

Abstract
The synthetic ciguatoxin CTX3C has been shown to activate tetrodotoxin (TTX)-sensitive sodium channels (Nav1.2, Nav1.4, and Nav1.5) by accelerating activation kinetics and shifting the activation curve toward hyperpolarization (Yamaoka, K., Inoue, M., Miyahara, H., Miyazaki, K., and Hirama, M. (2004) Br. J. Pharmacol. 142, 879–889). In this study, we further explored the effects of CTX3C on the TTX-resistant sodium channel Nav1.8. TTX-resistant channels have been shown to be involved in transducing pain and related sensations (Akopian, A. N., Sivilotti, L., and Wood, J. N. (1996) Nature 379, 257–262). Thus, we hypothesized that ciguatoxin-induced activation of the Nav1.8 current would account for the neurological symptoms of ciguatera poisoning. We found that 0.1 μm CTX3C preferentially affected the activation process of the Nav1.8 channel compared with those of the Nav1.2 and Nav1.4 channels. Importantly, without stimulation, 0.1 μm CTX3C induced a large leakage current (IL). The conductance of the IL calculated relative to the maximum conductance (Gmax) was 10 times larger than that of Nav1.2 or Nav1.4. To determine the molecular domain of Nav1.8 responsible for conferring higher sensitivity to CTX3C, we made two chimeric constructs from Nav1.4 and Nav1.8. Chimeras containing the N-terminal half of Nav1.8 exhibited a large response similar to wild-type Nav1.8, indicating that the region conferring high sensitivity to ciguatoxin action is located in the D1 or D2 domains.
Ciguatoxin is a lipophilic cyclic polyether with 13 ether rings (Fig. 1) derived from the dinoflagellate Gambierdiscus toxicus (1) and is a cause of ciguatera, which is a widespread fish poisoning that presents a variety of neurological symptoms, including hyperalgesia and allodynia. These symptoms might be a consequence of the direct interaction of ciguatoxin with voltage-dependent Na+ channels expressed in the peripheral nervous system. CTX1B2 suppressed fast sodium channel inactivation, evoked spontaneous action-potential firing of frog nodal membranes (2), and induced a tetrodotoxin-sensitive (TTX-S) persistent leakage current of rat DRG neurons (3). Furthermore, it has been shown that ciguatoxin competes with brevetoxin binding to site 5 of Nav1.2 channels by radioactive photolabeling techniques (4, 5). However, it is still uncertain how ciguatoxin causes pain or how it modulates sensory inputs by changing the activity of sodium channels expressed in DRG neurons. Multiple sodium channel isoforms are expressed in DRG neurons, including both TTX-S (Nav1.1, Nav1.6, and Nav1.7) and TTX-resistant (TTX-R) (Nav1.8 and Nav1.9) Na+ channels (6). Among these, Nav1.8 might be of great importance for transmitting pain signals, because it is expressed exclusively by primary afferent neurons (7, 8), and it also carries more than 80% of the current at the rising phase of the action potential of small DRGs (9). In addition, deletion of the Nav1.8 gene in mice produced mutants that were resistant to noxious mechanical stimuli and refractory to the development of inflammatory hyperalgesia (7, 8). Similarly, antisense oligonucleotides directed against Nav1.8 reversed neuropathic pain behavior (10). Acute and selective pharmacological blockade of Nav1.8 sodium channels in vivo produces significant antinociception in animal models of neuropathic and inflammatory pain (11). Thus, ciguatoxin might preferentially attack Nav1.8 compared with TTX-S Na+ channels, explaining the neurological symptoms of ciguatera. We tested the effects of synthetic ciguatoxin CTX3C on heterologously expressed Nav1.2, Nav1.4, and Nav1.8 channels in either HEK293 or ND7-23 cells. To confirm the preferential effects of CTX3C on Nav1.8 channels, we also tested the effects on chimeric channels formed from the N-terminal domains of Nav1.8 and the C-terminal domains of Nav1.4, or vice versa. Nav1.8 expressed in ND7-23 cells had an apparently increased sensitivity to ciguatoxins compared with that of Nav1.2 and Nav1.4 expressed in HEK293 cells. Data from chimeric channels indicated that high sensitivity to ciguatoxins of Nav1.8 was derived of the N-terminal half of the Nav1.8 molecule.
FIGURE 1.

Structures of CTX3C, 51-OH-CTX3C, and CTX1B.
EXPERIMENTAL PROCEDURES
All procedures involving animal handling and experimental protocols were approved by the Institutional Animal Care Committee (Hiroshima International University and Hiroshima University) and carried out in accordance with the guidelines issued by the National Institutes of Health.
