

Abstract
The mechanisms remain uncertain by which mutations in CFTR cause lung disease in cystic fibrosis (CF). Teichgräber et al. recently reported increased ceramide in CF lungs, which was proposed to result from defective lysosomal acidification in airway epithelial cells and consequent impairment of pH-dependent ceramide-metabolizing enzymes (Teichgräber, V., Ulrich, M., Endlich, N., Reithmüller, J., Wilker, B., Conceição Ce Olivereira-Munding, C., van Heeckeren, A. M., Barr, M. L., von Kürthy, G., Schmid, K. W., Weller, M., Tümmler, B., Lang, F., Grassme, H., Döring, G., and Gulbins, E. (2008) Nat. Med. 14, 382–391). Here, we measured lysosomal pH in several CFTR-expressing and -deficient cell lines, freshly isolated airway epithelial cells from non-CF and CF mice and humans, and well-differentiated primary cultures of human non-CF and CF airway epithelial cells. Lysosomal pH was measured by ratio imaging using a fluorescent pH indicator consisting of 40-kDa dextran conjugated to Oregon Green 488 and tetramethylrhodamine. In all cell types, lysosomal pH was ∼4.5, unaffected by the thiazolidinone CFTR inhibitor CFTRinh-172, and increased to ∼6.5 following bafilomycin inhibition of the vacuolar proton pump. Lysosomal pH did not differ significantly in airway epithelial cells from non-CF versus CF humans or mice. Our results provide direct evidence against the conclusions of Teichgräber et al. that lysosomal acidification is CFTR-dependent, impaired in CF, or responsible for ceramide accumulation. As such, alternative mechanisms are needed to explain increased ceramide in CF airways. Non-CFTR mechanisms, such as ClC-type chloride channels, are likely involved in maintaining electroneutrality during organellar acidification.
Chronic lung infection, inflammation, and deterioration of lung function cause morbidity and mortality in cystic fibrosis (CF)2 (1). Although the genetic cause of CF mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator protein (CFTR) (2) was identified nearly 20 years ago, the mechanisms by which CFTR mutations cause lung disease remain unclear. Various mechanisms have been proposed to link defective CFTR function to CF lung disease, including impaired organellar acidification, reduced airway submucosal gland function, abnormal immune cell response, and altered airway surface liquid (ASL) composition and volume (1, 3, 4). Determination of the mechanisms linking defective CFTR function to CF pathology is of great importance in developing rational therapies to treat CF.
Teichgräber et al. (5) recently reported a new pathological manifestation of CF with the potential to link CFTR mutation to lung disease. They reported that the sphingolipid ceramide was increased by 4–10-fold in respiratory tract epithelial cells, submucosal gland cells, and alveolar macrophages in murine models of CF and in airway epithelial cells from human CF subjects. In CF mice, reduction of ceramide levels by amitriptyline (an inhibitor of the ceramide-producing enzyme acid sphingomyelinase) or knock-out of the acid sphingomyelinase gene reduced several key parameters of CF disease including airway inflammation, phagocyte recruitment, and susceptibility to infection by Pseudomonas aeruginosa. To relate defective CFTR function to ceramide accumulation, Teichgräber et al. (5) reported greatly impaired acidification in lysosomes in airway epithelial cells from CF mice with pH ∼6, compared with pH ∼5 in cells from non-CF mice. The acidification defect in CF was proposed to involve impaired CFTR-dependent chloride entry into the lumen of lysosomes, preventing electrogenic H+ entry. The relatively alkaline lysosome in CF was proposed to greatly impair the activity of ceramide-metabolizing enzymes resulting in ceramide accumulation. Defective lysosomal acidification was thus proposed to be the critical link between CFTR mutation and ceramide accumulation.
Motivated by three concerns, we re-examined the central observation of Teichgräber et al. (5) that lysosomal acidification is CFTR-dependent and defective in CF airway epithelial cells. First, the observation recapitulates the “defective organellar acidification hypothesis” proposed in 1991 (6) and subsequently refuted by multiple laboratories using various CF models (see “Discussion”). Second, recent data suggest that members of the ClC family of Cl-/H+ antiporters are responsible for electrogenic chloride transport in lysosomes (7). Last, we believe that the lysosomal pH measurements by Teichgräber et al. (5) made using LysoSensor Green (Invitrogen), are technically flawed and invalid (see “Results” and “Discussion”).
