

Abstract
PEP-19 is a small calmodulin (CaM)-binding protein that greatly increases the rates of association and dissociation of Ca2+ from the C-domain of CaM, an effect that is mediated by an acidic/IQ sequence in PEP-19. We show here using NMR that PEP-19 is an intrinsically disordered protein, but with residual structure localized to its acidic/IQ motif. We also show that the kon and koff rates for binding PEP-19 to apo-CaM are at least 50-fold slower than for binding to Ca2+-CaM. These data indicate that intrinsic disorder confers plasticity that allows PEP-19 to bind to either apo- or Ca2+-CaM via different structural modes, and that complex formation may be facilitated by conformational selection of residual structure in the acidic/IQ sequence.
PEP-19 (purkinje cell protein 4, pcp4) and RC32 (neurogranin, Ng) are small IQ-motif calmodulin (CaM)-binding proteins originally identified in the central nervous system (for reviews see Refs. 1–4). They are present at high concentrations (up to 40–50 μm), but have no known activity other than binding to CaM in the presence or absence of Ca2+. We showed that PEP-19 and RC3 have unique effects on Ca2+ binding to the C-domain of CaM. Specifically, PEP-19 greatly increases the Ca2+ binding kon and koff rates, but does not alter Ca2+ binding affinity (5), whereas RC3 increases koff but has a lesser effect on the kon thereby decreasing the affinity of Ca2+ binding to the C-domain of CaM (6). These data point to PEP-19 and RC3 as modulators or regulators of CaM signaling that act at the level of Ca2+ binding to CaM.
PEP-19 is of particular interest because its expression is not restricted to the central nervous system, and its pattern of expression suggests a link to Ca2+ metabolism. For example, the NCBI Gene Expression Omnibus shows PEP-19 is expressed in neuroendocrine organs such as prostate, uterus, and kidney, which require high Ca2+ for secretory and transport functions. PEP-19 is induced in lactating breast (7) and during osteogenic differentiation of bone marrow stem cells (8), which also have a need for high Ca2+ levels.
A protective role for PEP-19 against high Ca2+ levels is suggested by overexpression of PEP-19, which inhibits apoptosis in PC-12 cells (9), and protects HEK293T cells against death due to Ca2+ overload (10). Indeed, a general protective role for PEP-19 was put forth previously based on the fact that purkinje cells of the cerebellum and granule-cell neurons in the dentate gyrus, which have high levels of PEP-19, are largely spared from the effects of Alzheimer disease, whereas PEP-19 negative cells are severely affected (11). Conversely, cell types that are most affected by Huntington disease exhibit a significant loss of expression of PEP-19 (11).
A protective role for PEP-19 against the high Ca2+ in normal or pathogenic conditions emphasizes the importance of characterizing its biophysical properties and its interaction with CaM. We show here that PEP-19 is intrinsically disordered, but with residual structure localized to the acidic/IQ region that regulates Ca2+ binding to CaM. Moreover, this region remains partially structured even when bound to CaM. These data point to structural disorder as a dominant feature of PEP-19 that compliments the conformational plasticity of CaM to allow binding in the presence and absence of Ca2+, and facile transition of the CaM·PEP-19 complex from apo to Ca2+-bound states.
EXPERIMENTAL PROCEDURES
Recombinant Proteins and Peptides—Recombinant CaM and PEP19 were cloned, expressed, and purified as described previously (5, 12, 13). Labeling CaM(T110C) with IAEDANS was previously reported (13, 14). PEP-19 with a C-terminal Gly-Cys extension was labeled with DDPM as described previously (15).
Circular Dichroism Spectroscopy—CD measurements were performed on an Aviv 62ADS spectropolarimeter. Free protein, or protein mixtures at 1:1 molar ratio, were scanned at a concentration of 20 μm in a buffer containing 10 mm MES, 100 mm KCl, at pH 6.3 in the presence of either 1 mm EGTA or 1 mm CaCl2 in a quartz cuvette with a path length of 1 mm. Wavelength scans were performed at 25 °C, except experiments involving PEP-19 were performed at 25, 50, and 70 °C. Recording time was 10 s in 1-nm steps in the range of 190–260 nm with the data averaged over 3 experiments. The Savitzky-Golay smoothing algorithm (16) was applied to the curve derived from the averaged data.
NMR Methodology—Backbone assignment of [13C,15N]PEP-19 was carried out using a Varian Inova 800 MHz and Bruker DRX 800 MHz spectrometers using three-dimensional HNCACB, CBCA(CO)NH, HN(CA)CO, and HNCO experiments at 298 K. For experiments requiring decalcified proteins, proteins and buffers were passed over a calcium sponge (Molecular Probes). The decalcified proteins was lyophilized and resuspended to 0.5 mm in decalcified buffer containing 10 mm imidazole, pH 6.3, 100 mm KCl, 1 mm EGTA, and 5% D2O. For Ca2+ samples, 1 mm EGTA was replaced by 5 mm CaCl2 in the buffer. Experiments involving CaM or CaM·PEP-19 complex were performed on a Bruker DRX 600 MHz spectrometer equipped with a 5-mm TXI CryoProbe at 298 K. The data were processed using FELIX 2004 software. Resonance assignments for apo-CaM were made by comparison with literature reports (17) and confirmed by three-dimensional CBCA(CO)NH, CBCAHN, HNCO, HN(CA)CO, and 15N-edited NOESY experiments (18). Titration of [15N]apo-CaM with PEP-19 was made using a 10 mm stock solution of PEP-19 and a 0.5 mm sample of apo-CaM. PEP-19 was added in 1/8 molar eq to the NMR sample. Correction in protein concentration was taken into effect during data analysis. The average amide chemical shift change was calculated using the following formula,
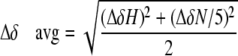 |
(Eq. 1) |
where ΔδH = change in 1H chemical shift; and Δδn = change in 15N chemical shift.