Construction of Chimeras—The α-subunits of rat skeletal muscle Na+ channel (Nav1.4) (12) and rat brain type II Na+ channel (Nav1.2) (13) cDNAs were cloned into mammalian expression vectors of pcDNA3.1(–) (Invitrogen) and pCI-neo (Promega, Madison, WI), respectively. The sensory neuron-specific (SNS) TTX-R Na+ channel (Nav1.8) cDNA (7) was cloned into pcDNA3.1(–). Chimeras were constructed using the two cDNAs by swapping domains; the N-terminal half-domains (D1D2) of the SNS cDNA were flanked by the C-terminal half-domains (D3D4) of the μ1 cDNA resulting in SS/MM (the first two domains derived from SNS Na+ channels followed by the last two domains derived from μ1Na+ channels), and vice versa resulting in MM/SS, at the junction of the intracellular loop between D2 and D3, corresponding to Pro-918 for μ1 cDNA or Pro-1000 for SNS cDNA. Briefly, the insertion of five bases, GGCCG, at the proline coding codon, C/CC, created the NotI site, GC/GGCCGC, using the QuikChange mutagenesis kit (Stratagene, La Jolla, CA). Domain swapping was performed at the created NotI sites. Finally, the artificially inserted five bases were deleted using the QuikChange mutagenesis kit. All of the resulting constructs were confirmed with restriction mapping and sequencing using an ABI PRISM™ 310 Genetic Analyzer (Applied Biosystems, Foster City, CA).
Isolation of Rat p11—Total cellular RNA was isolated from myometrial tissue dissected from adult Sprague-Dawley rats following the TRIzol protocol (Invitrogen). mRNA was purified by a FirstTrack mRNA purification kit (Invitrogen). First-strand and second-strand cDNA were synthesized with a Marathon cDNA kit using poly(dT) primer (BD Biosciences). The final cDNA product was used as a myometrium marathon cDNA template for PCR amplification with rat DRG p11-specific forward (5′-ATGCCATCCCAAATGGAGCAT-3′) and reverse primers (5′-CTACTTCTTCTGCTTCATGTGTACTAC-3′). The rat p11 sequence has been deposited with DNA Data Bank of Japan (DDBJ) data libraries under the accession number AB453238.
Transient Transfection and Cell Culture—The wild-type rat Nav1.4 and rat Nav1.2 cDNAs were transiently transfected into HEK293 cells with the S65A bright green fluorescent protein mutant in the vector pCA-GFP (14) to allow the detection of transfected cells. Chimeric mutants of SS/MM and MM/SS were cotransfected into HEK293 cells with the Naβ1 (15) subunit subcloned into pIRES-hrGFP-1a (Stratagene) to increase the expression levels as well as for the detection of transfected cells. Nav1.8 was poorly expressed in HEK293 cells. Thus, we employed a more suitable heterologous expression system for Nav1.8 Na+ channels. The wild-type SNS cDNA was transiently transfected into ND7-23 cells purchased from Sigma with p11, subcloned into pIRES-hrGFP-1a. John et al. (16) reported that the ND7-23 cell line allowed stable expression of rat Nav1.8 channels with biophysical properties that closely resemble the native TTX-resistant currents in DRG neurons. Also, p11 was reported to promote the translocation of Nav1.8 to the plasma membrane, producing functional channels (17). The transfection of cDNAs into HEK293 cells or ND7-23 cells was performed with Effectine reagent (Qiagen K. K., Tokyo, Japan). Cells were grown to 50% confluence in Dulbecco's modified Eagle's medium (Invitrogen), containing 10% fetal bovine serum (BioWhittaker, Walkersville, MD), 30 units/ml penicillin G (Invitrogen), and 30 μg/ml streptomycin (Invitrogen), in a humidified atmosphere of 5% CO2 and 95% air at 37 °C. The transfected cells were used for electrophysiological experiments through post-transfection day 3. Transfection-positive cells were identified by epifluorescence microscopy performed using a 100-watt mercury lamp and a standard fluorescein isothiocyanate filter set (Nikon, Tokyo, Japan). Single isolated green fluorescent cells were then studied as described below.
Solutions and Chemicals—The bath solution contained 70 mm NaCl, 67 mm N-methyl-d-glucamine, 1 mm CaCl2, 1.5 mm MgCl2, 10 mm glucose, and 5 mm HEPES (pH 7.4). TTX (0.3 μm) was added only when currents from ND7-23 cells were recorded to eliminate INa other than Nav1.8. The pipette solution contained 70 mm CsF, 60 mm CsCl, 12 mm NaF, 5 mm EGTA, and 5 mm HEPES (pH 7.4). Ciguatoxin congeners, CTX3C, and 51-hydroxy-CTX3C (51-OH-CTX3C) were synthesized de novo by the method described previously (18–20). The stock solution of the ciguatoxin congeners (1 mm) in DMSO was diluted with bath solution to a final concentration of 0.1–10 μm. Bath solution was continuously supplied at a rate of 2 ml min–1. Separately, as described previously (21), CTX3C solution was directly infused into the bath chamber with siphon and vacuum tubing in a Y configuration, so that the cells under study were positioned in the center of the stream.
