, Rohner T, Staab D, Wiederhold KH. MALDI MS imaging of amyloid. Methods Enzymol. 2006;412:94–106. doi: 10.1016/S0076-6879(06)12007-8. [DOI] [PubMed] [Google Scholar]
- 87.Hsieh Y, Chen J, Korfmacher WA. Mapping pharmaceuticals in tissues using MALDI imaging mass spectrometry. J Pharmacol Toxicol Methods. 2007;55(2):193–200. doi: 10.1016/j.vascn.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 88.Deutsch EW, Lam H, Aebersold R. PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows. EMBO Rep. 2008;9(5):429–434. doi: 10.1038/embor.2008.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mallick P, Schirle M, Chen SS, Flory MR, Lee H, Martin D, Ranish J, Raught B, Schmitt R, Werner T, Kuster B, Aebersold R. Computational prediction of proteotypic peptides for quantitative proteomics. Nat Biotechnol. 2007;25(1):125–131. doi: 10.1038/nbt1275. [DOI] [PubMed] [Google Scholar]
- 90.Ishihama Y, Sato T, Tabata T, Miyamoto N, Sagane K, Nagasu T, Oda Y. Quantitative mouse brain proteomics using culture-derived isotope tags as internal standards. Nat Biotechnol. 2005;23(5):617–621. doi: 10.1038/nbt1086. [DOI] [PubMed] [Google Scholar]
- 91.Beynon RJ, Doherty MK, Pratt JM, Gaskell SJ. Multiplexed absolute quantification in proteomics using artificial QCAT proteins of concatenated signature peptides. Nat Methods. 2005;2(8):587–589. doi: 10.1038/nmeth774. [DOI] [PubMed] [Google Scholar]
- 92.Pratt JM, Simpson DM, Doherty MK, Rivers J, Gaskell SJ, Beynon RJ. Multiplexed absolute quantification for proteomics using concatenated signature peptides encoded by QconCAT genes. Nat Protoc. 2006;1(2):1029–1043. doi: 10.1038/nprot.2006.129. [DOI] [PubMed] [Google Scholar]
- 93.Mirzaei H, McBee JK, Watts J, Aebersold R. Comparative evaluation of current peptide production platforms used in absolute quantification in proteomics. Mol Cell Proteomics. 2008;7(4):813–823. doi: 10.1074/mcp.M700495-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lin S, Shaler TA, Becker CH. Quantification of intermediate-abundance proteins in serum by multiple reaction monitoring mass spectrometry in a single-quadrupole ion trap. Anal Chem. 2006;78(16):5762–5767. doi: 10.1021/ac060613f. [DOI] [PubMed] [Google Scholar]
- 95.Whiteaker JR, Zhang H, Zhao L, Wang P, Kelly-Spratt KS, Ivey RG, Piening BD, Feng LC, Kasarda E, Gurley KE, Eng JK, Chodosh LA, Kemp CJ, McIntosh MW, Paulovich AG. Integrated pipeline for mass spectrometry-based discovery and confirmation of biomarkers demonstrated in a mouse model of breast cancer. J Proteome Res. 2007;6(10):3962–3975. doi: 10.1021/pr070202v. [DOI] [PubMed] [Google Scholar]
- 96.Whiteaker JR, Zhao L, Zhang HY, Feng LC, Piening BD, Anderson L, Paulovich AG. Antibody-based enrichment of peptides on magnetic beads for mass-spectrometry-based quantification of serum biomarkers. Anal Biochem. 2007;362(1):44–54. doi: 10.1016/j.ab.2006.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bjorhall K, Miliotis T, Davidsson P. Comparison of different depletion strategies for improved resolution in proteomic analysis of human serum samples. Proteomics. 2005;5(1):307–317. doi: 10.1002/pmic.200400900. [DOI] [PubMed] [Google Scholar]