Equilibrium Binding of PEP-19 to CaM—A FRET-based assay to measure equilibrium binding of PEP-19 to CaM was described previously (15). Briefly, a solution of 0.5 μm IAEDANS-labeled CaM(T110C) was prepared in 20 mm MOPS, pH 7.5, 100 mm KCl, and 1 mm dithiothreitol with either 0.1 mm CaCl2 or 0.1 mm EGTA. Concentrated stock solutions of DDPM-labeled PEP-19 in the same solution were then used to titrate CaM. The final increase in volume was less than 10%. We assessed potential nonspecific FRET effects using DDPM coupled to free Cys, and found a linear 5% decrease in fluorescence from donor-labeled CaM per increment of 25 μm Cys-DDPM. The FRET effect between donor-labeled CaM and acceptor-labeled PEP-19 was corrected for this nonspecific effect, and the upper concentration of DDPM-labeled PEP-19 was limited to 50 μm. Dissociation constants (Kd) were derived by fitting titration curves to the following equation,
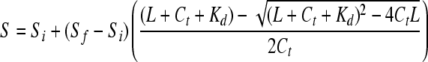 |
(Eq. 2) |
where S = fluorescence signal at a given titration point; Si = initial signal in the absence of ligand; Sf = final signal in the presence of excess ligand; L = total ligand added at a given titration point; Ct = total CaM concentration; and Kd = dissociation constant. The equation was used to fit plots of S versus L with Si, Sf, and Kd as fitted variables.
Stopped-flow Measurements—Stopped-flow fluorescence experiments were performed at 23 °C using an Applied Photophysics Ltd. (Leatherhead, UK) model SX.18 MV sequential stopped-flow spectrofluorimeter with a 150 watt Xe/Hg lamp, and a dead time of 1.7 ms. All solutions contained a base buffer of 20 mm MOPS, pH 7.5, 100 mm KCl. The concentrations of other reagents in stopped-flow mixing solutions A and B are defined in the figure legends. The final concentration of these reagents in the optical chamber was one-half of these values, because the mixing ratio was 1:1.
Computational Model—A model for cooperative Ca2+ binding to the C-domain of CaM was reported previously (15). This was used to determine microscopic Ca2+ binding constants in the presence of PEP-19 for use in a model that predicts the effect of PEP-19 on the rate of association of the C-domain of CaM with a high affinity CaM target protein in response to an increase in free Ca2+. Details of the model are provided under supplemental materials.
RESULTS
Circular Dichroism and NMR Show PEP-19 Is an Intrinsically Unstructured Protein—CD spectroscopy was used to characterize the secondary structure in PEP-19 as a function of CaCl2, temperature, and trifluoroethanol (TFE). Fig. 1A shows that the far UV spectrum of PEP-19 is characteristic of a random-coil with a large negative ellipticity at 200 nm in the absence or presence of CaCl2. The mean residual ellipticity at 222 nm indicates that 3% of the residues are helical (19) at 25 °C. Increasing the temperature to 70 °C did not change the random coil nature of the protein (data not shown). CD spectrum collected in the presence of the helix-promoting solvent TFE indicates substantial α-helix content from the negative bands at 208 and 222 nm. A maximal helical content of about 50% was estimated from CD spectra collected in 40% TFE. Thus, the CD data shows that PEP-19 is highly disordered and unstructured but has the potential to form regions of α-helix.
FIGURE 1.

PEP-19 is intrinsically disordered. Panel A shows the far UV CD spectra for PEP-19 in the presence of 1 mm EDTA (closed diamonds), 1 mm CaCl2 (open diamonds), or in the presence of 30% TFE (closed triangles). Panel B shows the 1H-15N HSQC spectrum of PEP-19 at 298 K in a buffer containing 10 mm imidazole, 100 mm KCl at pH 6.8. The lack of chemical shift dispersion and narrow line widths seen for the amide protons are characteristic of intrinsically disordered proteins.
Fig. 1B shows the two-dimensional 1H-15N HSQC spectrum of uniformly 15N-labeled PEP-19. All protein backbone amides are clearly visible, but chemical shifts are restricted to a window of 0.8 ppm in the 1H dimension, and the resonances show very narrow line widths. Both of these observations are hallmarks of proteins that lack stable long-range interactions, and indicate that PEP-19 is composed of unstructured or highly flexible regions. This is similar to that seen for RC3 (20). Additionally, the 6 Gly resonances in PEP-19 (circled in the spectrum) have chemical shifts that are characteristic of proteins denatured in 8 m urea (21), which also supports the conclusion that PEP-19 is disordered.
Primary Sequence Analysis Indicates PEP-19 Has Residual Helical Content—We used a combination of structure prediction algorithms to identify potential variations in order/disorder along the primary sequence of PEP-19 (22, 23). Fig. 2A shows that both PONDR VLS1 (open circles) and RONN (closed circles) predict PEP-19 to be disordered, but its C-terminal half is disordered to a lesser extent than the N-terminal half. The sequence of PEP-19 was next analyzed using AGADIR (24, 25) to predict localized regions of residual helical structure as shown in Fig. 2B. AGADIR predicts that PEP-19 has an overall helical content of less than 1% under conditions used in the NMR experiments, which is punctuated by two distinct regions of residual helicity. One region spans residues 38 to 54, including part of the IQ motif. The second region shows lower helicity and is upstream between residues 20 to 30. Fig. 2C shows that TFE has the greatest effect on amides for those residues predicted in Fig. 2B to have residual helical structure.
FIGURE 2.

