

Abstract
A homomer is formed by self-interacting copies of a protein unit. This is functionally important1,2, as in allostery3-5, and structurally crucial because mis-assembly of homomers is implicated in disease6,7. Homomers are widespread, with 50–70% of proteins with a known quaternary state assembling into such structures8,9. Despite their prevalence, their role in the evolution of cellular machinery10,11 and the potential for their use in the design of new molecular machines12,13, little is known about the mechanisms that drive formation of homomers at the level of evolution and assembly in the cell9,14. Here we present an analysis of over 5,000 unique atomic structures and show that the quaternary structure of homomers is conserved in over 70% of protein pairs sharing as little as 30% sequence identity. Where quaternary structure is not conserved among the members of a protein family, a detailed investigation revealed well-defined evolutionary pathways by which proteins transit between different quaternary structure types. Furthermore, we show by perturbing subunit interfaces within complexes and by mass spectrometry analysis15, that the (dis)assembly pathway mimics the evolutionary pathway. These data represent a molecular analogy to Haeckel's evolutionary paradigm of embryonic development, where an intermediate in the assembly of a complex represents a form that appeared in its own evolutionary history. Our model of self-assembly allows reliable prediction of evolution and assembly of a complex solely from its crystal structure.
Although homomers are central to biology, only anecdotal knowledge exists on their principles of evolution and assembly, and no unifying theory has been proposed. Large increases in structural data in recent years, however, have enabled us to study quaternary structure or spatial arrangement of subunits on a data set of 5,375 unique structures. This data set is ∼tenfold greater than any studied previously16 (Methods). On the basis of this data set, we quantify how often proteins change their quaternary structure, and identify the evolutionary routes taken to do so. Subsequently, as evolution of a complex can be viewed as assembly over a long timescale, we compare evolutionary routes with (dis)assembly routes probed by mass spectrometry.
Homomers can be separated into two main classes of open or closed symmetry. The first class corresponds to open structures that would polymerize to infinity in the absence of limiting factors. Such assemblies (for example, tubulin and actin) are rare in our data set (3%), probably because their innate dynamic character renders them difficult to crystallize. In contrast, closed symmetries are finite in space, and most homomers adopt either cyclic or dihedral symmetry (Fig. 1a), with only a small fraction (1%) having cubic symmetry (not shown). Throughout we denote Cn as a cyclic complex containing n subunits, and Dn as a dihedral complex containing 2n subunits.
Figure 1. Abundance and properties of cyclic and dihedral symmetries.
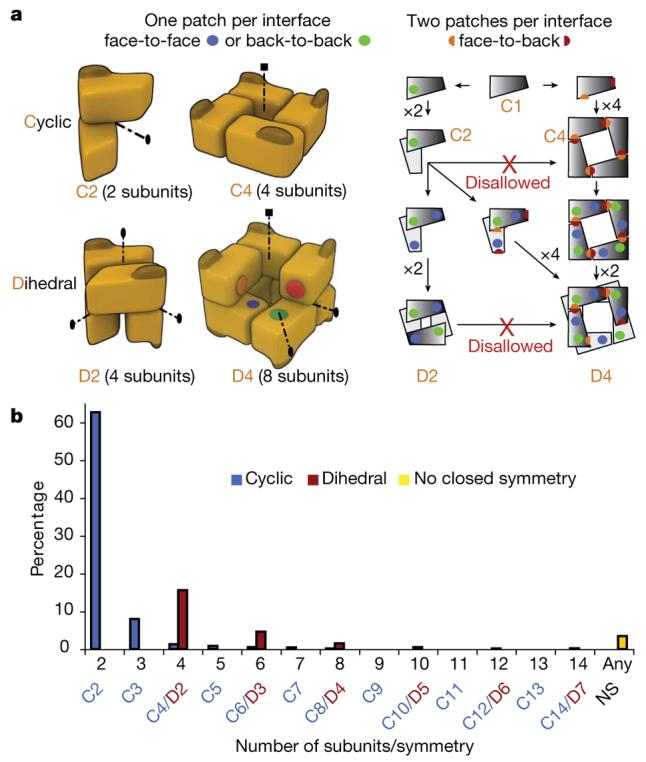
a, n subunits in a cyclic complex are related by a single n-fold symmetry axis (dotted lines); ellipses and squares represent two- and four-fold axes, respectively. For a monomer to evolve towards a cyclic tetramer (C4), two complementary surfaces have to evolve simultaneously (red and orange patches). For a dihedral tetramer (D2), two different and self-complementary surfaces (green and blue patches) can evolve serially with an intermediate dimer (C2). b, The abundance of homomers with cyclic, dihedral, or no symmetry (3.5%). 62.7% are cyclic dimers (C2), 8% are cyclic trimers (C3), and 3.2% have higher order cyclic symmetry (from C4 to C14). Dihedral complexes dominate (22.6%) among complexes with ≥4 subunits.