Using pH-sensitive fluorescent dextran conjugates and standard “pulse-chase” protocols (8), we used ratio imaging fluorescence microscopy to measure lysosomal pH in cell cultures that express and do not express CFTR, and primary murine and human airway epithelial cells from non-CF and CF subjects. In each epithelial cell type studied, lysosomal pH was not altered by CFTR inhibition (by CFTRinh-172) or CFTR mutation. Our findings provide direct evidence against the conclusions of Teichgräber et al. (5) that lysosomal acidification is CFTR-dependent and impaired in CF and, therefore, indicates that defective lysosomal acidification cannot account for the elevated ceramide.
EXPERIMENTAL PROCEDURES
Cell Lines—MDCK type II cells (kidney proximal tubule epithelial cells that do not express CFTR (9)), HT29 cells (CFTR-expressing colorectal adenocarcinoma epithelium, ATCC HTB-38) and Calu-3 cells (CFTR-expressing lung adenocarcinoma, ATCC HTB-55) were cultured using standard methods at 37 °C in 5% CO2/95% air in medium without phenol red. For microscopy, cells were grown on 18-mm diameter round coverglasses for 2–4 days prior to imaging, or, in the case of polarized Calu-3 cells, for 8–14 days on Costar Snapwell permeable supports having a 0.4-μm pore diameter polyester membrane.
Isolation of Primary Murine Epithelial Cells—Murine tracheal epithelial cells from age-matched wild type and ΔF508-CFTR homozygous mice in a CD1 genetic background were isolated and studied without culturing. Mice were euthanized by lethal injection of Avertin, the trachea excised, and epithelial cells were obtained using a 5-mm diameter cytology brush (Olympus Medical Systems). All procedures were approved by the UCSF Institutional Animal Care and Use Committee.
Isolation of Primary Human Epithelial Cells—Human nasal epithelial cells (non-CF and CF (homo- or heterozygous for the ΔF508 allele)) were obtained freshly from consenting volunteers and studied without culturing. Following topical application of lidocaine the anterior-inferior turbinate was brushed using a 5-mm diameter cytology brush. Alternatively, brushings were obtained from consenting subjects undergoing an unrelated surgical procedure. All procedures were approved by the UCSF Committee on Human Research.
Primary Human Epithelial Cell Cultures—For some studies freshly isolated primary human airway epithelial cells were cultured at an air-liquid interface at 37 °C in 5% CO2, 95% air as described (10). Nasal epithelial cells from non-CF and CF subjects were obtained as described above, and non-CF human bronchial epithelial cells were obtained from human lungs that were either rejected for lung transplantation (as part of the California Transplant Program) or removed during lung transplantation. Briefly, after brushing cells were dissociated by enzymatic digestion and suspended in a 1:1 mixture of Dulbecco's modified Eagle's medium and Ham's F-12 medium containing 5% fetal calf serum, gentamicin (50 μg/ml), penicillin (100 units/ml), streptomycin (1 μg/ml), and fungizone (2.5 μg/ml), and seeded at a density of 106 cells/cm2 onto Costar Snapwell permeable supports overlaid with a thin coat (15 μg/cm2) of human placental collagen. After 24 h, cells were rinsed with PBS, and the medium was replaced with ALI medium (11) containing gentamicin, penicillin, and streptomycin. Medium was subsequently changed every 3 days, and cultures were used 12–24 days after plating at which time transepithelial resistance was ∼1000 Ω/cm2, a thin airway surface liquid layer was observed, and full mucociliary differentiation was confirmed by histology (performed in parallel cultures).
Synthesis of Fluorescent pH Probe—A fluorescent conjugate was synthesized using standard succinimidyl ester chemistry in which amino dextran (40 kDa) was covalently labeled with Oregon Green 488 (Invitrogen) and tetramethylrhodamine (OG488/TMR-dextran). Briefly, 40-kDa amino dextran was reacted with TMR-succinimidyl ester in 200 mm sodium bicarbonate buffer (pH 8.2) for 1 h at room temperature, purified by dialysis (yielding TMR-dextran), and reacted with Oregon Green 488 succinimidyl ester. The conjugate was dialyzed extensively and confirmed to be free of unreacted dye by size exclusion chromatography.
