

Abstract
Hyperaldosteronism is associated with impaired endothelium-dependent vascular reactivity owing to increased reactive oxygen species and decreased bioavailable nitric oxide (NO·); however, the effects of aldosterone on vasodilatory signaling pathways in vascular smooth muscle cells (VSMC) remain unknown. Soluble guanylyl cyclase (GC) is a heterodimer that is activated by NO· to convert cytosolic GTP to cGMP, a second messenger required for normal VSMC relaxation. Here, we show that aldosterone (10-9-10-7 mol/liter) diminishes GC activity by activating NADPH oxidase in bovine aortic VSMC to increase reactive oxygen species levels and induce oxidative posttranslational modification(s) of Cys-122, a β1-subunit cysteinyl residue demonstrated previously to modulate NO· sensing by GC. In VSMC treated with aldosterone, Western immunoblotting detected evidence of GC β1-subunit disulfide bonding, whereas mass spectrometry analysis of a homologous peptide containing the Cys-122-bearing sequence exposed to conditions of increased oxidant stress confirmed cysteinyl sulfinic acid (m/z 435), sulfonic acid (m/z 443), and disulfide (m/z 836) bond formation. The functional effect of these modifications was examined by transfecting COS-7 cells with wild-type GC or mutant GC containing an alanine substitution at Cys-122 (C122A). Exposure to aldosterone or hydrogen peroxide (H2O2) significantly decreased cGMP levels in cells expressing wild-type GC. In contrast, aldosterone or H2O2 did not influence cGMP levels in cells expressing the mutant C122A GC, confirming that oxidative modification of Cys-122 specifically impairs GC activity. These findings demonstrate that pathophysiologically relevant concentrations of aldosterone increase oxidant stress to convert GC to an NO·-insensitive state, resulting in disruption of normal vasodilatory signaling pathways in VSMC.
Elevated levels of the mineralocorticoid hormone aldosterone are associated with impaired vascular reactivity in patients with an aldosterone-producing adenoma, hypertension, and congestive heart failure that is remediable following surgical resection of the tumor or treatment with a mineralocorticoid receptor antagonist (1–5). It has been suggested that aldosterone-induced vascular dysfunction is a consequence of a vasculopathy that results from the propensity for aldosterone to generate ROS2 and decrease bioavailable NO· (6). In the vascular endothelium, aldosterone has been shown to promote endothelial dysfunction by inducing an acquired antioxidant-deficient state that disrupts cellular redox homeostasis to increase ROS accumulation and diminish NO· levels. In vivo, this results in diminished endothelium-dependent vascular reactivity (7). The relationship between aldosterone-induced oxidant stress and NO·-stimulated vasodilatory signaling pathways in VSMC, however, remains unknown.
The influence of oxidant stress on NO·-activated GC signaling in VSMC has been the subject of investigation for over three decades (8–12). Guanylyl cyclase is a heterodimeric enzyme that requires coexpression of the α1-, or α2-, and β1-subunits to achieve catalytic activity and convert cytosolic GTP to cGMP, which, in turn, induces VSMC relaxation (13). When GC is functionally deficient, vasodilatory signaling is disrupted and vascular compliance is decreased, an effect that is associated with myocardial infarction, stroke, and progression of coronary artery disease (3, 6, 14).
The mechanism by which oxidant stress adversely affects GC function remains controversial. NO· is the primary, biologically active stimulator of GC. In the presence of excess ROS, it has been suggested that GC enzymatic function may be decreased by: (i) a peroxynitrite (ONOO-)-mediated decrease in GC specific activity (15); (ii) oxidation of the β1-subunit-associated prosthetic heme group that converts GC to an NO·-insensitive state (14); (iii) oxidation-induced disulfide formation of β1-subunit-thiol groups (16, 17); or (iv) NO·-dependent posttranslational modification (e.g. S-nitrosation) of a β1-subunit-thiol (18). Based on these observations, we hypothesized that aldosterone-mediated ROS formation promotes oxidative posttranslational modification of GC and conversion of the enzyme to an NO·-insensitive state, thereby disrupting a key pathway essential for normal VSMC relaxation.
EXPERIMENTAL PROCEDURES
Cell Culture and Treatments—Bovine aortic VSMC (Genlantis) were grown to confluence using phenol-free Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin, at 37 °C, 5% CO2. Cells were passaged twice weekly using 0.5% trypsin/EDTA, and experiments were performed on cells from passages 4–10. Aldosterone (Steraloids) and apocynin (Cayman) were dissolved in dimethyl sulfoxide (10 nmol/liter), which also served as the vehicle control. Cells were treated with aldosterone (10-9-10-7 mol/liter) for 24 h, and in selected experiments, coincubated with apocynin (3 × 10-5 mol/liter) or 2-amino-5,6-dihydro-6-methyl-4H-1,2-thiazin (AMT) (100 μmol/liter)(Cayman) for 24 h. NOS2 was induced using the combination of irradiated lipopolysaccharide (30 μg/ml)-(Sigma), interleukin 1-β (50 ng/ml)(Endogen), and interferon-γ (50 ng/ml)(Endogen) as described previously (19).
Dichlorodihydrofluorescein Fluorescence—ROS generation was assessed using 6-carboxy-2′,7′ dichlorodihydrofluorescein diacetate ester fluorescence (5 μmol/liter) (Molecular Probes) as described previously (7).
Amplex Red Activity Assay—Hydrogen peroxide was measured using the horseradish peroxidase-linked Amplex Red assay per kit instructions (Invitrogen). Phenol-red free tissue culture medium from experimental samples was incubated in 100 μm Amplex Red reagent, and absorbance was measured at an excitation wavelength of 560 nm and an emission wave-length of 590 nm in a microplate fluorometer (Spectramax Gemini XPS, Molecular Devices). Data are standardized to sample protein concentration.
NADPH Oxidase Activity—NADPH oxidase activity was measured using lucigenin (5 μmol/liter) chemiluminescence. Photon emission was measured every 10 s for 5 min in a luminometer (Turner Biosystems 20/20n), and the rate of enzyme activity was calculated as described previously (20).