It has long been observed that smaller complexes are more abundant than larger ones, and even numbers of subunits are favoured over odd numbers8,9,17. Here we confirm this observation, with 62% of complexes being dimers. We quantify the different types of symmetries found in homomers and show that the abundance of complexes with even numbers of subunits is due to the prevalence of dihedral complexes. Whenever an option exists for cyclic or dihedral, on average we find an 11-fold preference for dihedral complexes (Fig. 1b). There is an evolutionary explanation for this preference, as the probability that a dihedral complex evolved by random mutation should be higher than the probability for a cyclic complex for at least two reasons: first, at the level of individual interfaces, in dihedral complexes most interfaces are face-to-face (or back-to-back), whereas all interfaces in cyclics are face-to-back (Fig. 1a) and these are less likely to form by random mutation5,18; and second, at the level of whole complexes, evolution of dihedral complexes can take place in multiple steps (C1→C2→D2) whereas cyclics must evolve in one step (C1→C4, Fig. 1a).
Notably, dihedral and cyclic symmetries are geometrically related: a complex with Dn symmetry can be formed from n dimers with C2 symmetry or from two n-mers with Cn symmetry19 (Fig. 1a). If a protein complex has a particular symmetry, we find that homologues are likely to have the same symmetry type. More specifically, for sequence identities >90%, conservation is nearly 100%, whereas in the range of 30–40% sequence identities, conservation is ∼70% (Supplementary Fig. 1). Proteins with different degrees of quaternary structure conservation are illustrated in Fig. 2a. Thymidylate synthase always exists as a dimer, adenylyltransferase is a dimer in Bacillus subtilis and a hexamer in human (trimer of B. subtilis dimers), whereas two phospholipase A2s have geometrically very distant quaternary structures.
Figure 2. Routes for homomer evolution.