Labeling of Lysosomes with Fluorescent Dextrans—A standard pulse-chase protocol was used to label lysosomes in which cells were incubated with 5–12 mg/ml OG488/TMR-dextran (30–45 min, 37 °C) in PBS (supplemented with 6 mm glucose and 1.1 mm pyruvate; PBSgp), washed 3–4 times in PBSgp, and incubated (2–3 h, 37 °C) to chase OG488/TMR-dextran to the lysosomal compartment (8). In some experiments, TMR-dextran was used in an identical manner to label lysosomes. For cells grown on coverglasses, OG488/TMR-dextran was included in the bathing medium. Polarized cells grown on permeable supports were labeled with OG488/TMR-dextran from the basolateral side (as endocytosis is more efficient from this surface (12)). After labeling, the permeable support was excised with a scalpel and cell layers were mounted on polylysine-coated coverglasses (MatTek) with apical surface down for imaging. Freshly isolated epithelial cells were washed three times in PBSgp by centrifugation (5 min, 1000 × g), labeled in suspension, washed, and plated on polylysine- or Cell-Tak (BD Biosciences)-coated coverglasses for imaging. In some experiments, after the 2–3-h chase period, the CFTR inhibitor CFTRinh-172 (10 μm, 30 min) or the vacuolar ATPase inhibitor bafilomycin (100 nm, 30 min) were added prior to imaging. Because CFTRinh-172 interacts at the cytoplasmic surface of CFTR with amino acids in its sixth transmembrane domain (13), it is anticipated that CFTRinh-172 would inhibit CFTR in lysosomes because both plasma membrane and lysosomal CFTR present the same surface to the cytoplasm. For all cell preparations and maneuvers, 3–5 separate experiments were done (each representing a different cell culture, different murine sample, or separate human sample) and 15–96 lysosomal regions were analyzed per data set.
Fluorescence Microscopy, Image Analysis, and Calibration Procedures—Quantitative fluorescence ratio imaging was done on a Nikon Eclipse TE2000E microscope equipped with a Nikon 100×, NA 1.49 Apo TIRF objective lens, Exfo X-cite light source (attenuated by neutral density filters to prevent photobleaching) and Photometrics QuantEM:512SC CCD camera (∼95% quantum efficiency). Cells were maintained at 37 °C during imaging with a micro-incubator. Sequential, wide-field images of cells were captured using 31001 (for Oregon Green 488 fluorescence) and 31002 (TMR) filter sets (Chroma). As reported in prior studies of endosomal pH and chloride concentration (14–16), image analysis was performed by computing the ratio of area-integrated, background-subtracted fluorescence intensities for specified regions-of-interest in Oregon Green 488 and TMR images (typically containing 1–4 lysosomes) using NIH ImageJ software.
In situ calibrations were performed to relate OG488/TMR-dextran fluorescence ratio to lysosomal pH using methods described previously (15, 16). Briefly, lysosomes were loaded as above and pH in all cellular compartments was clamped by incubation in high K+ solutions containing inhibitors and ionophores (5 mm Hepes, 5 mm potassium citrate, 120 mm KCl, 20 mm NaCl, 1 mm CaCl2, 1 mm MgCl2, 100 nm bafilomycin A1, 10 μm nigericin, 10 μm valinomycin, and 10 μm CCCP) over the pH range 2.5–7.43. A full pH calibration is shown for MDCK II cells, and partial calibrations for other cell types confirmed similar responses. Background-corrected fluorescence ratios at different pH values were fitted to the equation: Fgreen/Fred = A+B ([1 + 10(pKa-pH)nH]-1) where pKa is the acid dissociation constant, nH is the Hill coefficient and A and B are offset and amplitude factors, respectively.
Immunostaining of Cells with TMR-Dextran-labeled Lysosomes—Cells were labeled with TMR-dextran (which contains free amino groups) as described above, fixed (PBS containing 4% paraformaldehyde, 10 min), permeabilized (PBS containing 0.1% Triton X-100, 3 min) and blocked (PBS containing 1% bovine serum albumin, 20 min) prior to immunostaining and imaging. Cells were immunostained with a primary rabbit polyclonal antibody against lysosomal-associated membrane protein 1 (LAMP-1, Abcam) and secondary donkey anti-rabbit IgG-labeled with Alexa Fluor 488 (Invitrogen).