Sequence analysis predicts residual structure in PEP-19. Panel A shows plots of disorder probability per residue for PEP-19 predicted from PONDR VLS1 (open circles) and RONN (closed circles). Panel B shows an AGADIR prediction of the fractional α-helical population in PEP-19. Panel C shows the average weighted change in backbone amide chemical shift perturbation observed upon addition of 15% TFE to PEP-19. Chemical shift differences were calculated as described under “Experimental Procedures.”
We next used a variety of NMR experiments to identify regions of residual structure in PEP-19 under native conditions. Secondary shifts, which are deviations of 13Cα and 13Cβ chemical shifts from random coil values, can be used as indicators of secondary structure due to their dependence on backbone dihedral angles. Relative to random coils values, 13Cα resonances are typically shifted downfield ∼3.1 ppm for residues in stable α-helices, and shifted upfield (∼1.5 ppm) in stable β-sheets (26–28). The 13Cβ resonances are less sensitive to helical environments, but are shifted downfield by about 2.5 ppm in β sheets. Residues in less ordered regions of proteins will have smaller deviations from random coil values as a result of rapid conformational averaging. Positive 13Cα secondary shifts, and weaker negative 13Cβ secondary shifts for Glu38 to Lys52 in Fig. 3, A and B, indicate residual helical structures in this region of PEP-19. As indicated by the shaded regions, these residues coincide with the region predicated by AGADIR to have the greatest extent of residual helix. Based on an empirical relationship between fractional helicity and 13Cα chemical shifts (26), PEP-19 is calculated to have an overall helicity of 2.2%, whereas residues 38 to 52 have a fractional helicity of about 18%.
FIGURE 3.

NMR of PEP-19 indicates residual structure. Secondary chemical shifts for 13Cα (panel A) and 13Cβ (panel B) were calculated by subtracting random coil shifts corrected for sequence-dependent variations (27) from the experimental chemical shifts. Panel C shows solvent accessibility of PEP-19 backbone amides indicated by the HN-H2O exchange cross-peak intensity extracted for each residue from the three-dimensional 15N-edited NOESY-HSQC spectrum. Panel D shows the effect of ionic strength on backbone amide chemical shifts in PEP-19. The weighted average change in N and HN chemical shifts (Δδ) in the presence and absence of 100 mm KCl was calculated for each residue as described under “Experimental Procedures.”
Another indicator of potential residual structure is the intensity of the water peak derived from three-dimensional 15N-edited NOESY-HSQC spectra. Residues in PEP-19 that populate residual secondary structures would be predicted to have greater protection from solvent, and lower exchange rates. Indeed, Fig. 3C shows that the region spanning Val25 to Ser61 has weaker exchange peaks (small dNH,H2O exchange peaks) relative to residues Arg3 to Lys24, indicating potential residual secondary structure in the C-terminal half of PEP-19. The AGADIR program predicts that the helical content of the C-terminal region of PEP-19 will decrease as the ionic strength increases. Indeed, Fig. 3D shows addition of KCl causes small but detectable changes in backbone amides in the C-terminal portion of the protein. These small chemical shift changes correlate well with the presence of the residual α-helical structure in the C-region of PEP-19 determined by 13Cα chemical shifts in Fig. 3A. Together, data in Figs. 1, 2, 3 provide computational and experimental evidence that PEP-19 is an intrinsically unstructured protein, but with regions of predicted residual helical structure localized to the C-terminal portion of the protein.
Effect of Complex Formation on CD Spectra—Fig. 4 compares the CD spectra of CaM·PEP-19 complexes to summed spectra of free proteins in the presence or absence of Ca2+. The difference between the measured data and the summation spectra allow an estimation of secondary structure induced by binding PEP-19 to CaM. We found that the CD spectra for the CaM·PEP-19 complex was similar in the presence or absence of Ca2+ (closed and open triangles), with a difference in calculated helical content of only 1.4%. However, there is a decrease in molar ellipticity at 222 nm for the complexes relative to the summed spectra of the free proteins, and this difference is especially pronounced in the absence of Ca2+. This suggests an increase in overall α-helical content upon complex formation, possibly due to changes in the secondary structure of PEP-19 or rearrangement of α-helices in apo-CaM.
FIGURE 4.

Far UV circular dichroism of PEP-19·CaM complexes. CD spectra of equal molar mixtures of PEP-19 and CaM were collected in the absence (open triangles) and presence of Ca2+ (closed triangles). Mathematical sums of the spectra of the individual free proteins are shown in the absence (open squares) and presence of Ca2+ (closed squares). Samples were scanned at a concentration of 20 μm in a buffer containing 10 mm MES, 100 mm KCl, at pH 6.3.
Effect of CaM on PEP-19 Amide Chemical Shifts—We used backbone amide chemical shifts to identify residues in PEP-19 that are affected by binding CaM. Similar results were obtained using either apo- or Ca2+-CaM. Fig. 5A shows a portion of the two-dimensional 1H-15N HSQC of 15N-labeled PEP-19 collected in the absence (gray), or presence of apo-CaM (black). Upon binding CaM, PEP-19 residues were unaffected (e.g. Lys24), slightly shifted (e.g. Val26), or severely broadened (e.g. Asp31, Glu40, Gln47, Lys57, and Lys55). Fig. 5B summarizes these effects relative to the primary sequence of PEP-19. A striking observation is that only the C-terminal region of PEP-19 appears to participate in binding CaM. Residues 24–61 and 26–61 in PEP-19 are affected upon binding apo-CaM and Ca2+-CaM, respectively. The majority of these residues, including those associated with the core IQ motif, are broadened beyond detection.
FIGURE 5.