NO· Metabolites—Nitrite
(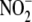 ) and nitrate
(
) and nitrate
(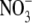 ) were measured from cell culture
medium containing 2% fetal bovine serum and l-arginine (1
mmol/liter)(Sigma) by 2,3-diaminonaphthalene fluorescence (Cayman).
) were measured from cell culture
medium containing 2% fetal bovine serum and l-arginine (1
mmol/liter)(Sigma) by 2,3-diaminonaphthalene fluorescence (Cayman).
3-Nitrotyrosine Immunohistochemistry—Cells were grown to confluence on glass chamber slides, exposed to anti-3-nitrotyrosine antibody (Santa Cruz Biotechnology), and stained using the 3,3′-diaminobenzidine substrate method (Vector laboratories) as described previously (21). Images were viewed under 200× magnification with the Olympus BX51™ microscope and acquired by Picture Taker™.
cGMP Measurement—Confluent cells were washed twice with ice-cold phosphate-buffered saline and then exposed to 6% trichloroacetic acid. Cells were collected and centrifuged at 1,500 × g for 10 min at 4 °C, cGMP formation was measured by immunoassay according to the manufacturer's instructions (Cayman), and bicinchoninic acid (Bio-Rad) was used for protein determination.
Soluble Guanylyl Cyclase Activity Assay—Intracellular GC activity was measured using the methodology of Waldman et al. (22) with minor modifications. All samples were incubated with 0.5 mm 3-isobutyl-1-methylxanthine and DETA NONOate (10 mm, Cayman) at 37° for 10 min, and the reaction was initiated by the addition of 4 mm MgCl2 and 1 mm GTP (Sigma). After 10 min, 6% trichloroacetic acid was added, supernatant fractions were extracted using water-saturated ethyl ether (Fisher), and samples were dried under nitrogen gas. cGMP levels were measured by immunoassay according to the manufacturer's instructions (Cayman).
Immunoblotting—Proteins were size-fractionated electrophoretically using SDS-PAGE and transferred to polyvinylidene fluoride membranes. The membranes were incubated with anti-NOS2 (Santa Cruz Biotechnology) and anti-GC α1-subunit and anti-GC β1-subunit (Cayman) antibodies overnight at 4 °C and visualized using the ECL detection system (Amersham Biosciences).
Western Blot to Detect GC Disulfide Bond Formation— VSMC were lysed in an alkylating buffer (0.1 m Tris-HCl, pH 6.8, 1% SDS, 100 mm iodoacetamide, 100 mm N-ethylmaleimide) and sonicated on ice. Following a 30-min incubation at 25 °C, alkylated proteins were precipitated with acetone. Proteins were resuspended in 50 μl of 0.1 m Tris-HCl, pH 7.4, 1% SDS, and disulfides were reduced by adding a final concentration of 5 mm Tris(2-carboxyethyl)phosphine hydrochloride. Following a 20-min incubation at 25 °C, Tris(2-carboxyethyl)phosphine hydrochloride was removed with a Micro Bio-Spin column 6 (Bio-Rad), and 1% SDS was added to the eluant. The previously oxidized (now reduced) cysteines were labeled with 1 mm polyethylene glycol-conjugated maleimide (molecular mass 10 kDa, Fluka). After a 1-h incubation at 25 °C, proteins were precipitated with acetone, resuspended in 50 μl of non-reducing SDS electrophoresis buffer, size-fractionated electrophoretically using SDS-PAGE, and transferred to a polyvinylidene fluoride membrane (23). To detect disulfides specific to the sGC β1-subunit, the membrane was immunoblotted with an sGC β1-subunit antibody (Santa Cruz Biotechnology).
Site-directed Mutagenesis and Transfection—Expression constructs in pCMV5 were used to overexpress α1-, β1-, and β1-C122A subunits of rat GC as described previously (18). COS-7 cells, which do not express endogenous GC, were plated in 6-well tissue culture dishes and transfected with 5 μg of DNA for 4 h with Lipofectamine 2000™ (Invitrogen) (24). After this time, medium was replaced with full growth media, and experiments were performed after 48 h.
Mass Spectrometry—In-gel trypsin digestion of immunoprecipitated GC from bovine VSMC whole cell extracts was performed according to the method by Shevchenko et al. (25). Extracted peptides were subjected to liquid chromatographymass spectrometry (LC-MS) analysis, and modified cysteinyl thiols in digested peptides were monitored using MS1 analysis. A doubly charged ion of m/z 435 corresponding to a Cys-122-containing peptide (CTDADKGK) with the addition of two oxygens was identified. A model peptide containing CTDADKGK was synthesized in the Biopolymers Laboratory at Harvard Medical School (Boston, MA). To study the effect of oxidant stress on Cys-122, the peptide (0.1 mm) was incubated with 200 μmol/liter H2O2 and 0.2 mmol/liter ferrous sulfate (Sigma) in 50 mm ammonium acetate, pH 5.9, for 30 min at 22 °C. Reaction products were mixed with 10 volumes of 30% acetonitrile and 0.1% formic acid in water and analyzed with an LTQ linear ion trap mass spectrometry system (Thermo Electron Co., San Jose, CA) equipped with a static nanospray ion source probe. The spray voltage was set to 1.4 kV, and the heated capillary was set to 200 °C. MS1 scanning was performed using an enhanced scan mode to monitor the size and charge state of the ions. MS-MS scanning was acquired for definitive identification of the oxidation products of the peptide.
Statistical Analysis—Continuous data were expressed as means ± S.E. Comparison between groups was performed by Student's paired two-tailed t test, and p < 0.05 is considered significant.
RESULTS
Aldosterone Induces Oxidant Stress in VSMC—To evaluate the effect of aldosterone on ROS accumulation, bovine aortic VSMC were exposed to aldosterone (10-9-10-7 mol/liter) for 24 h. When compared with vehicle-treated cells, there was a concentration-dependent increase in ROS levels in aldosterone-treated cells (117.9 ± 5.7 versus 124.5 ± 6.1 versus 144.9 ± 4.6% control, p < 0.01, n = 3) (Fig. 1A). As no further increase in ROS formation was observed after 24 h, experiments were performed following this incubation period in VSMC exposed to 10-7 mol/liter aldosterone, a pathophysiologically relevant concentration comparable with levels achieved in patients with primary or secondary hyperaldosteronism (3).