Electrophysiological Recording and Data Analysis—Macroscopic INa from transfected cells was measured using the whole-cell patch clamp method. Whole-cell patch pipettes with a resistance of 1.5–3 megohms were used to achieve optimum voltage control. Whole-cell currents (filtered at 10 kHz) were recorded using an Axopatch 200B amplifier (Axon Instruments; Foster City, CA), and more than 80% of the series resistance was compensated to minimize voltage errors. Recordings were started 10 min after establishing a whole-cell recording configuration. Whole-cell membrane currents were digitized at a sampling rate of 50–100 kHz with a 12-bit analogue-to-digital converter (DigiData 1321A interface; Axon Instruments), controlled by pClamp software (version 8; Axon Instruments). INa was determined by subtraction of linear capacitative and leak current with the P/4 protocol (22) in all experiments, except for the case where a relatively large background current was induced with CTX3C or those that were undertaken to measure the voltage dependence of steady-state inactivation. All experiments were conducted at room temperature (22–24 °C). Data are presented as mean ± S.E. along with the number of observations (n), unless otherwise stated. Statistically significant differences between group means were determined by the Student's t test for paired or unpaired observations, as appropriate. The criterion for statistical significance was p < 0.05.
RESULTS
Ciguatoxin-induced Background Currents through Na+ Channels—It has been demonstrated that at least 1 μm CTX3C is necessary to modulate Nav1.2, Nav1.4, and Nav1.5 channels (21) when the holding potential is kept at –140 mV. In this study, the holding potential was set at –100 mV. Under these conditions, 0.1 μm CTX3C was sufficient to induce activation of Nav1.2 (data not shown) and Nav1.4 channels expressed in HEK293 cells, as demonstrated by a negative shift of threshold potential, a shortening of time to peak, and a suppression of the amplitude of the sodium current INa (Fig. 2B). In the presence of TTX (0.3 μm), we recorded the INa of Nav1.8 channels expressed in ND7-23 cells (23), the functional expression of which is enhanced with cotransfection of p11 as reported previously (16, 17). Nav1.8 channels were activated and reached maximum currents at more depolarized potentials (from –50 to –30 mV for threshold and from –10 to +10 mV for peak potentials) and were inactivated with a 10 times slower time course than those of Nav1.4 (Fig. 2C). The most prominent difference between Nav1.4 and Nav1.8 channels was that background (or leak) currents were induced in the latter without depolarizing pulses at –100 mV (Fig. 3). CTX3C (0.1 μm) induced a large current of more than 400 pA in Nav1.8, but not in Nav1.4 (∼6 pA), when no depolarizing pulses were applied from a holding potential of –100 mV, although both cells exhibited comparable maximum peak currents (–2,650 pA versus –2,370 pA for Nav1.8 and Nav1.4, respectively) in the absence of CTX3C.
FIGURE 2.

Typical effects of 0.1 μm CTX3C on the current-voltage relationships of Nav1.4 and Nav1.8 Na+ channels. A, depolarizing test pulses (20 ms) from a holding potential of –100 to 60 mV in 10-mV increments were given every 1 s, before (left) and 5 min after (right) perfusion with 0.1 μm CTX3C. For clarity, the current records of INa are displayed only in 20-mV steps. The peak INa at each voltage step was measured and plotted against the membrane potential (shown at the bottom of B and C) in the absence (▪) and presence (○) of 0.1 μm CTX3C. Slower activation and incomplete inactivation of Nav1.8 channels compared with Nav1.4 channels are clearly shown in the current records shown above each I-V graph. Voltage steps near the resting potential evoked earlier activation in the presence of CTX3C in both channels. Note the suppression of the peak INa by CTX3C.
FIGURE 3.

Spontaneous induction of sustained current by CTX3C without depolarized steps. The membrane was continuously clamped at –100 mV during the perfusion of 0.1 μm CTX3C. This induced a spontaneous leak current of ∼6 pA in Nav1.4 (A), but a large current in Nav1.8 (B). Further depolarization to –80 mV (the bottom traces in each panel indicate the membrane potential applied) increased the currents in both Nav1.4 and Nav1.8 channels, but the Nav1.8 channels were already susceptible to inactivation at –80 mV.
Quantification of Ciguatoxin-induced Resting Sodium Conductance—As the number of functionally expressed channels varies from cell to cell, it is not appropriate simply to compare the amplitude of ciguatoxin-induced currents between cells. Maximum conductance in the absence of CTX3C (Gmax-control) is a good measure of the number of functionally expressed channels in each cell. Thus, we evaluated the potency of CTX3C to induce background conductance (resting sodium conductance) at –100 mV in the presence of 0.1 μm CTX3C relative to Gmax-control. CTX3C (0.1 μm) induced a resting sodium conductance of 0.84 ± 0.27% (n = 7) relative to the Gmax-control in Nav1.8, which was significantly larger than that in Nav1.4 (0.08 ± 0.03%, n = 5; p < 0.05; Fig. 4).
FIGURE 4.

Structure/activity relationships of wild-type and chimeric Nav1.8/1.4 channels. A, schematic presentation of chimeric constructs. B, effects of CTX3C on resting Na+ conductance of three wild-type Na+ channel subtypes and two chimeras. The ciguatoxin-induced leakage current magnitudes were quantitatively evaluated by dividing the ciguatoxin-induced resting conductance by the maximum conductance (Gmax) in the absence of CTX3C. The Nav1.8 and SS/MM showed 10 times larger conductance than those of Nav1.2, -1.4, and MM/SS.