LysoSensor Green DND-189 Studies—Experiments were performed to evaluate the properties of LysoSensor Green DND-189 (Invitrogen), the fluorescent probe molecule used by Teichgräber et al. (5) to measure lysosomal (vesicular) pH. Cells were loaded by 15-min incubation with 1 μm LysoSensor Green (per the manufacturer's protocol); in some experiments cells were loaded with 70 nm LysoSensor Green as described by Teichgräber et al. (5). To determine the cellular location of LysoSensor, lysosomes were labeled with TMR-dextran prior to staining with LysoSensor Green and visualized by fluorescence microscopy. Two approaches were applied to attempt to calibrate a cellular pH response for LysoSensor Green (as described in Fig. 2 of Teichgräber et al. (5)). In a first approach, lysosomes were labeled with TMR-dextran, cellular pH was clamped using ionophores (as described under “Fluorescence Microscopy, Image Analysis, and Calibration Procedures”) and cells were labeled with 1 μm LysoSensor Green prior to fluorescence imaging. In a second approach, lysosomes were labeled with TMR-dextran, cells were treated with PBS containing 0.005% digitonin (with pH adjusted in the range 4–7.4), and cells were labeled with 1 μm LysoSensor Green prior to fluorescence imaging.
FIGURE 2.

Lysosomal acidification in epithelial cell cultures does not depend on CFTR. Lysosomal pH was measured by ratio imaging of OG488/TMR-dextran fluorescence. Shown are pH under control conditions (blue circles), and in the presence of CFTRinh-172 (10 μm, gray circles) or bafilomycin (100 nm, red circles). Filled circles are mean ± S.D. for individual sets of experiments, and open circles are overall mean ± S.E. Data are shown for MDCK II, HT29, non-polarized Calu-3, and polarized (permeable support-grown) Calu-3 epithelial cell cultures. Insets show fluorescence micrographs of Oregon Green 488 fluorescence in lysosomes (bar, 5 μm). For all data sets, no significant difference from control following CFTR inhibition (by ANOVA with Scheffe multiple comparison posthoc test). p < 0.005 for bafilomycin treatment in all cases.
RESULTS
To measure lysosomal pH, a fluorescent pH indicator consisting of Oregon Green 488 (OG488, pKa ∼4.7), and TMR was synthesized (Fig. 1, inset). The Oregon Green 488 moiety provides excellent sensitivity at or near the pH of lysosomes, and the pH-insensitive TMR fluorophore allows pH measurement by quantitative ratio imaging. To calibrate the pH probe, cells were incubated with the OG488/TMR-dextran for 30–45 min for cellular uptake by fluid-phase endocytosis (pulse) and then further incubated for 2–3 h for trafficking of probe molecules to lysosomes (chase). Cells were then incubated in solutions at specified pH containing high-K+, ionophores and bafilomycin to equalize pH in all cellular compartments. The pKa determined for Oregon Green 488 by ratio imaging of cells was the same as that determined in vitro (Fig. 1A), indicating that OG488/TMR-dextran reports lysosomal pH accurately and that the lysosomal environment does not alter the properties of OG488/TMR-dextran. Although the pulse-chase protocol used here is an accepted, standard method, we confirmed that lysosomes were labeled with dextran conjugates by colocalization with LAMP-1. Cells were labeled with TMR-dextran by 30-min pulse and 2-h chase, fixed, and immunostained for LAMP-1, which predominantly localizes to lysosomes (17–19). The punctate labeling by TMR-dextran (Fig. 1B, top) colocalized very well with LAMP-1 (Fig. 1B, middle and bottom, merge shown in orange, ∼90% co-localization).
FIGURE 1.

In situ calibration of Oregon Green 488 and TMR-labeled dextran in lysosomes. A, calibration of Oregon Green 488/TMR-dextran (OG488/TMR-dextran, inset)-labeled lysosomes in MDCK II epithelial cells. Cells were labeled using a standard pulse-chase protocol and fluorescence ratio (Fgreen/Fred) measured at specified pH using perfusates containing high K+/ionophores (see “Experimental Procedures”). The average data (±S.E.) are shown for 4–5 separate measurements at each calibration point. The average lysosomal pH from one data set is overlaid on the calibration curve (dashed lines at ∼pH 4.5). B, fluorescence micrographs of fixed and permeabilized MDCK II cells labeled with TMR-dextran by pulse-chase (red, top; bar, 5 μm) and immunostained for the lysosomal marker LAMP-1 (green, middle). A merged image with orange indicating co-localization is also shown (bottom).