FIGURE 1.

Aldosterone increases oxidant stress in VSMC. A, vascular smooth muscle cells were exposed to aldosterone (ALDO) (10-9-10-7 mol/liter) or vehicle control (V) for 24 h, and ROS accumulation was measured by 6-carboxy-2′,7′ dichlorodihydrofluorescein diacetate ester (DCF) fluorescence (n = 3). B, hydrogen peroxide (H2O2) generation was evaluated by Amplex Red assay fluorescence (n = 3) *, p < 0.01 versus vehicle control. Data are presented as mean ± S.E.
Aldosterone-mediated ROS generation resulted from activation of NADPH
oxidase, a predominant source of ROS in VSMC. Aldosterone increased NADPH
oxidase activity nearly 2-fold when compared with vehicle-treated cells (61.7
± 1.5 versus 117.9 ± 5.7 nmol
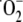 /min/mg of
protein, p < 0.001, n = 3). Coincubation of
aldosterone-treated cells with apocynin (30 μmol/liter), an NADPH oxidase
inhibitor, decreased ROS levels significantly (145.1 ± 1.8
versus 115.6 ± 8.6% control, p < 0.04, n
= 3), confirming that the majority of aldosterone-mediated ROS formation
resulted from NADPH oxidase activation.
/min/mg of
protein, p < 0.001, n = 3). Coincubation of
aldosterone-treated cells with apocynin (30 μmol/liter), an NADPH oxidase
inhibitor, decreased ROS levels significantly (145.1 ± 1.8
versus 115.6 ± 8.6% control, p < 0.04, n
= 3), confirming that the majority of aldosterone-mediated ROS formation
resulted from NADPH oxidase activation.
Next, the effect of aldosterone on H2O2 levels was assessed as H2O2 has been shown to modify protein function by reacting with free thiol moieties and GC contains several cysteinyl thiols. When compared with vehicle, aldosterone increased H2O2 formation significantly as determined by Amplex Red fluorescence (5.24 ± 0.02 versus 9.31 ± 1.06 μm/μg of protein, p < 0.03, n = 3) (Fig. 1B). Coincubation with apocynin decreased aldosterone-induced H2O2 production by 66.1% (p < 0.01, n = 3). Together, these studies demonstrate that aldosterone activates NADPH oxidase to increase oxidant stress in VSMC.
Aldosterone and NO·
Metabolism—Under basal conditions in VSMC, bioavailable
NO· has one of two metabolic fates. It may either remain
unoxidized to bind the Fe2+ center of the prosthetic heme group to
activate GC and increase cGMP levels or undergo oxidation to nitrite
(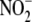 ) and nitrate
(
) and nitrate
(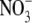 ). Therefore, to assess the
influence of aldosterone on endogenously produced NO·
bioactivity, we induced iNOS expression to increase
). Therefore, to assess the
influence of aldosterone on endogenously produced NO·
bioactivity, we induced iNOS expression to increase
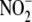 production (0.0 ± 0.0
versus 0.08 ± 0.01 μm/μg of protein,
p < 0.01, n = 4) (Fig.
2, A and B). AMT (100 μmol/liter), a specific
iNOS inhibitor, decreased
production (0.0 ± 0.0
versus 0.08 ± 0.01 μm/μg of protein,
p < 0.01, n = 4) (Fig.
2, A and B). AMT (100 μmol/liter), a specific
iNOS inhibitor, decreased 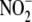 formation
to non-detectable levels in iNOS-expressing cells, indicating that iNOS was
the primary source of
formation
to non-detectable levels in iNOS-expressing cells, indicating that iNOS was
the primary source of 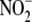 . Exposure of
iNOS-expressing cells to aldosterone had no further influence on either iNOS
protein expression or
. Exposure of
iNOS-expressing cells to aldosterone had no further influence on either iNOS
protein expression or 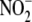 levels. To
demonstrate that these findings were not unique to iNOS-expressing cells, we
treated VSMC with the exogenous NO· donor, DETA NONOate (1
mmol/liter). DETA NONOate-treated cells exhibited a significant increase in
levels. To
demonstrate that these findings were not unique to iNOS-expressing cells, we
treated VSMC with the exogenous NO· donor, DETA NONOate (1
mmol/liter). DETA NONOate-treated cells exhibited a significant increase in
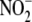 formation (0.0 ± 0.0
versus 1.34 ± 0.02 μm/μg of protein,
p < 0.03, n = 3), and aldosterone had no additional
effect on these levels (1.35 ± 0.02 μm/μg of protein,
p = not significant, n = 3).
formation (0.0 ± 0.0
versus 1.34 ± 0.02 μm/μg of protein,
p < 0.03, n = 3), and aldosterone had no additional
effect on these levels (1.35 ± 0.02 μm/μg of protein,
p = not significant, n = 3).
FIGURE 2.

Aldosterone and NO· byproduct formation.
A, vascular smooth muscle cells were exposed to ALDO (10-7
mol/liter) or vehicle control (V) in the presence or absence of
cytokines (lipopolysaccharide (LPS) = 30 μg/ml, interleukin
1-β (IL-1-β) = 50 ng/ml, interferon-γ
(IFN-γ) = 50 ng/ml) for 24 h to induce iNOS expression. The
effect of ALDO on iNOS protein expression was determined by Western blotting
(n = 3), and as shown in B, the influence of ALDO on
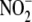 formation (n = 4) was
measured by 2,3 diaminonaphthalene fluorescence. To confirm iNOS as the source
of
formation (n = 4) was
measured by 2,3 diaminonaphthalene fluorescence. To confirm iNOS as the source
of 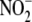 formation, cells were treated
with AMT (100 μmol/liter), a specific iNOS inhibitor. ND,
non-detectable. C, the influence of aldosterone and NADPH oxidase on
the
formation, cells were treated
with AMT (100 μmol/liter), a specific iNOS inhibitor. ND,
non-detectable. C, the influence of aldosterone and NADPH oxidase on
the
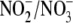 ratio in iNOS-expressing cells was assessed (n = 6). Apo,
apocynin. D, peroxynitrite formation was assessed by examining
anti-3-nitrotyrosine immunohistochemistry (n = 3). *,
p < 0.01 versus vehicle control, **,
p < 0.001 versus ALDO; Representative images are shown.