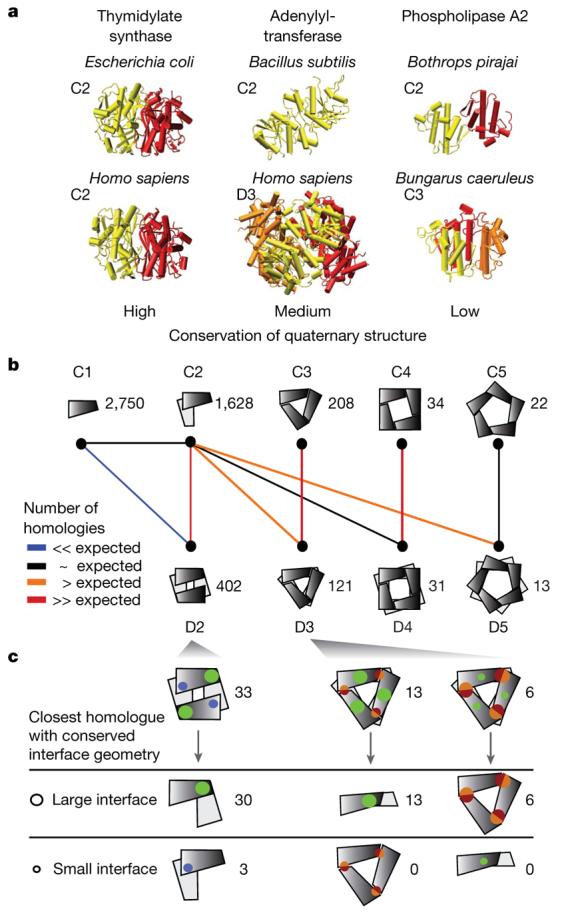
a, Illustrative examples of different levels of quaternary structure conservation (termed high, medium and low). Thymidylate synthases (PDB accessions 1ajm and 1hw3) are both dimers. Adenylyltransferases (PDB accessions 1kam and 1kr2) differ in their number of subunits, with similar dimers (yellow) common to both quaternary structures. The dimer and trimer of venom-toxin phospholipase A2s are not related geometrically and the quaternary structures are therefore less conserved. b, Schematic illustration of large-scale analysis of quaternary structures; the number of unique complexes is indicated. Coloured lines indicate the significance in over-representation of shared homologous complexes (Methods). c, In 49 out of 52 cases, the largest interface is present in the dimeric or trimeric homologue, illustrated by size of interface patches.
When quaternary structure is not conserved, we speculate that pathways linking geometrically related symmetries represent both evolutionary and assembly routes. For example, a dihedral tetramer (D2) can be described as a dimer of dimers, where a back-to-back dimerization patch forms a first dimer, and a second face-to-face dimerization patch forms the dimer of dimers. This is not true of a cyclic tetramer (C4), where subunits interact in a face-to-back manner, such that two different surface patches are involved in forming an interface (Fig. 1a). Therefore, we expect many more dihedral than cyclic tetramers to share evolutionary relationships with dimers. This is illustrated by the pathway from a dimer to a dihedral tetramer (Fig. 1a) and the disallowed transition from a dimer to a cyclic tetramer.
Following this idea, we looked at evolutionary relationships in terms of sequence similarity between different quaternary structures to unveil the routes most commonly taken to build larger complexes (Fig. 2b). Each quaternary structure is represented schematically with the numbers of proteins of each type. Pairs of quaternary structures are connected according to the statistical significance of the number of evolutionary transitions between them. Most pairs have fewer transitions between them than expected in a random model (Methods) as exemplified by monomers (C1) and dihedral tetramers (D2). Other pairs with insignificant numbers of transitions are shown in Supplementary Fig. 2. We find that cyclic dimers, trimers and tetramers share notable numbers of transitions with their dihedral counterparts, supporting the stepwise evolutionary scenario where homomers with dihedral symmetry evolve through cyclic intermediates (Fig. 1a).
Notably in this stepwise scenario, two evolutionary routes lead to a dihedral complex (Dn): either from n dimers or from two cyclic n-mers (Fig. 2b). This raised the question as to whether it was possible to identify which of these two routes was taken by a given dihedral complex. On the basis of energetic considerations (Supplementary Information 1), we propose that a hierarchy of interface sizes exists within dihedral complexes, and that the larger interface is conserved in evolution. To test this hypothesis, we looked for tetramers homologous to a dimer, as well as hexamers homologous to a dimer or trimer. In this data set (Fig. 2c and Supplementary Table 1) we examined whether the interface within the dimer or trimer corresponded to the largest interface in the homologous tetramer or hexamer. Among 33 tetramers and 19 hexamers studied, 49 complexes conserve the larger interface with the dimeric or trimeric homologue, whereas only 3 conserve their smaller interface (Fig. 2c and Supplementary Table 1). This result implies that the evolutionary route of a homomer can be predicted solely from its interface sizes. Our predictions for the evolutionary pathways of D3, D4 and D5 complexes (Supplementary Fig. 3a) have led us to formulate a general model of homomer evolution (Supplementary Fig. 3b).