Lysosomal pH was first measured in epithelial cell lines expressing CFTR (Calu-3 and HT29) or lacking CFTR (MDCK II). Fig. 2 summarizes many measurements done under control conditions (blue circles), with CFTR inhibition (gray circles), and with proton pump inhibition (red circles). Lysosomes in each of these cell lines acidified to ∼pH 4.5. Lysosomal pH was sensitive to bafilomycin treatment, confirming that lysosomal acidification is dependent on vacuolar H+-ATPase activity. However, lysosomal acidification was not altered by the thiazolidinone CFTR inhibitor CFTRinh-172. Also, lysosomal acidification was independent of cell polarization in Calu-3 cells (cells grown on permeable supports versus glass coverslips).
Lysosomal pH was also measured in freshly isolated and cultured airway epithelial cells from non-CF and CF mice and humans. In freshly isolated murine tracheal epithelial cells, lysosomal pH was ∼4.5, insensitive to CFTR inhibition, and comparable in cells from wild type and CF (ΔF508-CFTR) mice (Fig. 3A). Lysosomal acidification in human nasal epithelial cells that were freshly excised (Fig. 3B) or cultured at an airliquid interface (Fig. 3C, right) was insensitive to CFTR inhibition and not altered by CFTR mutation (ΔF508). Finally, lysosomal acidification in human bronchial epithelial cells cultured at an air-liquid interface was insensitive to CFTR inhibition (Fig. 3C, left). In each cell type lysosomal acidification was sensitive to bafilomycin.
FIGURE 3.

Lysosomal acidification does not depend on CFTR in freshly isolated murine tracheal and human nasal epithelial cells, or in well-differentiated cultures of human nasal and bronchial epithelial cells from non-CF and CF subjects. Lysosomal pH was measured by ratio imaging of OG488/TMR-dextran fluorescence. Data are presented as in Fig. 2 for freshly isolated epithelial cells from murine trachea (A) and human nasal (B) sources, and cultured bronchial and nasal epithelial cells from humans (C). Insets show fluorescence micrographs of Oregon Green 488 fluorescence in lysosomes (bar, 5 μm). For all data sets, no significant difference from control with CFTR inhibition (except murine tracheal cells [n = 2 for CFTRinh-172]) or CFTR mutation (by ANOVA with Scheffe posthoc test). p < 0.005 for bafilomycin treatment in all cases.
The data in Figs. 2 and 3 provide direct evidence that lysosomal acidification is not affected by CFTR mutation or inhibition in murine and human airway epithelial cells, and not influenced by CFTR expression or inhibition in various epithelial cell lines. Our data thus do not agree with the data of Teichgräber et al. (5) that lysosomal acidification in airway epithelial cells from mice is CFTR-dependent and inhibited by CFTRinh-172. Consequently, our data do not support the conclusions that lysosomal acidification is CFTR-dependent, impaired in CF, and responsible for ceramide accumulation.
To examine possible reasons for these different findings, we studied the properties of LysoSensor Green (DND-189), the probe used by Teichgräber et al. to measure lysosomal pH. LysoSensor Green is a weak base that accumulates in acidic organelles in response to both pH gradients and absolute organellar pH, and whose fluorescence is increased upon protonation. To determine which organelles are labeled by LysoSensor Green, lysosomes were labeled with TMR-dextran by pulse-chase and then labeled with LysoSensor Green for 15 min using 1 μm (per the manufacturer's specifications) or 70 nm (as used by Teichgräber et al. (5)) concentrations. TMR-dextran colocalized with LysoSensor at both concentrations of LysoSensor Green (Fig. 4A), albeit with ∼5–10-fold brighter staining at the higher concentration. LysoSensor Green fluorescence is thus predominantly derived from lysosomes and presumably reflects both increased partitioning to acidic lysosomes and fluorescence enhancement upon protonation. We next carried out pH calibration as done by Teichgräber et al. (5). Although complete details of the calibration method were not provided (including the mechanism of cell permeabilization), two protocols were used. In a first protocol, lysosomes were initially labeled with TMR-dextran, pH was equalized in all cellular compartments with solutions containing high [K+], ionophores and bafilomycin (as done in the calibration of OG488/TMR-dextran), followed by labeling with 1 μm LysoSensor Green (Fig. 4B). Although punctate TMR-dextran fluorescence was observed at all pH values in the range 4–7, verifying lysosomal integrity, no LysoSensor Green fluorescence was observed, consistent with the requirement of a pH gradient for organellar uptake of LysoSensor Green. In a second protocol, lysosomes were labeled with TMR-dextran, and then cells were treated with PBS containing 0.005% digitonin in the range pH 4–7 (to permeabilize the plasma membrane and set cytoplasmic pH), followed by labeling with 1 μm LysoSensor Green (Fig. 4C). Using this protocol, lysosomal integrity was again confirmed via TMR-dextran fluorescence; however, LysoSensor Green fluorescence could not be detected at low pH and was greatly reduced at higher pH. These results indicate that generation of a valid pH calibration curve for LysoSensor Green is not possible, as probe fluorescence is influenced by multiple factors including pH gradients and absolute compartmental pH. Quantitative determination of lysosome pH without a calibration curve is not possible.