Molecular Determinants of Nav1.8 Exhibiting Larger Resting Sodium Conductance in Response to CTX3C—To test whether the effects of CTX3C on Nav1.8 expressed in ND7-23 cells are derived from ND7-23 cell environment or Nav1.8 molecule itself, we made two chimeras from Nav1.4 and Nav1.8 channels that could be successfully expressed in HEK293 cells: MM/SS and SS/MM. The former contained the two N-terminal domains of Nav1.4 followed by the C-terminal two domains of Nav1.8, and vice versa for the latter (Fig. 4A). CTX3C (0.1 μm) induced a resting sodium conductance of 0.85 ± 0.12% (n = 6) in SS/MM-expressing cells, relative to the Gmax-control, which was significantly larger (p < 0.01) than that of the MM/SS-expressing cells (0.05 ± 0.03%; n = 5), but similar to Nav1.8 in ND7-23 cells. We further tested whether the property of SS/MM could be preserved in ND7-23 cells. SS/MM in ND7-23 cells responded to 0.1 μm 51-OH-CTX3C producing a resting sodium conductance of 0.40 ± 0.20% (n = 4) relative to the Gmax-control, which are comparable with data from SS/MM in HEK293 cells in the presence of 0.1 μm 51-OH-CTX3C, 1.91 ± 1.33% (n = 3). We used 51-OH-CTX3C here, because of a lack of availability of CTX3C. 51-OH-CTX3C had a quantitatively similar effect on resting sodium conductance of SS/MM in HEK293 cells to that of CTX3C (see Fig. 4B).
Changes in Sodium Channel Kinetics Induced by CTX3C—Induction of resting sodium conductance by CTX3C might be an extreme form of leftward shift of the activation curve that can be observed using the conductance-voltage relationship (Fig. 5). The voltage dependences of activation of Nav1.4, Nav1.8, and the related chimera (SS/MM and MM/SS) channels in the absence of CTX3C were adequately described by a single Boltzmann distribution; however, the sum of two Boltzmann distributions was required for ciguatoxin-modified Nav1.8 (Fig. 5B) and SS/MM channels. The voltages required for half-maximal activation (Vact1/2; midpoint) and kact (slope factor) for each construct are shown in Table 1. The Vact1/2 for Nav1.8 and SS/MM in control cells was more depolarized by ∼20–30 mV than those for Nav1.4 and MM/SS, respectively, indicating that the high threshold nature of Nav1.8 is derived from the N-terminal domains of the molecule (D1/D2). Ciguatoxin application induced a leftward shift of the Vact1/2 of Nav1.4 to a limited extent (∼3 mV), but more substantially in MM/SS, although the slopes of the two constructs were not notably changed (Table 1). A significant effect on the Vact1/2 was seen in both Nav1.8 and SS/MM; an extra component of the activation curve (the low threshold component) was created in a more hyperpolarized range. The Vact1/2 values for the high threshold component for Nav1.8 and SS/MM, respectively, were depolarized by 11 and 8 mV compared with that of the single component in each respective control (Table 1). In addition, the slope factors of the ciguatoxin-created low threshold components of Nav1.8-expressing and SS/MM-expressing cells were remarkably large (–18.3 ± 2.8 and –17.8 ± 1.4 mV, respectively). The creation of this Nav1.8-selective component may be related to the induction of resting sodium conductance.
FIGURE 5.

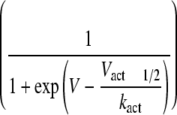 |
 |
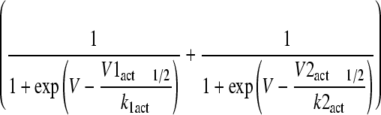 |
TABLE 1.
Ciguatoxin-induced changes in activation properties measured with conductance-voltage relationship

* p < 0.05, statistical comparisons were performed using the paired two-tailed Student's t test.
Depolarizing square pulses to –10 mV from a holding potential of –100 mV in a voltage clamp condition activated the channels and let their currents reach peak values within 0.44 ± 0.05 ms (n = 3) in Nav1.2-expressing cells and 0.43 ± 0.04 ms (n = 5) in Nav1.4-expressing cells; however, this was much slower in Nav1.8-expressing cells as the time to peak for Nav1.8 was 1.51 ± 0.28 ms (n = 6). The values of the time to peaks of the related constructs, SS/MM and MM/SS, fell between these limits (1.01 ± 0.05 ms, n = 3, for SS/MM and 0.76 ± 0.16 ms, n = 4, for MM/SS). CTX3C (0.1 μm) accelerated the activation process of the Na+ channels by shortening the time to peaks by 26.0 ± 8.2% (n = 3) in Nav1.2 and by 11.3 ± 3.3% (n = 5) in Nav1.4, but this effect was significantly larger (p < 0.05) in Nav1.8 (by 31.9 ± 6.9%, n = 6). Again, the related constructs, SS/MM and MM/SS, showed values between those of Nav1.4 and Nav1.8.
The suppressive effect of CTX3C on the Na+ channels can be seen in the changes in maximum conductance (Gmax) estimated by fitting conductance-voltage relationships using the Boltzmann equation (Fig. 5). When the activation curve was made of the sum of two components, the Gmax was treated as the sum of each amplitude (Gmax = Gmax1 + Gmax2). CTX3C (0.1 μm) equally reduced the Gmax of Nav1.2, Nav1.4, Nav1.8, and SS/MM by around 18–22%, except in MM/SS-transfected cells, where it was reduced by 37% (Table 1).