Data are presented as mean ± S.E.
ratio in iNOS-expressing cells was assessed (n = 6). Apo,
apocynin. D, peroxynitrite formation was assessed by examining
anti-3-nitrotyrosine immunohistochemistry (n = 3). *,
p < 0.01 versus vehicle control, **,
p < 0.001 versus ALDO; Representative images are shown.
Data are presented as mean ± S.E.
In addition to the observed lack of effect of aldosterone on
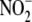 levels, aldosterone had no
influence on total NO· metabolites
(NOx:
levels, aldosterone had no
influence on total NO· metabolites
(NOx: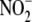 +
+
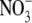 ) in iNOS-expressing cells
(Fig. 2C). It has been
shown previously that following iNOS induction in vitro, the
) in iNOS-expressing cells
(Fig. 2C). It has been
shown previously that following iNOS induction in vitro, the
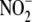 to
to
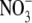 ratio is ∼2:1; however, in the
presence of increased ROS generation, enhanced peroxynitrite
(ONOO-) formation may shift this ratio in favor of
ratio is ∼2:1; however, in the
presence of increased ROS generation, enhanced peroxynitrite
(ONOO-) formation may shift this ratio in favor of
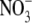 (26). Here, in iNOS-expressing
cells, the
(26). Here, in iNOS-expressing
cells, the
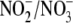 ratio was found to be 1.86:1, and aldosterone decreased this ratio by 31.7%
(p < 0.04, n = 6). To explain the observed shift in the
ratio was found to be 1.86:1, and aldosterone decreased this ratio by 31.7%
(p < 0.04, n = 6). To explain the observed shift in the
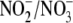 ratio, we examined ONOO- formation
(Fig. 2D). In
iNOS-expressing cells, aldosterone induced a 54% rise in ONOO-
formation when compared with untreated cells as assessed by 3-nitrotyrosine
immunostaining (p < 0.02, n = 3). Inhibition of NADPH
oxidase activity with apocynin in aldosterone-treated cells fully restored the
ratio, we examined ONOO- formation
(Fig. 2D). In
iNOS-expressing cells, aldosterone induced a 54% rise in ONOO-
formation when compared with untreated cells as assessed by 3-nitrotyrosine
immunostaining (p < 0.02, n = 3). Inhibition of NADPH
oxidase activity with apocynin in aldosterone-treated cells fully restored the
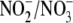 ratio, increasing it modestly to ∼3:1
(Fig. 2C). Taken
together, these studies reveal that aldosterone increases ONOO-
formation in iNOS-expressing cells and offer an explanation for the observed
decrease in the
ratio, increasing it modestly to ∼3:1
(Fig. 2C). Taken
together, these studies reveal that aldosterone increases ONOO-
formation in iNOS-expressing cells and offer an explanation for the observed
decrease in the
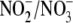 ratio.
ratio.
Aldosterone Decreases GC Activity and cGMP Production— Next, the influence of aldosterone on NO·-GC signaling was examined. Although there was no effect of aldosterone on GC α1-or β1-subunit protein expression (Fig. 3A), we observed a 46% decrease in basal GC activity in aldosterone-treated when compared with vehicle-treated cells (0.016 ± 0.001 versus 0.008 ± 0.002 pmol cGMP/10 min/μg of protein, p < 0.02, n = 4) (Fig. 3B). Similarly, in iNOS-expressing cells, aldosterone decreased GC activity by 35.3% (p < 0.001, n = 4) (Fig. 3C) and cGMP levels by 50.7% (0.014 ± 0.0019 versus 0.007 ± 0.002 cGMP pmol/μg of protein, p < 0.05, n = 4) (Fig. 3D). This finding was confirmed in DETA NONOate-treated cells where aldosterone decreased cGMP levels by 54% when compared with DETA NONOate alone (2.80 ± 0.43 versus 1.28 ± 0.25 fmol/μg of protein, p < 0.02, n = 4).
FIGURE 3.

Aldosterone and GC subunit expression and activity. A, GC α1- and β1-subunit protein expression in VSMC exposed to either ALDO(10-7 mol/liter) or vehicle control (V) for 24 h was assessed by Western blotting (n = 3). B and C, the effect of aldosterone (10-7 mol/liter) on GC activity under basal conditions (B) and iNOS-expressing cells in the presence or absence of NADPH oxidase inhibition with apocynin (Apo)(3 × 10-5 mol/liter) (n = 4) (C). D, cGMP levels were determined in iNOS-expressing cells (n = 4). *, p < 0.02 versus vehicle control, **, p < 0.04 versus iNOS, #, p < 0.03 versus ALDO. Representative blots are shown. Data are presented as mean ± S.E.
The role of aldosterone-stimulated NADPH oxidase activity as the mediator of decreased GC activity was investigated. When compared with aldosterone alone, inhibition of NADPH oxidase activity with apocynin restored iNOS-stimulated GC activity (Fig. 3C) and increased cGMP formation in both iNOS-expressing (Fig. 3D) and DETA NONOate-treated cells (1.28 ± 0.25 versus 2.3 ± 0.16 fmol/μg of protein, p < 0.02, n = 4).
Disulfide Bond Formation in GC β-Subunit—To determine whether oxidative modification of redox-susceptible cysteinyl thiols was one mechanism by which aldosterone impairs GC activity, we assessed disulfide bond formation in aldosterone-treated cells. The presence of disulfide bond formation may occur as a consequence of increased oxidant stress and has been suggested previously to influence adversely normal NO·-GC signaling (17). To determine whether aldosterone induced disulfide bond formation in the β1-subunit of GC, we treated VSMC with either vehicle or aldosterone and blocked free thiols with iodoacetamide and N-ethylmaleimide. We then reduced any potential disulfide bonds and labeled the previously oxidized (and now reduced) thiols with polyethylene glycol-conjugated maleimide (molecular mass 10 kDa). In this manner, two cysteinyl thiols involved in a disulfide bond increase the apparent molecular mass of the GC β1-subunit by a total of 20 kDa (23). The vehicle-treated cells demonstrated only the fully reduced GC β1-subunit; however, following aldosterone treatment, bands at 85 and 105 kDa were apparent, suggesting that aldosterone oxidatively modified cysteinyl thiols in the GC β1-subunit to induce the formation of two disulfide bonds (Fig. 4A).