The effect of apo- or Ca2+-CaM on the 1H-15N HSQC spectra of 15N-labeled PEP-19. Panel A shows a selected region of the spectrum of 15N-enriched PEP-19 (gray contours) overlaid on spectra collected in the presence of 3 molar eq of apo-CaM (black contours). Numbers correspond to assigned residues in PEP-19. Panel B uses a bar diagram to summarize the effect of CaM on PEP-19 backbone amide chemical shifts. The position of the consensus IQ motif in PEP-19 is indicated (residues 46–59). Panel C shows the 1H-15N spectra for selected amides in free PEP-19 (light gray contours), and after sequential addition of 15% TFE (dark gray contours) and Ca2+-CaM (black contours shown at a 2-fold lower contour level).
Severe broadening of resonances at substoichiometric levels of ligand is characteristic of chemical exchange that is intermediate on the NMR time scale, but resonances for PEP-19 do not refocus in the presence of excess CaM. This suggests that exchange broadening is not driven simply by rate constants of the binary interaction between CaM and PEP-19. One possibility is that PEP-19 is not fully ordered when bound to CaM, but rather exists in relatively slow exchange between multiple states, possibly helical and disordered states. If true, then addition of TFE may stabilize the structure of PEP-19 when bound to CaM and allow detection of amide resonances. Fig. 5C compares a region of the 1H-15N HSQC spectra for free PEP-19 (light gray), with spectra collected after sequential additions of 15% TFE (dark gray) and Ca2+-CaM (black). TFE induces a general upfield shift in amides of free PEP-19, which is characteristic of helix formation, and is consistent with the effect of TFE on the CD spectra of PEP-19 shown in Fig. 1. Addition of Ca2+-CaM in the presence of TFE allows observation of amides for all residues in PEP-19, with the greatest change seen for residues in the C-terminal, as shown for Thr16 and Thr10 versus Ala45 and Ala58.
Interestingly, some residues in the C-terminal region of PEP-19, such as Ala45 and Ala58 in Fig. 5C, show chemical shift changes upon addition of Ca2+-CaM that are a linear extension of the initial shift caused by TFE. This most likely indicates that Ca2+-CaM causes a further increase of the percent helical content of this region. In contrast, residues such as Ile46 of the IQ dipeptide, show a distinct bidirectional change in chemical shift upon sequential addition of TFE and Ca2+-CaM, which likely indicates a direct interaction of these residues with CaM.
Effect of PEP-19 on CaM Amide Chemical Shifts—We previously reported that PEP-19 bound primarily to the C-domain of Ca2+-CaM and that amide chemical shifts showed characteristics of fast exchange on the NMR time scale (5). The rate of dissociation of PEP-19 from Ca2+-CaM was as least 154 s–1 based on the largest 1H change.
Similar to Ca2+-CaM, amide chemical shifts for residues in the N-domain of apo-CaM, such as Asp58 in Fig. 6A, are unaffected by PEP-19, but show an average decrease in intensity of about 30% due to the increased mass of the CaM·PEP-19 complex. In contrast to Ca2+-CaM, amide chemical shifts in the central linker of apo-CaM show characteristics of slow exchange upon binding to PEP-19. For example, Fig. 6, B and C, shows that two resonances are observed for Thr79 in the presence of 0.5 eq of PEP-19, but a single resonance corresponding to the bound form is observed at 1 eq. A rate of dissociation of PEP-19 from apo-CaM of about 5 s–1 can be estimated from line broadening associated with this slow exchange phenomenon.
FIGURE 6.

Calcium affects the chemical exchange rate between CaM and PEP-19. Panels A–D compare selected backbone amide chemical shifts for 15N-labeled apo-CaM at PEP-19 molar ratios of 0:1 (panel A), 0.5:1 (panel B), 1:1 (panel C), and 10:1 (panel D). The two cross-peaks corresponding to Thr79 at 0.5:1 ratio of PEP19 to CaM are enclosed by a rectangle. The boxed cross-peaks in panel D correspond to C-domain residues that appear at high concentrations of PEP-19. Panel E shows the effect of PEP-19 on cross-peak intensity of backbone amide chemical shifts across the primary sequence of apo-CaM. Relative intensity is defined as the ratio of cross-peak intensity in the presence (I) or absence (I0) of 0.5 molar eq of PEP-19. Residues 2, 64, 66, 92, 98, and 112 were not detected at this concentration of PEP-19.
Residues in the C-domain of apo-CaM are severely broadened upon binding PEP-19. Fig. 6, A–C, shows line broadening of Met109 and Met145 upon addition of 0.5 eq of PEP-19, and broadening beyond detection at 1 eq. Resonances that account for the majority of the C-domain residues are observed at higher concentrations of PEP-19 as shown by the boxed cross-peaks in Fig. 6D. These amides could not be assigned by titration due to the slow exchange limit, and they showed weak intensity, suggesting broadening due to a chemical exchange process.
The selective effect of PEP-19 on the central and C-region of apo-CaM is emphasized by Fig. 6E, which shows the ratio of amide resonance intensity for each amino acid in the presence and absence of 0.5 eq of PEP-19 (I/Io). It is clear from this analysis that the relative intensities of amides in the C-domain are decreased to a much greater extent than those in the N-domain, and that this is a global effect on the C-domain. Thus, PEP-19 binds preferentially to the C-domain of both apo and Ca2+-CaM.