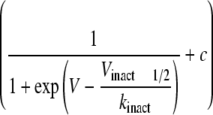
The steady-state voltage dependence of fast inactivation was evaluated by applying a 500-ms conditioning prepulse to various potentials from a holding potential of –100 mV, followed by a brief repriming interval (500 μs) and finally a test pulse to –20 mV. The relationship between the test INa and membrane potential in the presence and absence of CTX3C was fitted by the Boltzmann equation with two parameters, Vinact1/2 (half-inactivation voltage) and slope factor (kinact). The values of Vinact1/2 in the controls indicated a depolarized voltage-dependent relationship in the steady-state inactivation of Nav1.8, SS/MM, and MM/SS compared with that of Nav1.4 (Table 2). In contrast to activation-curve studies, ciguatoxin did not create an extra component of steady-state inactivation. The slope factors were relatively constant. However, the voltage dependence was partly affected by 0.1 μm CTX3C, as the Vinact1/2 values were significantly hyperpolarized in Nav1.2, Nav1.4, and Nav1.8 (Table 2). The Vinact1/2 was less affected in SS/MM, whereas a large shift of Vinact1/2 was observed in MM/SS (Table 2).
TABLE 2.
Ciguatoxin-induced changes in inactivation properties measured with steady-state inactivation curves

* p < 0.05.
** p < 0.01; statistical comparisons were performed using the paired two-tailed Student's t test.
Effects on Rate of Recovery from Slow Inactivation—It has been demonstrated that TTX-R sodium channels show faster repriming kinetics from inactivation than TTX-S sodium channels, and CTX1B further shortened their recovery time, with consequent increases in excitability, such as an easy transition to repetitive firing (3). We did similar experiments with a standard two-pulse protocol and a variable interpulse interval (5, 10, 20, 40, 128, 256, 512, 1,000, and 2,000 ms). A conditioning prepulse to 0 mV was used to inactivate sodium channels, after which a 4-ms depolarizing test pulse to 0 mV was applied. A 500-ms prepulse duration was used to fully inactivate the sodium channels. Typical recovery time courses from inactivation for Nav1.4 and Nav1.8 are shown in Fig. 6. In the control, the Nav1.8 channels recovered with two time constants of τ1 = 15.1 ± 3.5 ms and τ2 = 670 ± 238 ms (n = 4), and the relative amplitude between the two components, Aτ2/Aτ1, was 0.27 ± 0.1; the corresponding values for Nav1.4 were τ1 = 32.4 ± 15.6 ms, τ2 = 580 ± 113 ms, and Aτ2/Aτ1 = 1.15 ± 0.24 (n = 5). CTX3C (0.1 μm) slowed the time constants to τ1 = 18.4 ± 4.0 ms, τ2 = 17,500 ± 14,200 ms, and Aτ2/Aτ1 = 0.23 ± 0.03 for Nav1.8 (n = 4), and τ1 = 21.0 ± 9.2 ms, τ2 = 706 ± 252 ms, and Aτ2/Aτ1 = 1.48 ± 0.34 for Nav1.4 (n = 5), respectively (Table 3). From these data, it is difficult to determine which channel has the faster recovery kinetics. For clarity, the available, noninactivated fraction of channels at the 5-ms (R5) and 128-ms (R128) interval are indicated in Table 3. Nav1.8 recovered by 52 ± 4% for 5 ms and 93 ± 0.2% for 128 ms in the control, but this was slowed by CTX3C to 43 ± 7 and 87 ± 13% (n = 3), respectively. Nav1.4 recovered more slowly than Nav1.8 and to a more limited extent (by 78 ± 1.9% in the control and 58 ± 8.3% in the presence of CTX3C during 128 ms). These results do not support the contribution of change in repriming kinetics by CTX3C to the hyperexcitability of DRG neurons, in contrast with the findings of Strachan et al. (3).
FIGURE 6.

Slowed and suppressed recovery from inactivation by CTX3C. A, pulse protocol with variable intervals (ΔT) between the conditioning pulse and the test pulse. Representative recovery data from Nav1.4 (B) and Nav1.8 (C) are shown. The duration of the conditioning depolarization was set to 500 ms. The recovered INa values evoked by the test pulse (It) relative to those without the conditioning pulse (I0) were plotted against the interpulse intervals in milliseconds (Δt in the axis). The recovery time courses were fitted by the sum of two exponentials as follows: (It/I0) = A1τ1·exp(–(t/τ1) + A2τ2·exp(–(t/τ2) + C. See Table 3 for the fitted parameters. Nav1.8 recovered slightly faster than Nav1.4. However, the slower time constants for Nav1.8 tended to be larger than that of Nav1.4.
TABLE 3.