FIGURE 4.

Disulfide bond formation and MS-MS spectra of peptide CTDADKGK oxidation products. A, vascular smooth muscle cells were exposed to 10-7 mol/liter ALDO or vehicle control (V) for 24 h, free thiols were blocked with iodoacetamide and N-ethylmaleimide, disulfide bonds were reduced, and previously oxidized (now reduced) cysteinyl thiols were labeled with polyethylene glycol-conjugated maleimide (molecular mass 10 kDa). Disulfide bond formation specific to the β1-subunit of GC is detected by Western immunoblot. For each disulfide bond, a 20-kDa increase in the molecular mass occurs, resulting in a shift in band location on Western immunoblot. Aldosterone treatment resulted in the formation of two disulfide bonds not present in the vehicle control (n = 3). A representative blot is shown. Cys122 oxidation products were examined by exposing the peptide CTDADKGK to H2O2 (200 μmol/liter) and FeSO4 (0.2 mmol/liter) at pH 5.9 for 30 min and mass spectrometry analysis. Three major oxidation products were identified by enhanced MS1 scan, and subsequently, MS-MS spectra were acquired for definitive identification of cysteinyl sulfinic acid ([M+2H]2+ = m/z 435) (B), cysteinyl sulfonic acid ([M+2H]2+ = m/z 443) (C), and peptide disulfide ([M+2H]2+ = m/z 836) formation (D).
Posttranslational Oxidative Modification of GC Decreases NO· Sensing—To support our hypothesis that aldosterone impairs GC activity by inducing oxidative modification of a key cysteinyl residue located within the NO·-sensing region of GC, we used liquid chromatography-mass spectrometry (LC-MS) to target three key β1-subunit associated cysteinyl residues (Cys-78, Cys-122, Cys-214). These cysteines were selected for analysis based on prior studies that implicated them as integral to normal NO· sensing by GC (10, 18). Immunoprecipitation of GC using a β1-subunit antibody was performed on VSMC exposed to aldosterone for 24 h, and after separation of peptides by capillary column liquid chromatography, MS1 scanning suggested potential oxidative modification of Cys-122 (peptide sequence: CTDADKGK). To examine the effect of oxidant stress on Cys-122, a model peptide containing the above amino acid sequence was synthesized. After exposure of this peptide to H2O2 (200 μmol/liter) for 30 min, MS1 scanning identified three doubly charged ions at m/z 435, m/z 443, and m/z 836 that corresponded to the CTDADKGK oxidation products cysteinyl sulfinic acid, cysteinyl sulfonic acid, and cysteinyl disulfide, respectively. MS-MS scanning confirmed the identity of these products (Fig. 4, B–D). Collision-induced dissociation of ion m/z 435 resulted in a spectrum with a characteristically predominant [M-81] ion (m/z = 394 at doubly charged state), representing the loss of NH3 and SO2 in the parent ion after a rearrangement of the N-terminal cysteinyl sulfinic acid in the peptide (for details of this reaction, see Ref. 27) (Fig. 4B). In addition, fragmentation of ion m/z 836 resulted in a spectrum with the most abundant daughter ion being derived from the homolytic cleavage of the disulfide bond present in the parent ion (Fig. 4D). These mass spectrometry analyses reveal that oxidation of CTDADKGK results in oxidation products by either the addition of oxygen or disulfide bond formation.
To determine whether oxidative modification of Cys-122 has functional implications for normal NO· sensing by GC, we transiently transfected COS-7 cells with rat GC DNAs coding for the wild-type (WT) α1- and β1-subunit of GC or the wild-type α1-subunit and a mutant β1-subunit containing a substitution of alanine for cysteine at position 122 (α1/β1-C122A). Although a single amino acid dissimilarity exists at position 126 between rat (glutamate) and bovine (aspartate) GC DNA, this difference is conservative and unlikely to affect the microenvironment of Cys-122. Expression of the transiently transfected WT α1-subunit and WT or mutant β1-subunit GC DNA in COS-7 cells was confirmed by Western blotting (Fig. 5A). GC activity was determined in COS-7 cells expressing the WT or α1/β1-C122A mutant DNA exposed to H2O2 (250 μmol/liter) for 5 min to induce oxidant stress or vehicle control. Although exposure to H2O2 decreased NO·-stimulated cGMP levels by 66.3% in the WT-transfected cells when compared with vehicle-treated cells (100 ± 13.7 versus 33.7 ± 10.6% control, p < 0.005, n = 3), H2O2 had no effect on NO·-stimulated cGMP levels in α1/β1-C122A mutant-transfected cells (100 ± 6.1 versus 93.5 ± 2.8% control, p = not significant, n = 3) (Fig. 5B). These findings confirm that oxidative modification of Cys-122 renders GC insensitive to NO·.
FIGURE 5.

Oxidation of β1-subunit Cys-122 results in GC desensitization to NO·. A, COS-7 cells were transiently transfected with GC WT α1-subunit DNA and WT β1-subunit DNA or WT α1-subunit DNA and mutant β1-subunit DNA containing a cysteine to alanine substitution at position 122 (β1-C122A), and protein expression was confirmed by Western blotting (n = 3). UnTx, untransfected. B, COS-7 cells expressing WT (α1/β1 subunits) or α1/β1-C122A were stimulated with sodium nitroprusside (1 mmol/liter) for 15 min in the presence or absence of H2O2 (250 μmol/liter) and cGMP levels were measured (n = 3). *, p < 0.005 versus vehicle control (V). Representative blots are shown. Data are presented as mean ± S.E.