Binding of PEP-19 to Apo-CaM Exhibits Slow kon and koff Rates—The data in Fig. 6 show that CaM amide chemical shifts exhibit slow to intermediate exchange in the absence of Ca2+. This could be explained if PEP-19 bound with higher affinity to apo-CaM relative to Ca2+-CaM, however, PEP-19 was shown to bind to CaM-Sepharose with comparable affinity in the presence or absence of Ca2+ (29). To further explore this phenomenon, we designed a binding assay based on FRET between donor-labeled CaM (CaMD) and acceptor-labeled PEP-19 (PEP-19A) (15). Fig. 7A shows that PEP-19A binds to CaMD with apparent dissociation constants (Kd) of 20 and 13 μm in the presence and absence of Ca2+, respectively. We next determined the effect of Ca2+ on the rates of dissociation (koff) and association (kon) of PEP-19 with CaM. In the presence of Ca2+ both koff and kon were too great to measure using a stopped-flow fluorimeter with a dead time of 1.7 ms. This means the koff is at least 400 s–1, and that the kon is at least 20 μm–1 s–1, given a Kd of 20 μm for binding PEP-19 to Ca2+-CaM. In striking contrast, Fig. 7B shows a slow rate of dissociation of PEP-19A from apo-CaMD of 5.9 s–1, and Fig. 7C shows that a pseudo first-order kon of 1.0 μm–1 s–1 was easily measured for the association of PEP-19A with apo-CaMD.
FIGURE 7.

Ca2+ significantly affects the rates of binding PEP-19 to CaM. Panel A shows equilibrium binding of PEP-19 to apo-CaM (open circles) or Ca2+-CaM (closed circles) measured using a FRET-based assay with donor-labeled CaM and acceptor-labeled PEP-19 (see “Experimental Procedures” for details). Data were corrected for dilution and the effect of nonspecific FRET. Kd values averaged from three separate experiments were 13 ± 2 and 20 ± 4 μm in the absence and presence of Ca2+, respectively. Panel B shows the rate of dissociation of acceptor-labeled PEP-19 (10 μm) from donor-labeled apo-CaM (0.5 μm) upon rapid mixing with excess unlabeled apo-CaM (25 μm). Panel C shows the rates of association of apo-CaM with PEP-19 by rapidly mixing 0.5 μm donor-labeled CaM with 0, 2.5, 5, 10, 15, or 20 μm acceptor-labeled PEP-19. The inset in panel C shows the plot of rate versus PEP-19 concentration to determine the pseudo-first order rate constant. koff and kon values averaged from three separate experiments were 5.9 ± 0.8 s–1 and 1.0 ± 0.3 s–1 μm–1, respectively.
DISCUSSION
PEP-19 Is Intrinsically Disordered but with Residual Structure—Our previous studies focused on the effects of PEP-19 on the Ca2+ binding properties of calmodulin (5, 15). In this report we investigate the biophysical properties of isolated PEP-19, and its interaction with apo- and Ca2+-CaM. Sequence analysis, CD, and NMR data all demonstrate that PEP-19 is a member of the large family of intrinsically disordered proteins, many of which have regulatory roles (30, 31). It is thought that disorder allows regulatory proteins to bind specifically, but with low affinity, to multiple partners. This structural design is ideal for PEP-19 because it must bind to apo- and Ca2+-CaM, but with low affinity such that it does not competitively inhibit activation of other Ca2+-CaM-dependent proteins. Intrinsic disorder also raises the possibility that PEP-19 binds to other EF-hand regulatory proteins that are particularly abundant in neuronal tissue.
From a general perspective, intrinsic disorder appears to be an integral aspect of CaM signaling. Disorder in a CaM target was first demonstrated for melittin, which transitions from a random coil to a helix when bound to Ca2+-CaM (32). This paradigm extends to other small synthetic CaM-binding peptides (33–37), and CaM binding domains (38–40). Computational approaches support the idea that intrinsic disorder is a general feature of many, if not most, CaM binding domains (41). CaM also has a significant degree of backbone plasticity that allows variations in interhelical angles (42). This combination of intrinsic disorder and conformational plasticity in both CaM and its binding proteins allows recognition of multiple targets with high specificity (43, 44).
Although disordered, free PEP-19 exhibits two discrete regions of residual, or nascent structure, that center on Pro37 and span the acidic/IQ motif that binds to CaM and is essential for modulating Ca2+ binding (15). Interestingly, RC3 is also disordered, but exhibits residual structure in the region that binds to CaM (20). We propose that the residual structure plays a role in initiating binding of PEP-19 to CaM by selection of a population of PEP-19 conformers that display helical structure. Support for such a mechanism was recently reported for the intrinsically disordered γ-subunit of phosphodiesterase 6. In this example, the free γ-subunit exhibits a transient structure that resembles the structure of the protein when bound to the α-subunit of transducin (45).
PEP-19 Appears Partially Structured When Bound to CaM—The effects of CaM on PEP-19 backbone amide chemical shifts are very similar in both the presence and absence of Ca2+. Under both conditions, only the regions of residual structure in the C-terminal half of PEP-19 are affected upon binding to CaM, with the majority of residues undergoing a chemical exchange process that broadens the amide chemical shifts beyond detection. Interestingly, PEP-19 resonances remain broadened beyond detection at high concentrations of CaM, and significant broadening is seen even in the presence of TFE. This is consistent with a high degree of conformational exchange in CaM-bound PEP-19.
At least two processes could contribute to conformational exchange of bound PEP-19. The first is that weak binding of PEP-19 to the C-domain does not provide a sufficient decrease in free energy to stabilize a defined conformation of PEP-19. A second potential source of amide broadening is transient interactions with the N-domain that are sensed as a chemical exchange process by bound PEP-19 due to its highly adaptable intrinsically disordered nature. The first mechanism is supported by a recent crystal structure showing that Tyr1675 and Phe1676 anchor the IQ peptide from the human voltage-dependent Ca2+ channel (CaV1.2, Swiss_Prot Q13936) to the C-domain of Ca2+-CaM (46, 47), and that increased disorder in the bound peptide is observed if Phe1676 is changed to Ala (46, 47). Similarly, disorder in CaM-bound PEP-19 may exist because its acidic/IQ motif lacks a corresponding stabilizing Tyr.