Ciguatoxin-induced changes in recovery time course from slow inactivation
| n | R5 | R128 | τ1 | τ2 | Aτ2/Aτ1 | |
|---|---|---|---|---|---|---|
| ms | ms | |||||
| Control | ||||||
| Nav1.2 | 3 | 0.60 ± 0.07 | 0.84 ± 0.05 | 30.5 ± 27 | 475 ± 171 | 0.79 ± 0.26 |
| Nav1.4 | 5 | 0.55 ± 0.02 | 0.78 ± 0.02 | 32.4 ± 15.6 | 580 ± 113 | 1.15 ± 0.24 |
| Nav1.8 | 4 | 0.52 ± 0.04 | 0.93 ± 0.02 | 15.3 ± 3.5 | 670 ± 238 | 0.27 ± 0.1 |
| CTX3C | ||||||
| Nav1.2 | 3 | 0.26 ± 0.08 | 0.59 ± 0.13 | 30.6 ± 16.0 | 1970 ± 397 | 1.23 ± 0.5 |
| Nav1.4 | 5 | 0.40 ± 0.11 | 0.58 ± 0.09 | 21.0 ± 9.2 | 706 ± 252 | 1.48 ± 0.34 |
| Nav1.8 | 4 | 0.43 ± 0.07 | 0.87 ± 0.02 | 18.7 ± 4.0 | 17,500 ± 14,200 | 0.23 ± 0.03 |
DISCUSSION
Typical and persistent signs of ciguatera induced by ciguatoxin are neurological symptoms such as hyperalgesia or allodynia. Recent studies on TTX-R Na+ channels by deletion of the gene encoding Nav1.8 (7, 8, 24–26), by ablating Nav1.8-mRNA by antisense oligodeoxynucleotides (10, 27–30), or by evaluating changes in the expression levels of Nav1.8 after nerve injury (10, 27, 29) have revealed the role of Nav1.8 channels in pain-sensing pathways. Thus, it is reasonable to suggest that ciguatoxin might have profound effects on Nav1.8 channels in particular. Yet ciguatoxin has not been documented to exhibit a clear preference for Na+ channel subtypes. In our previous study, we found no significant difference in the effects of CTX3C on Nav1.2, Nav1.4, and Nav1.5 channels (21). By contrast, Strachan et al. (3) have reported differential actions of CTX1B (P-CTX-1) between TTX-S and TTX-R Na+ channels of DRG neurons. CTX1B induced a leakage current for TTX-S, although it hastened the repriming kinetics (recovery from inactivation) of TTX-R Na+ channels. These data suggest a greater contribution of TTX-S currents to increased neuronal excitability. In this study, our results were to the contrary. First, CTX3C (0.1 μm) shifted the activation curves to the more hyperpolarized direction or created extra low threshold components with a larger kact (less steep) in Nav1.8. Second, CTX3C exhibited an ability to induce 10 times larger resting sodium conductance in the Nav1.8-expressing cells than in the others (Nav1.2, Nav1.4, and MM/SS). Third, CTX3C slowed the repriming kinetics of both Nav1.4 and Nav1.8 channels. The first two properties of ciguatoxin modulation listed above are in good accordance with the neurological symptoms of ciguatera, assuming that Nav1.8 is the main regulator of the pain-sensing pathway in DRG neurons.
In this study, three subgroups of wild-type sodium channel α-subunits were transiently expressed in HEK293 cells for Nav1.2 and Nav1.4 and in ND7-23 cells for Nav1.8 (these currents could not be recorded in HEK293 cells possibly because of a lack of some cofactors). These environmental differences in ND7-23 cells might therefore favor ciguatoxin actions, resulting in larger effects on Nav1.8. However, this was not the case in our study. We were able to express chimeric channels of Nav1.4 and Nav1.8 (SS/MM and MM/SS) in HEK293 cells. The SS/MM chimera mimicked the wild-type Nav1.8 high sensitivity to CTX3C by showing a similar large resting sodium conductance. In addition, the ciguatoxin-sensitive chimera, SS/MM expressed in ND7-23 cells exhibited a large resting sodium conductance comparable with that in HEK293 cells, in the presence of 0.1 μm 51-OH-CTX3C. These findings suggested that the ND7-23 environment of the cell was not a major factor in inducing higher sensitivity to CTX3C; in other words, it indicated a critical role of the N-terminal domains of Nav1.8 for preferential CTX3C binding.
Our data, in contrast to those reported by Strachan et al. (3), suggest that the primary means of ciguatoxin modulation of pain sensing is mediated by TTX-R and not TTX-S Na+ channels. The TTX-S currents recorded by Strachan et al. (3) could be mainly from Nav1.7, which has been reported to be expressed abundantly throughout the peripheral nervous system (31). Nav1.7 might behave differently from Nav1.2 or Nav1.4 against ciguatoxin, as knock-out mice with nociceptor-specific gene deletion of Nav1.7 exhibited reduced sensitivity to noxious mechanical stimuli and inflammation-induced hyperalgesia (32). Also, mutations in Nav1.7 channels have been found to be the cause of human erythermalgia, characterized by intermittent burning pain with redness and heat in the extremities (33). Examining the effects of ciguatoxin on Nav1.7 in comparison with Nav1.8 would resolve this question. In severely intoxicated cases of ciguatera, autonomic dysfunction might appear as bradycardia or hypotension. These symptoms imply that Nav1.7 predominantly expressed in the autonomic nervous system might be affected, but they need a higher dose of ciguatoxin than Nav1.8. Another possibility to explain the discrepancy between our results and those reported by Strachan et al. (3) is that chemically modified states of Na+ channels from freshly isolated DRG might differ from our heterologously expressed Na+ channels. For example, in diabetic states, Nav1.8 currents were augmented by serine/threonine phosphorylation (15). It is well known that chemically mediated inflammation leads to alteration in both TTX-S (34, 35) and TTX-R Na+ channels (8, 36–38). A GTP-induced TTX-R Na+ current has been reported (39), implying that differences in experimental conditions, including the internal solutions in patch pipettes, might affect the results.