Aldosterone Decreases GC NO· Sensing by Oxidative Modification of Cys-122—To determine whether aldosterone had similar functional implications for normal NO· sensing by GC as H2O2, we first demonstrated that COS-7 cells express a functional mineralocorticoid receptor and NADPH oxidase enzyme (supplemental Fig. 1A). Similar to VSMC, we found that COS-7 cells treated with aldosterone for 24 h increased significantly p67phox protein expression when compared with vehicle control (p < 0.05, n = 3) (supplemental Fig. 1A). Next, we assessed the effect of aldosterone on NADPH oxidase activation and ROS generation. Here, we observed that aldosterone increased ROS levels by 51% (p < 0.02, n = 3) (supplemental Fig. 1B) and significantly increased H2O2 levels by Amplex Red fluorescence (0.31 ± 0.3 versus 2.02 ± 0.26 μm/μg of protein, p < 0.02, n = 3) when compared with vehicle control (supplemental Fig. 1C). Thus, when stimulated with aldosterone, COS-7 cells activate NADPH oxidase to increase ROS and H2O2 levels.
Next, we investigated the functional effect of these findings on GC activity. Although WT-transfected COS-7 cells treated with aldosterone demonstrated decreased NO·-stimulated cGMP formation when compared with vehicle-treated cells (100 ± 8.1 versus 67.6 ± 8.8% control, p < 0.05, n = 3), aldosterone failed to decrease NO·-stimulated cGMP levels in α1/β1-C122A mutant-transfected cells (100 ± 13.4 versus 120.1 ± 5.1% control, p = not significant, n = 3) (supplemental Fig. 1D). These findings confirm that aldosterone-mediated oxidative modification of Cys-122 limits GC NO· sensing.
DISCUSSION
These studies demonstrate that pathophysiologically relevant concentrations of aldosterone activate NADPH oxidase in bovine VSMC to increase ROS levels, which, in turn, create a redox milieu that favors oxidative posttranslational modification(s) of the catalytically active β1-subunit of GC. Furthermore, these studies implicate oxidative modification of Cys-122, a key cysteine moiety resides in the NO·-sensing domain of the GC β1-subunit (28), as a mechanism by which oxidant stress impairs GC enzyme activity. Our functional studies of GC support this observation. NO· sensing by WT (α1/β1)GCwas inhibited significantly in cells exposed to either H2O2 or aldosterone; however, a mutant form of GC, with a substitution of alanine for cysteine at position 122 (α1/β1-C122A), was resistant to oxidative modification under the same conditions and exhibited normal NO· sensing. Furthermore, our studies reveal that aldosterone had no effect on total NO· metabolite levels, and, therefore, confirm the hypothesis that aldosterone-mediated ROS influences GC directly to limit GC activation rather than by decreasing levels of bioavailable NO·.
Aldosterone has been shown to increase both H2O2
levels and ONOO- formation in hearts and peripheral blood
mononuclear cells isolated from rats supplemented with dietary salt
(29,
30). Our data are in concert
with these observations; VSMC exposed to aldosterone demonstrated elevated
levels of H2O2 and ONOO-. This finding has
consequence for our studies of GC activity in a redox-active milieu as both
species have been suggested to modulate GC function. Elevated levels of
H2O2 may induce posttranslational oxidative
modification(s) of regulatory cysteinyl thiols to generate protein sulfenic
acid, which, in an oxygen-rich environment, is rapidly oxidized to yield
sulfinic or sulfonic acid or can form disulfide bonds
(31). Peroxynitrite has also
been shown to decrease GC activity in cytosolic extracts of rat aorta and
animal models of vascular disease
(14,
15). Although ONOO-
generation is associated with reduced levels of bioavailable
NO·, we found that aldosterone-induced increases in
ONOO- formation did not offset total NO·
oxidative product levels but merely altered the
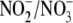 ratio. This result is not surprising as in the presence of ONOO-
(and CO2), preferential formation of
ratio. This result is not surprising as in the presence of ONOO-
(and CO2), preferential formation of
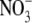 over
over
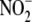 has been described owing to the
scavenging of
has been described owing to the
scavenging of 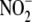 by
by
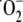 to form
peroxynitrate (O2NOO-)
(32). Our data are in
agreement with those others who have shown that by limiting the source of
to form
peroxynitrate (O2NOO-)
(32). Our data are in
agreement with those others who have shown that by limiting the source of
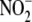 scavenging, accomplished here with
apocynin, an NADPH oxidase inhibitor and free radical antioxidant, the
scavenging, accomplished here with
apocynin, an NADPH oxidase inhibitor and free radical antioxidant, the
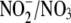 ratio
is restored (26,
33,
34).
ratio
is restored (26,
33,
34).
Prior work has suggested that, in the presence of increased ROS formation, decreased GC activity may be attributed to down-regulation of GC α1- and/or β1-subunit protein expression (35–37). In contrast, we found that aldosterone-induced oxidant stress had no influence on α1-or β1-subunit expression. These earlier investigations examined GC expression in the vasculature of aged hypertensive rat models, whereas our studies were performed in vitro in bovine vascular smooth muscle cells. Interestingly, other non-hypertensive animal models of impaired vascular reactivity, including hypercholesterolemic rabbits and nitrate-tolerant rats, demonstrated an increase in GC subunit expression (38). More recently, it was shown that rat aortic smooth muscle cells exposed to elevated levels of H2O2 (500 μmol/liter) for 24 h demonstrated decreased GC α1- and β1-subunit mRNA and protein expression; however, exposure to this concentration of H2O2 was associated with a 25% decrease in cell viability. In addition, when these investigators exposed cells to a lower concentration of H2O2 (150 μmol/liter), they saw no change in α1-subunit protein expression and an increase in β1-subunit expression (39). In our experiments, the levels of H2O2 generated by aldosterone were similar to the lower concentration of H2O2 studied by these investigators, and aldosterone treatment at these concentrations did not influence cell viability.