Conformational exchange in CaM-bound PEP-19 may have at least two functional advantages. The first is to allow transition of the PEP-19·CaM complex from apo to Ca2+-bound forms. The second is that rapid exchange of bound PEP-19 between multiple conformations may increase rates of Ca2+ association and dissociation in the C-terminal Ca2+ binding loops of CaM via allosteric coupling and conformational gating. It has been proposed that intrinsic disorder enhances allosteric effects (48), and we showed that allosteric coupling between the acidic and IQ region of PEP-19 and between PEP-19 and CaM are necessary to modulate Ca2+ binding to CaM (15). It has also been demonstrated that rates of ligand binding and release can be gated by intermolecular conformational exchange (49), and the rates of conformational exchange in the C-domain of CaM mutant correlate with the Ca2+ off-rate (50). Together these observations support a mechanism in which conformational exchange of CaM-bound PEP-19 exerts an allosteric effect that gates, or regulates the rates of association and dissociation of Ca2+.
Kinetics of Binding PEP-19 to CaM—Although the effects of CaM on the amides of PEP-19 are very similar in the presence and absence of Ca2+, this is not the case for the effect of PEP-19 on amides of Ca2+-CaM versus apo-CaM. Amide chemical shifts of Ca2+-CaM show characteristics of fast exchange on the NMR time scale when titrated with PEP-19 (5). In contrast, binding of PEP-19 causes severe broadening of amides throughout the C-domain of apo-CaM even though PEP-19 binds with similar affinity to both apo- and Ca2+-CaM. We show here that this is due, at least in part, to low koff and kon rates for binding PEP-19 to apo-CaM. Thus, under the conditions used for the experiments in Fig. 6, the kex would be 30 s–1 and >1000 s–1 when apo- and Ca2+-CaM, respectively, are half-saturated with PEP-19.
The underlying structural basis for greatly different rates of binding PEP-19 to apo versus Ca2+-CaM likely resides in the Ca2+-dependent structural dynamics of the C-domain. The Ca2+-bound C-domain is generally well structured, with a defined hydrophobic core and restricted backbone dynamics (42, 51). In contrast, the apo C-domain has an ill-defined aromatic hydrophobic core (51, 52), multiple thermal melting transitions (53), with regions of intrinsic disorder (51, 54), and a high degree of backbone conformational entropy that allows global conformation exchange between at least two conformations (51, 55, 56). Thus, the slow rate of association of PEP-19 with CaM in the absence of Ca2+ may be due, in part, to a low probability of presentation of complimentary transient structures in disordered PEP-19 and the poorly ordered apo C-domain. The slow rate of dissociation may reflect an intrinsically stable apo-CaM·PEP-19 complex, whereas the low overall affinity is driven by the slow association rate.
Computational Model for Effect of PEP-19 on CaM/Target Binding—The data in Fig. 7 suggest that slow release of apo-CaM from PEP-19 could be a rate-limiting step in the transfer of CaM to high affinity target proteins upon elevation of Ca2+ levels. However, PEP-19 greatly increases the intrinsically slow kon rate for Ca2+ binding to the C-domain of CaM, and release of Ca2+-CaM from PEP-19 is also very fast. These factors may compensate for the slow release of apo-CaM from PEP-19, making this a minor pathway in the transition of apo-CaM to target-bound Ca2+-CaM.
We tested these concepts using a variation of the computational model described previously (15). The scheme shown in Fig. 8A incorporates cooperative Ca2+ binding to the C-domain of CaM, together with binding of PEP-19 to all forms of the C-domain, and binding a high affinity target to Ca2+-saturated C-domain. These reactions and corresponding rate constants were incorporated into a computational model to simulate the effect of PEP-19 on the rate of association of the C-domain of CaM with high affinity target proteins (see supplemental materials for details).
FIGURE 8.

Simulated binding of the C-domain of CaM to PEP-19 and a high affinity target protein. Panel A illustrates the linked equilibrium reactions associated with binding Ca2+, PEP-19, and a target protein to the C-domain of CaM. A description of the computational model based on panel A, together with rate and equilibrium binding constants, is given under supplemental materials. Panel B shows the predicted time course of association of target protein with CaM after an increase in free Ca2+ to 1 μm in the presence or absence of 30 mm PEP-19. Initial concentrations of various molecules and complexes are shown in the inset. Panel C shows the maximal rate of formation of Ca2+-CaM·Target complex at increasing concentrations of free Ca2+ in the presence (closed circles) or absence (open circles) of 30 μm PEP-19. The ordinate on the right shows the ratio of rates ± PEP-19 (closed squares).
The simulations shown in Fig. 8B predict that PEP-19 increases the rate of Ca2+-dependent association of target proteins with the C-domain of CaM. Fig. 8C shows that the effect of PEP-19 is greater at lower Ca2+ levels. Thus, the slow rate of release of apo-CaM from PEP-19 shown in Fig. 7 would not inhibit the association of Ca2+-CaM with target proteins due to compensatory effects of PEP-19 on the rate-limiting kinetics of Ca2+ binding to the C-domain. It must be noted that simulation does not take into account contribution of the N-domain of CaM to the rate of association with target proteins, but it would apply to targets that bind predominately to the C-domain, and it provides a framework for thinking about the general role of PEP-19 in CaM-protein interactions.
In summary, conformational plasticity in CaM, and transition of small peptides from coil to helix conformations upon binding to Ca2+-CaM is recognized as an integral aspect of CaM signaling that allows different modes of CaM-target interactions, and a multitude of targets to be recognized with high specificity (43, 44). Here we extend this general paradigm by showing that intact PEP-19 has global intrinsic disorder, and that it may remain partially structured even when bound to CaM. These characteristics support mechanisms by which PEP-19 can bind to either apo- or Ca2+-CaM, and can modulate Ca2+ binding to the C-domain of CaM.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants GM069611 and NS038310. This work was also supported by Robert A. Welch Foundation Grant AU1144. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1, Tables S1–S3, and Models 1 and 2.