It is notable that this signature of complex formation (hierarchy in interface sizes) is conserved throughout evolution. This can be interpreted in at least two different although not mutually exclusive ways: (1) once the complex is formed there is no need to dramatically change the interface size, analogous to the classical explanation for the marginal stability of proteins20 (that is, selective pressure becomes almost non-existent beyond the point where proteins fold); and (2) maintaining a hierarchy of interface strengths is important for a precise order during assembly21,22, in which case the largest interface would reflect the main intermediate species during assembly. To test this hypothesis we targeted ten complexes for study using electrospray mass spectrometry (Fig. 4a and Supplementary Table 2).
Figure 4. (Dis)assembly pathways in 16 complexes.
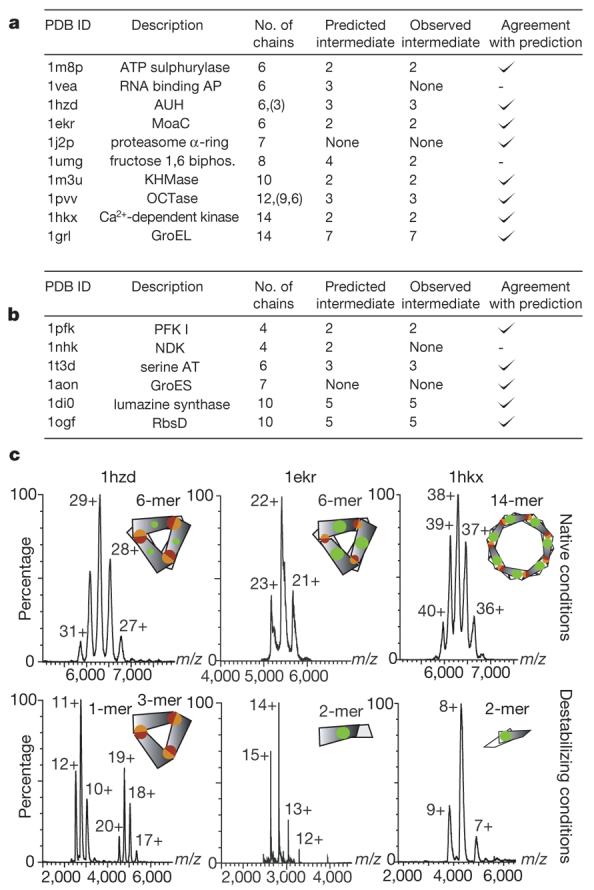
a, Homomers for which (dis)assembly was probed using electrospray mass spectrometry (Methods). ‘None’ denotes no intermediate detected. Complexes agree with our prediction where the subcomplex containing the larger interface is the most stable in eight out of ten cases. b, List of homomers for which: (1) information on (dis)assembly has been reported (Supplementary Table 3); (2) a crystal structure is known; and (3) the intermediate species observed during (dis)assembly could be mapped to a subcomplex in the structure. For these complexes, we found an agreement with our prediction in 5/6 cases. c, Mass spectra showing intact complexes (top panel) as well as subcomplexes obtained after destabilization in solution (bottom panel). AUH, an RNA binding protein; GroEL and GroES are chaperonins; KHMase, ketopantoate hydroxymethyltransferase; MoaC, molybdenum cofactor biosynthesis protein; RNA binding AP, RNA binding antitermination protein; serine AT, serine acyltransferase; OCTase, ornithine carbamoyltransferase.