- 98.Zhang H, Li XJ, Martin DB, Aebersold R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat Biotechnol. 2003;21(6):660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- 99.Liu T, Qian WJ, Gritsenko MA, Camp DG, Monroe ME, Moore RJ, Smith RD. Human plasma N-glycoproteome analysis by immunoaffinity subtraction, hydrazide chemistry, and mass spectrometry. J Proteome Res. 2005;4(6):2070–2080. doi: 10.1021/pr0502065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Barelli S, Canellini G, Thadikkaran L, Crettaz D, Quadroni M, Rossier JS, Tissot JD, Lion N. Oxidation of proteins: Basic principles and perspectives for blood proteomics. Proteomics Clin Appl. 2008;2:142–157. doi: 10.1002/prca.200780009. [DOI] [PubMed] [Google Scholar]
- 101.Gevaert K, Goethals M, Martens L, Van Damme J, Staes A, Thomas GR, Vandekerckhove J. Exploring proteomes and analyzing protein processing by mass spectrometric identification of sorted N-terminal peptides. Nat Biotechnol. 2003;21(5):566–569. doi: 10.1038/nbt810. [DOI] [PubMed] [Google Scholar]
- 102.Staes A, Van Damme P, Helsens K, Demol H, Vandekerckhove J, Gevaert K. Improved recovery of proteome-informative, protein N-terminal peptides by combined fractional diagonal chromatography (COFRADIC) Proteomics. 2008;8(7):1362–1370. doi: 10.1002/pmic.200700950. [DOI] [PubMed] [Google Scholar]
- 103.Krueger KE, Srivastava S. Posttranslational protein modifications: current implications for cancer detection, prevention, and therapeutics. Mol Cell Proteomics. 2006;5(10):1799–1810. doi: 10.1074/mcp.R600009-MCP200. [DOI] [PubMed] [Google Scholar]
- 104.Dennis JW, Granovsky M, Warren CE. Glycoprotein glycosylation and cancer progression. Biochim Biophys Acta. 1999;1473(1):21–34. doi: 10.1016/s0304-4165(99)00167-1. [DOI] [PubMed] [Google Scholar]
- 105.Kobata A, Amano J. Altered glycosylation of proteins produced by malignant cells, and application for the diagnosis and immunotherapy of tumours. Immunol Cell Biol. 2005;83(4):429–439. doi: 10.1111/j.1440-1711.2005.01351.x. [DOI] [PubMed] [Google Scholar]
- 106.Ono M, Hakomori S. Glycosylation defining cancer cell motility and invasiveness. Glycoconj J. 2004;20(1):71–78. doi: 10.1023/B:GLYC.0000018019.22070.7d. [DOI] [PubMed] [Google Scholar]
- 107.Chen R, Yi EC, Donohoe D, Pan S, Eng J, Crispin DA, Lane Z, Goodlett DA, Bronner MP, Aebersold R, Brentnall TA. Pancreatic Cancer Proteome: the Proteins that Underlie Invasion, Metastasis, and Immunologic Escape. Gastroenterology. 2005;129(4):1187–1197. doi: 10.1053/j.gastro.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 108.Anderson L. Candidate-based proteomics in the search for biomarkers of cardiovascular disease. J Physiol. 2005;563(Pt 1):23–60. doi: 10.1113/jphysiol.2004.080473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Barnidge DR, Goodmanson MK, Klee GG, Muddiman DC. Absolute quantification of the model biomarker prostate-specific antigen in serum by LC-Ms/MS using protein cleavage and isotope dilution mass spectrometry. J Proteome Res. 2004;3(3):644–652. doi: 10.1021/pr049963d. [DOI] [PubMed] [Google Scholar]
- 110.Dinges MM, Orwin PM, Schlievert PM. Exotoxins of Staphylococcus aureus. Clin Microbiol Rev. 2000;13(1):16–34. doi: 10.1128/cmr.13.1.16-34.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nicol GR, Han M, Kim J, Birse CE, Brand E, Nguyen A, Mesri M, FitzHugh W, Kaminker P, Moore PA, Ruben SM, He T. Use of an immunoaffinity-mass spectrometry based approach for the quantification of protein biomarkers from serum samples of lung cancer patients. Mol Cell Proteomics. 2008 doi: 10.1074/mcp.M700476-MCP200. on-line published. [DOI] [PubMed] [Google Scholar]