Footnotes
The abbreviations used are: RC3, neurogranin; CaM, calmodulin; Ca2+-CaM, Ca2+-bound calmodulin; FRET, fluorescence resonance energy transfer; MOPS, 4-morpholinepropanesulfonic acid; NOESY, nuclear Overhauser effect spectroscopy; IAEDANS, 5-((((2-iodoacetyl)amino)ethyl)amino)-naphthalene-1-sulfonic acid; DDPM, N-(4-dimethylamino-3,5-dinitrophenyl)maleimide; MES, 4-morpholineethanesulfonic acid; TFE, trifluoroethanol; HSQC, heteronuclear single quantrum coherence.
References
- 1.Rhoads, A., and Bahler, M. (2002) FEBS Lett. 513 107–113 [DOI] [PubMed] [Google Scholar]
- 2.Gerendasy, D. D. (1999) J. Neurosci. Res. 58 107–119 [PubMed] [Google Scholar]
- 3.Reck-Peterson, S. L., Provance, W. D., Jr., Mooseker, M. S., and Mercer, J. A. (2000) Biochim. Biophys. Acta 1496 36–51 [DOI] [PubMed] [Google Scholar]
- 4.Jurado, L. A., Sethu, P., Chockalingam, P. S., and Jarrett, H. W. (1999) Physiol. Rev. 79 661–681 [DOI] [PubMed] [Google Scholar]
- 5.Putkey, J. A., Kleerekoper, Q., Gaertner, T. R., and Waxham, M. N. (2003) J. Biol. Chem. 278 49667–49670 [DOI] [PubMed] [Google Scholar]
- 6.Gaertner, T. R., Putkey, J. A., and Waxham, M. N. (2004) J. Biol. Chem. 279 9374–9382 [DOI] [PubMed] [Google Scholar]
- 7.Rudolph, M. C., McManaman, J. L., Phang, T., and Russell, T. (2007) Physiol. Genomics 28 323–336 [DOI] [PubMed] [Google Scholar]
- 8.Xiao, J., Wu, Y., Chen, R., Lin, Y., Wu, L., Tian, W., and Liu, L. (2008) Mol. Cell. Biochem. 309 143–150 [DOI] [PubMed] [Google Scholar]
- 9.Erhardt, J. A., Legos, J. J., Johanson, R. A., Slemmon, J. R., and Wang, X. (2000) Neuroreport 11 3719–3723 [DOI] [PubMed] [Google Scholar]
- 10.Kanazawa, Y., Makino, M., Morishima, Y., Yamada, K., Nabeshima, T., and Shirakaki, Y. (2008) Neuroscience 154 473–481 [DOI] [PubMed] [Google Scholar]
- 11.Slemmon, J. R., Feng, B., and Erhardt, J. A. (2001) Mol. Neurobiol. 22 99–113 [DOI] [PubMed] [Google Scholar]
- 12.Putkey, J. A., Slaughter, G. R., and Means, A. R. (1985) J. Biol. Chem. 260 4704–4712 [PubMed] [Google Scholar]
- 13.Putkey, J. A., and Waxham, M. N. (1996) J. Biol. Chem. 271 29619–29623 [DOI] [PubMed] [Google Scholar]
- 14.Waxham, M. N., Tsai, A.-L., and Putkey, J. A. (1998) J. Biol. Chem. 273 17579–17584 [DOI] [PubMed] [Google Scholar]
- 15.Putkey, J. A., Waxham, M. N., Gaertner, T. A., Brewer, K. J., Goldsmith, M., Kubota, Y., and Kleerekoper, Q. K. (2008) J. Biol. Chem. 283 1401–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Savitzky, A., and Golay, M. J. E. (1964) Anal. Chem. 36 1627–1639 [Google Scholar]
- 17.Ikura, M., Krinks, M., Tochia, D. A., and Bax, A. (1990) FEBS Lett. 266 155–158 [DOI] [PubMed] [Google Scholar]
- 18.Cavanagh, J., Fairbrother, W. J., Palmer, A. G., III, and Skelton, N. J. (1996) Protein NMR Spectroscopy: Principles and Practice, Academic Press, San Diego
- 19.Pelton, J. T., and McLean, L. R. (2000) Anal. Biochem. 277 167–176 [DOI] [PubMed] [Google Scholar]
- 20.Ran, X., Miao, H. H., Sheu, F. S., and Yang, D. (2003) Biochemistry 42 5142–5150 [DOI] [PubMed] [Google Scholar]
- 21.Logan, T. M., Theriault, Y., and Fesik, S. W. (1994) J. Mol. Biol. 236 637–648 [DOI] [PubMed] [Google Scholar]
- 22.Obradovic, Z., Peng, K., Vucetic, S., Radivojac, P., and Dunker, A. K. (2005) Proteins 61 Suppl. 7, 176–182 [DOI] [PubMed] [Google Scholar]
- 23.Yang, Z. R., Thomson, R., McNeil, P., and Esnouf, R. M. (2005) Bioinformatics 21 3369–3376 [DOI] [PubMed] [Google Scholar]
- 24.Lacroix, E., Viguera, A. R., and Serrano, L. (1998) J. Mol. Biol. 284 173–191 [DOI] [PubMed] [Google Scholar]
- 25.Munoz, V., and Serrano, L. (1994) Nat. Struct. Biol. 1 399–409 [DOI] [PubMed] [Google Scholar]
- 26.Dyson, H. J., and Wright, P. E. (2001) Methods Enzymol. 339 258–270 [DOI] [PubMed] [Google Scholar]
- 27.Schwarzinger, S., Kroon, G. J., Foss, T. R., Chung, J., Wright, P. E., and Dyson, H. J. (2001) J. Am. Chem. Soc. 123 2970–2978 [DOI] [PubMed] [Google Scholar]
- 28.Wishart, D. S., and Case, D. A. (2001) Method Enzymol. 338 3–34 [DOI] [PubMed] [Google Scholar]