Initially we verified that the complexes could be generated intact and corresponded to the stoichiometry described in the protein data bank (PDB). The mass spectra recorded for two hexamers with D3 symmetry and one 14-mer with D7 symmetry revealed that the intact homomer is maintained in each case (Fig. 4c). We then induced the disassembly of each complex through the careful change in ionic strength or the stepwise addition of partial denaturants. We detected stable subcomplexes corresponding to trimers and dimers for hexameric AUH protein (an RNA binding protein), and MoaC (a molybdenum cofactor biosynthesis protein), respectively (Supplementary Table 2). Examination of the interface size shows that in both cases the larger interface is maintained. Similarly for the Ca2+-dependent kinase with D7 symmetry, a dimer is the principal dissociation product and buries the largest interface (Fig. 4c). For one complex (PDB entry 1vea) our results were ambiguous as no intermediate and only monomeric subunits were detected; for another complex (PDB entry 1umg) we predicted a tetramer and detected a dimer. In this case, both subcomplexes bury large surfaces (>5,000Å2), which may bias the use of interface size as a proxy for interface strength. For the remaining complexes, the predicted subcomplex containing the larger interface was observed. These results demonstrate that the largest interface is maintained consistently during disassembly.
To address whether the disassembly process was the reverse of the assembly pathway, we attempted to reassemble a subset of the complexes studied by dilution of the denaturant and/or manipulation of the ionic strength. In ∼50% of the complexes examined we were able to reassemble the original homomer. These results—together with previous studies where reassembly was found to be strongly dependent on factors such as ionic strength, temperature and concentration of denaturant23,24—indicate that disassembly is the reverse of assembly under the appropriate conditions.
To complement our experimental observations, we found six additional complexes for which (dis)assembly intermediates had been reported (Fig. 4b). Of these, five match our prediction and one (nucleoside diphosphate kinase) had no intermediate detected. This homomer may either assemble without forming subcomplexes, or subcomplexes may have escaped detection. Alternatively, formation of subcomplexes might involve factors absent from the experimental set-up25. Thus, although there are exceptions, we find agreement between the evolutionary pathway and (dis)assembly pathway in 81% of the cases we examined.
Overall, through analysis of a large set of homomers, we have shown that the evolutionary pathway of a homomer can be inferred from its atomic structure morphology. This allowed us to predict the (dis)assembly pathway of homomers in solution, and design mass-spectrometry-based experiments to validate our predictions. Results revealed that the (dis)assembly pathway, which takes place on a protein-folding timescale (∼seconds), mimics the evolutionary pathway that has taken place over a considerably longer timescale (∼millions of years). This is the first time that a general principle for formation and assembly of homomers has been demonstrated. We hope that this will stimulate further studies, as relationships between folding, complex formation and aggregation are only beginning to be explored.
METHODS SUMMARY
Data set of homomers
All data sets of homomers used were derived from the 3D complex database8. As the quaternary structure annotation in the PDB biological unit is erroneous in some cases, we used a manually curated data set26.
Randomization of evolutionary routes
To assess the significance of the number of evolutionary relationships between proteins with different quaternary structures, we compared the observed numbers to a random model of quaternary structure transitions where evolutionary relationships are reassigned randomly in proportion to the size of each quaternary structure type.
Prediction of evolutionary routes
The size of an interface is given by the number of amino acids in contact, as defined previously8. We predict evolutionary intermediates by taking the ‘closed’ subcomplex containing the largest interface. In cyclic complexes with three or more subunits, each subunit buries two equivalent surfaces. Thus, these interfaces are counted twice when compared to dimer interfaces.
Intact complexes
Complexes were donated by crystallographers and taken from a random selection from the PDB. For further details see Methods.
Generating subcomplexes
Intact complexes were disrupted through change in ionic strength or the stepwise addition of dimethylsulphoxide, methanol or acetonitrile. This process is illustrated in Supplementary Fig. 4 and solution conditions are summarized in Supplementary Table 2.
Supplementary Material
Figure 3. Prediction of evolutionary routes and link with (dis)assembly in solution.