Finally, differences in the ciguatoxin structure of CTX3C and CTX1B might affect their potency. CTX1B has almost the same ether ring skeleton as that of CTX3C (a small difference found at E-ring; CTX1B has 7-membered E-ring instead of 8-membered one for CTX3C, see Fig. 1) with the M-ring hydroxylated at the 51st position, and has a dihydroxylated extra-alkyl chain attached at position 1 (Fig. 1). We have observed that hydroxylation of the M-ring of CTX3C, resulting in 51-hydroxy-CTX3C, has no significant effect on its potency (data not shown). Hydroxy residues near the A-ring could be significant for ciguatoxin binding activity. This will be systematically tested in the near future.
This work was supported by Solution Oriented Research for Science and Technology, Japan Science and Technology Agency, and Grant 17390056 from the Ministry of Education and Culture of Japan (to K. Y.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The nucleotide sequence(s) reported in this paper has been submitted to the DDBJ/GenBank™/EBI Data Bank with accession number(s) AB453238.
Footnotes
The abbreviations used are: CTX1B, pacific CTX-1; TTX, tetrodotoxin; TTX-S, TTS-sensitive; TTX-R, TTX-resistant; DRG, dorsal root ganglion; SNS, sensory neuron-specific.
References
- 1.Yasumoto, T. (2001) Chem. Rec. 1228–242 [DOI] [PubMed] [Google Scholar]
- 2.Benoit, E., Legrand, A. M., and Dubois, J. M. (1986) Toxicon 24357–364 [DOI] [PubMed] [Google Scholar]
- 3.Strachan, L. C., Lewis, R. J., and Nicholson, G. M. (1999) J. Pharmacol. Exp. Ther. 288379–388 [PubMed] [Google Scholar]
- 4.Trainer, V. L., Thomsen, W. J., Catterall, W. A., and Baden, D. G. (1991) Mol. Pharmacol. 40988–994 [PubMed] [Google Scholar]
- 5.Trainer, V. L., Baden, D. G., and Catterall, W. A. (1994) J. Biol. Chem. 26919904–19909 [PubMed] [Google Scholar]
- 6.Rush, A. M., Cummins, T. R., and Waxman, S. G. (2007) J. Physiol. (Lond.) 5791–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akopian, A. N., Sivilotti, L., and Wood, J. N. (1996) Nature 379257–262 [DOI] [PubMed] [Google Scholar]
- 8.Akopian, A. N., Souslova, V., England, S., Okuse, K., Ogata, N., Ure, J., Smith, A., Kerr, B. J., McMahon, S. B., Boyce, S., Hill, R., Stanfa, L. C., Dickenson, A. H., and Wood, J. N. (1999) Nat. Neurosci. 2541–548 [DOI] [PubMed] [Google Scholar]
- 9.Renganathan, M., Cummins, T. R., and Waxman, S. G. (2001) J. Neurophysiol. 86629–640 [DOI] [PubMed] [Google Scholar]
- 10.Lai, J., Gold, M. S., Kim, C. S., Bian, D., Ossipov, M. H., Hunter, J. C., and Porreca, F. (2002) Pain 95143–152 [DOI] [PubMed] [Google Scholar]
- 11.Jarvis, M. F., Honore, P., Shieh, C. C., Chapman, M., Joshi, S., Zhang, X. F., Kort, M., Carroll, W., Marron, B., Atkinson, R., Thomas, J., Liu, D., Krambis, M., Liu, Y., McGaraughty, S., Chu, K., Roeloffs, R., Zhong, C., Mikusa, J. P., Hernandez, G., Gauvin, D., Wade, C., Zhu, C., Pai, M., Scanio, M., Shi, L., Drizin, I., Gregg, R., Matulenko, M., Hakeem, A., Gross, M., Johnson, M., Marsh, K., Wagoner, P. K., Sullivan, J. P., Faltynek, C. R., and Krafte, D. S. (2007) Proc. Natl. Acad. Sci. U. S. A. 1048520–8525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trimmer, J. S., Cooperman, S. S., Tomiko, S. A., Zhou, J. Y., Crean, S. M., Boyle, M. B., Kallen, R. G., Sheng, Z. H., Barchi, R. L., Sigworth, F. J., Goodman, R. G., Agnew, S. A., and Mandel, G. (1989) Neuron 333–49 [DOI] [PubMed] [Google Scholar]
- 13.Noda, M., Ikeda, T., Kayano, T., Suzuki, H., Takeshima, H., Kurasaki, M., Takahashi, H., and Numa, S. (1986) Nature 320188–192 [DOI] [PubMed] [Google Scholar]
- 14.Moriyoshi, K., Richards, L. J., Akazawa, C., O'Leary, D. D., and Nakanishi, S. (1996) Neuron 16255–260 [DOI] [PubMed] [Google Scholar]
- 15.Hong, S., Morrow, T. J., Paulson, P. E., Isom, L. L., and Wiley, J. W. (2004) J. Biol. Chem. 27929341–29350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.John, V. H., Main, M. J., Powell, A. J., Gladwell, Z. M., Hick, C., Sidhu, H. S., Clare, J. J., Tate, S., and Trezise, D. J. (2004) Neuropharmacology 46425–438 [DOI] [PubMed] [Google Scholar]