In the presence of increased ROS levels, decreased GC activity has been observed in isolated enzyme preparations and vascular cells (40, 41). This effect has been attributed to a ROS-mediated decrease in bioavailable NO· to limit enzyme activation, as well as the direct influence of ROS or ONOO- on GC. In our studies, we found that aldosterone-mediated ROS adversely modulates GC activity directly, supported by the observation that GC activity and cGMP levels were restored to basal levels when NADPH oxidase was inhibited by apocynin. Furthermore, we observed an aldosterone-induced uncoupling of iNOS-generated NO· bioactivity; aldosterone selectively decreased cGMP formation without influencing total NO· metabolite levels. Our observed effects of aldosterone on GC activity, therefore, cannot be explained by decreased levels of NO· available to activate the enzyme. These results are supported by studies that examined the influence of ROS on GC activity using both purified enzyme and VSMC stimulated with YC-1, an NO·-independent GC activator. In these studies, ROS generation via xanthine/xanthine oxidase inhibited YC-1-stimulated GC activity, an effect that could not be attributed to a ROS-mediated decrease in bioavailable NO· (40).
Our study is akin to work from others that implicates thiol redox status as a critical determinant of NO· sensing by GC (16–18). Early observations by Brandwein et al. (17) suggested disulfide bond formation as an explanation for decreased GC activity. Other investigators have shown that decreased GC activity associated with thiol oxidation was reversible by thiol reduction (16). Here, we demonstrate that cysteinyl thiol oxidation occurs in the presence of increased levels of H2O2 generated by aldosterone-induced NADPH oxidase activation followed by dismutation of superoxide to H2O2 to yield disulfide bond(s) as well as sulfinic and sulfonic acid oxidative modifications. In the LC-MS experiments, the Cys-122-containing peptide was also exposed to ferrous sulfate, a constituent present in GC under physiological conditions. Cys-122 is located near the His-105-associated prosthetic heme group, and ferrous sulfate by-reacts with H2O2 to yield OH- radicals (Fenton reaction), which, in turn, have been shown previously to oxidize cysteinyl thiols (42). Importantly, our studies do not exclude the possibility that aldosterone-mediated ROS oxidizes other key cysteinyl thiols or the His-105-associated prosthetic heme moiety to influence GC activity.
We also confirmed prior studies that implicate Cys-122 as a key regulatory element involved in NO· sensing by GC. Specifically, substitution of an alanine for cysteine at position 122 resulted in preserved GC activity under conditions of oxidant stress. As the crystal structure of mammalian GC has not yet been resolved, the precise mechanism by which oxidative modification of GC impairs NO· sensing remains controversial (43). Analysis of non-mammalian proteins that are structurally similar to GC in which Cys-122 is highly conserved has revealed that subtle conformational changes at Cys-122 may limit NO· binding at the sixth coordinating position of reduced heme (at His-105). This, in turn, would prevent the nitrosyl-heme formation that is necessary for GC activation (18, 28).
Taken together, our findings suggest a novel mechanism by which aldosterone-mediated ROS formation adversely influences vasodilatory signaling pathways in VSMC. These studies demonstrate that increased ROS levels, as occur in aldosterone-treated VSMC, induce oxidative posttranslational modification(s) of GC and subsequent conversion of the enzyme to an NO·-insensitive state, an effect that is abrogated by inhibition of NADPH oxidase. These data suggest further that impaired vascular reactivity associated with elevated levels of aldosterone may result, in part, from decreased GC activity and identify GC as a therapeutic target to ameliorate impaired vascular reactivity associated with hyperaldosteronism.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants HL089771 (to A. B.), HL61795, NO1HV28178, HL58976, and PO1HL81587 (to J. L.), and HL081110 (to J. A. L.). This work was also supported by an American Heart Association grant-in-aid (to J. A. L.) The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains a supplemental figure.
Footnotes
The abbreviations used are: ROS, reactive oxygen species; NO, nitric oxide; iNOS, inducible nitric oxide synthase; DETA NONOate, diethylenetriamine NONOate; GC, guanylyl cyclase; VSMC, vascular smooth muscle cells; AMT, 2-amino-5,6 dihydro-6-methyl-4H-1,3-thiazine; ALDO, aldosterone; LC-MS, liquid chromatography-mass spectrometry; MS-MS, tandem mass spectrometry; WT, wild type.
References
- 1.Swedberg, K., Eneroth, P., Kjekshus, J., and Wilhelmsen, L. (1990) Circulation 82 1730-1736 [DOI] [PubMed] [Google Scholar]
- 2.Duprez, D. A., De Buyzere, M. L., Rietzschel, E. R., Taes, Y., Clement, D. L., Morgan, D., and Cohn, J. N. (1998) Eur. Heart J. 19 1371-1376 [DOI] [PubMed] [Google Scholar]
- 3.Weber, K. T. (2001) N. Engl. J. Med. 345 1689-1697 [DOI] [PubMed] [Google Scholar]
- 4.Weber, K. T. (2003) J. Lab. Clin. Med. 142 71-82 [DOI] [PubMed] [Google Scholar]
- 5.Farquharson, C. A., and Struthers, A. D. (2000) Circulation 101 594-597 [DOI] [PubMed] [Google Scholar]
- 6.Struthers, A. D. (2004) Mol. Cell. Endocrinol. 217 239-241 [DOI] [PubMed] [Google Scholar]