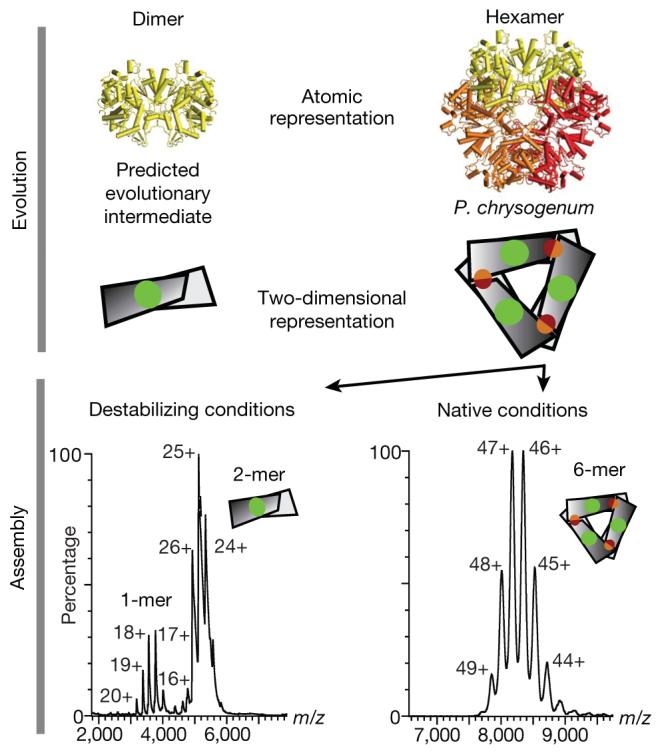
ATP sulphurylase is a hexamer in Penicillium chrysogenum, with a predicted dimeric evolutionary intermediate based on interface sizes (that is, the interface in the dimer (green patch) is larger than the trimeric interface (red and orange patch; top panel)). We perturb the hexameric ATP sulphurylase (PDB accession 1m8p, Supplementary Table 2) to disassemble it into subcomplexes probed using electrospray mass spectrometry (bottom panel) and determine whether a dimeric (with the larger interface) or a trimeric (small interface) subcomplex is detected. Both dimers and monomers were detected, with no evidence of trimers.
Acknowledgements
We thank the collaborators listed in Supplementary Table 2 for supplying the different complexes and acknowledge H. Hernandez, J. Freeke and L. Lane for assistance with mass spectrometry. We also thank C. Chothia, J. Clark and M. Babu for discussions. This work was supported by the Medical Research Council, the EMBO Young Investigators Programme, the Royal Society and the Waters Kundert Trust.
Footnotes
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Cabezon E, et al. Homologous and heterologous inhibitory effects of ATPase inhibitor proteins on F-ATPases. J. Biol. Chem. 2002;277:41334–41341. doi: 10.1074/jbc.M207169200. [DOI] [PubMed] [Google Scholar]
- 2.Hardy LW, et al. Atomic structure of thymidylate synthase: target for rational drug design. Science. 1987;235:448–455. doi: 10.1126/science.3099389. [DOI] [PubMed] [Google Scholar]
- 3.Iber D, Clarkson J, Yudkin MD, Campbell ID. The mechanism of cell differentiation in Bacillus subtilis. Nature. 2006;441:371–374. doi: 10.1038/nature04666. [DOI] [PubMed] [Google Scholar]
- 4.Marianayagam NJ, Sunde M, Matthews JM. The power of two: protein dimerization in biology. Trends Biochem. Sci. 2004;29:618–625. doi: 10.1016/j.tibs.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 5.Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 6.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 7.Hayouka Z, et al. Inhibiting HIV-1 integrase by shifting its oligomerization equilibrium. Proc. Natl Acad. Sci. USA. 2007;104:8316–8321. doi: 10.1073/pnas.0700781104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levy ED, Pereira-Leal JB, Chothia C, Teichmann SA. 3D complex: a structural classification of protein complexes. PLoS Comput. Biol. 2006;2:e155. doi: 10.1371/journal.pcbi.0020155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goodsell DS, Olson AJ. Structural symmetry and protein function. Annu. Rev. Biophys. Biomol. Struct. 2000;29:105–153. doi: 10.1146/annurev.biophys.29.1.105. [DOI] [PubMed] [Google Scholar]
- 10.Ispolatov I, Yuryev A, Mazo I, Maslov S. Binding properties and evolution of homodimers in protein-protein interaction networks. Nucleic Acids Res. 2005;33:3629–3635. doi: 10.1093/nar/gki678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pereira-Leal JB, Levy ED, Kamp C, Teichmann SA. Evolution of protein complexes by duplication of homomeric interactions. Genome Biol. 2007;8:R51. doi: 10.1186/gb-2007-8-4-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grueninger D, et al. Designed protein-protein association. Science. 2008;319:206–209. doi: 10.1126/science.1150421. [DOI] [PubMed] [Google Scholar]