- 17.Okuse, K., Malik-Hall, M., Baker, M. D., Poon, W. Y., Kong, H., Chao, M. V., and Wood, J. N. (2002) Nature 417653–656 [DOI] [PubMed] [Google Scholar]
- 18.Hirama, M., Oishi, T., Uehara, H., Inoue, M., Maruyama, M., Oguri, H., and Satake, M. (2001) Science 2941904–1907 [DOI] [PubMed] [Google Scholar]
- 19.Inoue, M., Miyazaki, K., Uehara, H., Maruyama, M., and Hirama, M. (2004) Proc. Natl. Acad. Sci. U. S. A. 10112013–12018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inoue, M., Miyazaki, K., Ishihara, Y., Tatami, A., Ohnuma, Y., Kawada, Y., Komano, K., Yamashita, S., Lee, N., and Hirama, M. (2006) J. Am. Chem. Soc. 1289352–9354 [DOI] [PubMed] [Google Scholar]
- 21.Yamaoka, K., Inoue, M., Miyahara, H., Miyazaki, K., and Hirama, M. (2004) Br. J. Pharmacol. 142879–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Armstrong, C. M., and Bezanilla, F. (1974) J. Gen. Physiol. 63533–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dunn, P. M., Coote, P. R., Wood, J. N., Burgess, G. M., and Rang, H. P. (1991) Brain Res. 54580–86 [DOI] [PubMed] [Google Scholar]
- 24.Kerr, B. J., Souslova, V., McMahon, S. B., and Wood, J. N. (2001) Neuroreport 123077–3080 [DOI] [PubMed] [Google Scholar]
- 25.Laird, J. M., Souslova, V., Wood, J. N., and Cervero, F. (2002) J. Neurosci. 228352–8356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roza, C., Laird, J. M., Souslova, V., Wood, J. N., and Cervero, F. (2003) J. Physiol. (Lond.) 550921–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gold, M. S., Weinreich, D., Kim, C. S., Wang, R., Treanor, J., Porreca, F., and Lai, J. (2003) J. Neurosci. 23158–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khasar, S. G., Gold, M. S., and Levine, J. D. (1998) Neurosci. Lett. 25617–20 [DOI] [PubMed] [Google Scholar]
- 29.Porreca, F., Lai, J., Bian, D., Wegert, S., Ossipov, M. H., Eglen, R. M., Kassotakis, L., Novakovic, S., Rabert, D. K., Sangameswaran, L., and Hunter, J. C. (1999) Proc. Natl. Acad. Sci. U. S. A. 967640–7644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoshimura, N., Seki, S., Erickson, V., Erickson, K., Kassotakis, L., Novakovic, S., Fraser, M. O., Chancellor, M. B., and de Groat, W. C. (2001) Urology 57116–117 [DOI] [PubMed] [Google Scholar]
- 31.Toledo-Aral, J. J., Moss, B. L., He, Z. J., Koszowski, A. G., Whisenand, T., Levinson, S. R., Wolf, J. J., Silos-Santiago, I., Halegoua, S., and Mandel, G. (1997) Proc. Natl. Acad. Sci. U. S. A. 941527–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nassar, M. A., Stirling, L. C., Forlani, G., Baker, M. D., Matthews, E. A., Dickenson, A. H., and Wood, J. N. (2004) Proc. Natl. Acad. Sci. U. S. A. 10112706–12711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang, Y., Wang, Y., Li, S., Xu, Z., Li, H., Ma, L., Fan, J., Bu, D., Liu, B., Fan, Z., Wu, G., Jin, J., Ding, B., Zhu, X., and Shen, Y. (2004) J. Med. Genet. 41171–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Black, J. A., Liu, S., Tanaka, M., Cummins, T. R., and Waxman, S. G. (2004) Pain 108237–247 [DOI] [PubMed] [Google Scholar]
- 35.Tanaka, M., Cummins, T. R., Ishikawa, K., Dib-Hajj, S. D., Black, J. A., and Waxman, S. G. (1998) Neuroreport 9967–972 [DOI] [PubMed] [Google Scholar]
- 36.Cardenas, L. M., Cardenas, C. G., and Scroggs, R. S. (2001) J. Neurophysiol. 86241–248 [DOI] [PubMed] [Google Scholar]
- 37.Gold, M. S., Reichling, D. B., Shuster, M. J., and Levine, J. D. (1996) Proc. Natl. Acad. Sci. U. S. A. 931108–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gold, M. S. (1999) Proc. Natl. Acad. Sci. U. S. A. 967645–7649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baker, M. D., Chandra, S. Y., Ding, Y., Waxman, S. G., and Wood, J. N. (2003) J. Physiol. (Lond.) 548373–382 [DOI] [PMC free article] [PubMed] [Google Scholar]