- 7.Leopold, J. A., Dam, A., Maron, B. A., Scribner, A. W., Liao, R., Handy, D. E., Stanton, R. C., Pitt, B., and Loscalzo, J. (2007) Nat. Med. 13 189-197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Melichar, V. O., Behr-Roussel, D., Zabel, U., Uttenthal, L. O., Rodrigo, J., Rupin, A., Verbeuren, T. J., Kumar, H. S. A., and Schmidt, H. H. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 16671-16676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Radi, R., Beckman, J. S., Bush, K. M., and Freeman, B. A. (1991) J. Biol. Chem. 266 4244-4250 [PubMed] [Google Scholar]
- 10.Friebe, A., Wedel, B., Harteneck, C., Foerster, J., Schultz, G., and Koesling, D. (1997) Biochemistry 36 1194-1198 [DOI] [PubMed] [Google Scholar]
- 11.Ignarro, L. J., Degnan, J. N., Baricos, W. H., Kadowitz, P. J., and Wolin, M. S. (1982) Biochim. Biophys. Acta 718 49-59 [DOI] [PubMed] [Google Scholar]
- 12.Murad, F., Lewicki, J. A., Brandwein, H. J., Mittal, C. K., and Waldman, S. A. (1981) Adv. Cyclic Nucleotide Res. 14 229-239 [PubMed] [Google Scholar]
- 13.Koesling, D. (1999) Methods (Amst.) 19 485-493 [DOI] [PubMed] [Google Scholar]
- 14.Stasch, J. P., Schmidt, P. M., Nedvetsky, P. I., Nedvetskaya, T. Y., H, S. A., Meurer, S., Deile, M., Taye, A., Knorr, A., Lapp, H., Muller, H., Turgay, Y., Rothkegel, C., Tersteegen, A., Kemp-Harper, B., Muller-Esterl, W., and Schmidt, H. H. (2006) J. Clin. Investig. 116 2552-2561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weber, M., Lauer, N., Mulsch, A., and Kojda, G. (2001) Free Radic. Biol. Med. 31 1360-1367 [DOI] [PubMed] [Google Scholar]
- 16.Mingone, C. J., Gupte, S. A., Chow, J. L., Ahmad, M., Abraham, N. G., and Wolin, M. S. (2006) Am. J. Physiol. 291 L337-L344 [DOI] [PubMed] [Google Scholar]
- 17.Brandwein, H. J., Lewicki, J. A., and Murad, F. (1981) J. Biol. Chem. 256 2958-2962 [PubMed] [Google Scholar]
- 18.Sayed, N., Baskaran, P., Ma, X., van den Akker, F., and Beuve, A. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 12312-12317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Welch, G. N., Upchurch, G. R., Jr., Farivar, R. S., Pigazzi, A., Vu, K., Brecher, P., Keaney, J. F., Jr., and Loscalzo, J. (1998) Proc. Assoc. Am. Physicians 110 22-31 [PubMed] [Google Scholar]
- 20.Ushio-Fukai, M., Zafari, A. M., Fukui, T., Ishizaka, N., and Griendling, K. K. (1996) J. Biol. Chem. 271 23317-23321 [DOI] [PubMed] [Google Scholar]
- 21.Walford, G. A., Moussignac, R. L., Scribner, A. W., Loscalzo, J., and Leopold, J. A. (2004) J. Biol. Chem. 279 4425-4432 [DOI] [PubMed] [Google Scholar]
- 22.Waldman, S. A., Leitman, D. C., and Murad, F. (1991) Methods Enzymol. 195 391-396 [DOI] [PubMed] [Google Scholar]
- 23.Mastroberardino, P. G., Orr, A. L., Hu, X., Na, H. M., and Greenamyre, J. T. (2008) Free Radic. Biol. Med. 45 971-981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Handy, D. E., Zhang, Y., and Loscalzo, J. (2005) J. Biol. Chem. 280 15518-15525 [DOI] [PubMed] [Google Scholar]
- 25.Shevchenko, A., Tomas, H., Havlis, J., Olsen, J. V., and Mann, M. (2006) Nat. Protoc. 1 2856-2860 [DOI] [PubMed] [Google Scholar]
- 26.Jiang, B., Haverty, M., and Brecher, P. (1999) Hypertension 34 574-579 [DOI] [PubMed] [Google Scholar]
- 27.Srikanth, R., Wilson, J., Bridgewater, J. D., Numbers, J. R., Lim, J., Olbris, M. R., Kettani, A., and Vachet, R. W. (2007) J. Am. Soc. Mass Spectrom. 18 1499-1506 [DOI] [PubMed] [Google Scholar]
- 28.Ma, X., Sayed, N., Beuve, A., and van den Akker, F. (2007) EMBO J. 26 578-588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun, Y., Zhang, J., Lu, L., Chen, S. S., Quinn, M. T., and Weber, K. T. (2002) Am. J. Pathol. 161 1773-1781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahokas, R. A., Sun, Y., Bhattacharya, S. K., Gerling, I. C., and Weber, K. T. (2005) Circulation 111 51-57 [DOI] [PubMed] [Google Scholar]
- 31.Saurin, A. T., Neubert, H., Brennan, J. P., and Eaton, P. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 17982-17987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldstein, S., Lind, J., and Merenyi, G. (2005) Chem. Rev. 105 2457-2470 [DOI] [PubMed] [Google Scholar]
- 33.Simons, J. M., Hart, B. A., Ip Vai Ching, T. R., Van Dijk, H., and Labadie, R. P. (1990) Free Radic. Biol. Med. 8 251-258 [DOI] [PubMed] [Google Scholar]
- 34.Heumuller, S., Wind, S., Barbosa-Sicard, E., Schmidt, H. H., Busse, R., Schroder, K., and Brandes, R. P. (2008) Hypertension 51 211-217 [DOI] [PubMed] [Google Scholar]
- 35.Bauersachs, J., Bouloumie, A., Mulsch, A., Wiemer, G., Fleming, I., and Busse, R. (1998) Cardiovasc. Res. 37 772-779 [DOI] [PubMed] [Google Scholar]
- 36.Kloss, S., Bouloumie, A., and Mulsch, A. (2000) Hypertension 35 43-47 [PubMed] [Google Scholar]
- 37.Ruetten, H., Zabel, U., Linz, W., and Schmidt, H. H. (1999) Circ. Res. 85 534-541 [DOI] [PubMed] [Google Scholar]
- 38.Munzel, T., Daiber, A., Ullrich, V., and Mulsch, A. (2005) Arterioscler. Thromb. Vasc. Biol. 25 1551-1557 [DOI] [PubMed] [Google Scholar]
- 39.Gerassimou, C., Kotanidou, A., Zhou, Z., Simoes, D. C., Roussos, C., and Papapetropoulos, A. (2007) Br. J. Pharmacol. 150 1084-1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mulsch, A., Bauersachs, J., Schafer, A., Stasch, J. P., Kast, R., and Busse, R. (1997) Br. J. Pharmacol. 120 681-689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mulsch, A., Busse, R., Winter, I., and Bassenge, E. (1989) Naunyn-Schmiedeberg's Arch. Pharmacol. 339 568-574 [DOI] [PubMed] [Google Scholar]
- 42.Shetty, V., Spellman, D. S., and Neubert, T. A. (2007) J. Am. Soc. Mass Spectrom. 18 1544-1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gladwin, M. T. (2006) J. Clin. Investig. 116 2330-2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.