- 13.Janin J. Biochemistry. Dicey assemblies. Science. 2008;319:165–166. doi: 10.1126/science.1152930. [DOI] [PubMed] [Google Scholar]
- 14.Blundell TL, Srinivasan N. Symmetry, stability, and dynamics of multidomain and multicomponent protein systems. Proc. Natl Acad. Sci. USA. 1996;93:14243–14248. doi: 10.1073/pnas.93.25.14243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hernandez H, et al. Subunit architecture of multimeric complexes isolated directly from cells. EMBO Rep. 2006;7:605–610. doi: 10.1038/sj.embor.7400702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brinda KV, Vishveshwara S. Oligomeric protein structure networks: insights into protein-protein interactions. BMC Bioinformatics. 2005;6:296. doi: 10.1186/1471-2105-6-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Monod J. Nobel Symposium 11: Symmetry and Function of Biological Systems at the Macromolecular Level. Almqvist & Wiksell; Stockholm: 1968. [Google Scholar]
- 18.Lukatsky DB, Shakhnovich BE, Mintseris J, Shakhnovich EI. Structural similarity enhances interaction propensity of proteins. J. Mol. Biol. 2007;365:1596–1606. doi: 10.1016/j.jmb.2006.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Claverie P, Hofnung M, Monod J. Sur certaines implications de l'hypothèse d'équivalence stricte entre les protomères des protéines oligomériques. C. R. Séanc. Acad. Sci. 1968;266:1616–1618. [Google Scholar]
- 20.DePristo MA, Weinreich DM, Hartl DL. Missense meanderings in sequence space: a biophysical view of protein evolution. Nature Rev. Genet. 2005;6:678–687. doi: 10.1038/nrg1672. [DOI] [PubMed] [Google Scholar]
- 21.Bahadur RP, Rodier F, Janin J. A dissection of the protein-protein interfaces in icosahedral virus capsids. J. Mol. Biol. 2007;367:574–590. doi: 10.1016/j.jmb.2006.12.054. [DOI] [PubMed] [Google Scholar]
- 22.Powers ET, Powers DL. A perspective on mechanisms of protein tetramer formation. Biophys. J. 2003;85:3587–3599. doi: 10.1016/S0006-3495(03)74777-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luke K, Wittung-Stafshede P. Folding and assembly pathways of co-chaperonin proteins 10: Origin of bacterial thermostability. Arch. Biochem. Biophys. 2006;456:8–18. doi: 10.1016/j.abb.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 24.Cheesman C, Ruddock LW, Freedman RB. The refolding and reassembly of Escherichia coli heat-labile enterotoxin B-subunit: analysis of reassembly-competent and reassembly-incompetent unfolded states. Biochemistry. 2004;43:1609–1617. doi: 10.1021/bi0354987. [DOI] [PubMed] [Google Scholar]
- 25.Kress W, Mutschler H, Weber-Ban E. Assembly pathway of an AAA+ protein: tracking ClpA and ClpAP complex formation in real time. Biochemistry. 2007;46:6183–6193. doi: 10.1021/bi602616t. [DOI] [PubMed] [Google Scholar]
- 26.Levy ED. PiQSi: Protein quaternary structure investigation. Structure. 2007;15:1364–1367. doi: 10.1016/j.str.2007.09.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.