- 29.Slemmon, J. R., Morgan, J. I., Fullerton, S. M., Danho, W., Hilbush, B. S., and Wengenack, T. M. (1996) J. Biol. Chem. 271 15911–15917 [DOI] [PubMed] [Google Scholar]
- 30.Dunker, A. K., Cortese, M. S., Romero, P., Iakoucheva, L. M., and Uversky, V. N. (2005) FEBS J. 272 5129–5148 [DOI] [PubMed] [Google Scholar]
- 31.Uversky, V. N., Oldfield, C. J., and Dunker, A. K. (2008) Annu. Rev. Biophys. 37 215–246 [DOI] [PubMed] [Google Scholar]
- 32.Seeholzer, S. H., Cohn, M., Putkey, J. A., Means, A. R., and Crespi, H. L. (1986) Proc. Natl. Acad. Sci. U. S. A. 83 3634–3638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Osawa, M., Tokumitsu, H., Swindells, M. B., Kurihara, H., Orita, M., Shibanuma, T., Furuya, T., and Ikura, M. (1999) Nat. Struct. Biol. 6 819–824 [DOI] [PubMed] [Google Scholar]
- 34.Prêcheur, B., Munier, H., Mispelter, J., Bârzu, O., and Craescu, C. T. (1992) Biochemistry 31 229–236 [DOI] [PubMed] [Google Scholar]
- 35.Craescu, C. T., Bouhss, A., Mispelter, J., Diesis, E., Popescu, A., Chiriac, M., and Barzu, O. (1995) J. Biol. Chem. 270 7088–7096 [DOI] [PubMed] [Google Scholar]
- 36.Kranz, J. K., Flynn, P. F., Fuentes, E. J., and Wand, A. J. (2002) Biochemistry 41 2599–2608 [DOI] [PubMed] [Google Scholar]
- 37.Gerendasy, D. D., Herron, S. R., Jennings, P. A., and Sutcliffe, J. G. (1995) J. Biol. Chem. 270 6741–6750 [DOI] [PubMed] [Google Scholar]
- 38.Permyakov, S. E., Millett, I. S., Doniach, S., Permyakov, E. A., and Uversky, V. N. (2003) Proteins 53 855–862 [DOI] [PubMed] [Google Scholar]
- 39.Zhou, N., Yuan, T., Mak, A. S., and Vogel, H. J. (1997) Biochemistry 36 2817–2825 [DOI] [PubMed] [Google Scholar]
- 40.Tapp, H., Al-Nagger, I. M., Yarmola, E. G., Harrison, A., Shaw, G., Edison, A. S., and Bubb, M. R. (2005) J. Biol. Chem. 280 9946–9956 [DOI] [PubMed] [Google Scholar]
- 41.Radivojac, P., Vucetic, S., O'Connor, T. R., Uversky, V. N., Obradovic, Z., and Dunker, A. K. (2006) Proteins 63 398–410 [DOI] [PubMed] [Google Scholar]
- 42.Chou, J. J., Li, S., Klee, C. B., and Bax, A. (2001) Nat. Struct. Biol. 8 990–997 [DOI] [PubMed] [Google Scholar]
- 43.Hoeflich, K. P., and Ikura, M. (2002) Cell 108 739–742 [DOI] [PubMed] [Google Scholar]
- 44.Vetter, S. W., and Leclerc, E. (2003) Eur. J. Biochem. 270 404–414 [DOI] [PubMed] [Google Scholar]
- 45.Song, J., Guo, L. W., Muradov, H., Artemyev, N. O., Ruoho, A. E., and Markley, J. L. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 1505–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fallon, J. L., Halling, D. B., Hamilton, S. L., and Quiocho, F. A. (2005) Structure 13 1881–1886 [DOI] [PubMed] [Google Scholar]
- 47.Van Petegem, F., Chatelain, F. C., and Minor, D. L., Jr. (2005) Nat. Struct. Mol. Biol. 12 1108–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shmukler, B. E., Bond, C. T., Wilhelm, S., Bruening-Wright, A., Maylie, J., Adelman, J. P., and Alper, S. L. (2001) Biochim. Biophys. Acta 1518 36–46 [DOI] [PubMed] [Google Scholar]
- 49.Palmer, A. G., III (2004) Chem. Rev. 104 3623–3640 [DOI] [PubMed] [Google Scholar]
- 50.Evenas, J., Malmendal, A., and Akke, M. (2001) Structure 9 185–195 [DOI] [PubMed] [Google Scholar]
- 51.Kuboniwa, H., Nico, T., Grzesiek, S., Ren, H., Klee, C. B., and Bax, A. (1995) Nat. Struct. Biol. 2 768–776 [DOI] [PubMed] [Google Scholar]
- 52.Zhang, M., Tanaka, T., and Ikura, M. (1995) Nat. Struct. Biol. 2 258–267 [DOI] [PubMed] [Google Scholar]
- 53.Tsalkova, T. N., and Privalov, P. L. (1985) J. Mol. Biol. 181 533–544 [DOI] [PubMed] [Google Scholar]
- 54.Lundstrom, P., Mulder, F. A. A., and Akke, M. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 16984–16989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rabl, C. R., Martin, S. R., Neumann, E., and Bayley, P. M. (2002) Biophys. Chem. 101–102 553–564 [DOI] [PubMed] [Google Scholar]
- 56.Chen, Y.-G., and Hummer, G. (2007) J. Am. Chem. Soc. 129 2414–2415 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.