FIGURE 4.

LysoSensor Green compartmentalization and pH response. A, compartmentalization of LysoSensor Green in MDCK II cells labeled at 1 μm and 70 nm. Cells were initially labeled with TMR-dextran by pulse-chase (red, left panels) and then with LysoSensor Green (right panels). B and C, LysoSensor Green response in pH calibration protocols using (B) ionophore or (C) digitonin (see “Experimental Procedures” and “Results”). Cells were initially labeled with TMR dextran using a pulse-chase protocol (red, left panels), and the cellular pH was clamped (at indicated pH values) prior to labeling with 1 μm LysoSensor Green (green, right panels). In all image sets, bar is 5 μm.
DISCUSSION
This study was motivated by a recent report indicating a critical role for CFTR in lysosomal acidification (5). Lysosomal acidification was reported to be CFTR-dependent in airway epithelial cells from mice, and this acidification defect was proposed to alter the activity of ceramide metabolizing enzymes resulting in ceramide accumulation. Using mouse models, increased ceramide was directly related to key manifestations of CF disease progression (5). Using the CFTR inhibitor CFTRinh-172, we found no evidence for CFTR involvement in lysosomal acidification in epithelial cell lines, freshly isolated murine tracheal and human nasal epithelial cells, and cultured human nasal and bronchial epithelial cells. Further, we found no evidence for defective lysosomal acidification in freshly isolated nasal epithelial cells from CF humans, tracheal epithelial cells from CF mice, and well-differentiated primary cultures of CF human nasal epithelial cells. As such, our findings provide direct evidence against the conclusions of Teichgräber et al. (5) that lysosomal acidification is CFTR-dependent and impaired in CF, and therefore indicate that defective lysosomal acidification cannot account for the elevated ceramide. These findings underscore the need for further evaluation of the mechanism of ceramide accumulation in CF lungs and whether ceramide accumulation contributes to the inflammatory state observed in other tissues affected in CF.
As described under “Results,” we believe the pH measurement method of Teichgräber et al. (5) was technically flawed, yielding incorrect lysosomal pH values. We were unable to generate a calibration curve for the LysoSensor Green pH response using protocols involving cellular permeabilization and pH equilibration prior to cell labeling with LysoSensor Green. Although a variety of methods are available to permeabilize cell membranes, all of these approaches either equilibrate pH across all cellular compartments (as with ionophores) or in the cytoplasm (as with digitonin). As such, it is not possible theoretically to construct a meaningful calibration curve for LysoSensor Green. Further, LysoSensor Green is not amenable to ratiometric determination of pH, so that interpretation of fluorescence would depend on multiple factors, including probe uptake, absolute organellar pH, cytoplasm-to-organelle pH gradient, and extent of probe protonation. Given that Teichgräber et al. (5) studied two murine CF models (complete and ∼95% reduction of CFTR), alternative explanations for discrepancies, such as residual CFTR activity in the presence of CFTRinh-172 or differential cellular trafficking of ΔF508-CFTR in murine versus human cells (20), are unlikely.
Several mechanisms can theoretically affect organellar pH including the
activities of H+-pump and H+-leak pathways, buffer
capacity, counterion conductances (Na+, K+,
H+, Cl-, and
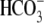 ), membrane potential, and
cytoplasmic ionic content. Presently, methods to measure each of these
parameters are not available; however, there is evidence using
chloride-sensitive fluorophores to indicate that chloride is the major ionic
species required for endosomal and Golgi acidification
(15,
21). Divergent roles for CFTR
in the regulation of organellar acidification, and consequent production of
the CF phenotype, have been proposed. In 1991, Al-Awqati and co-workers
(6) reported that CFTR
constitutes a prominent counterion conductance pathway required for organellar
acidification and that defective acidification in CF is responsible for
defective protein processing and consequent manifestations of CF disease.
Similarly, Nelson and co-workers
(22) reported that lysosomal
acidification in alveolar macrophages is CFTR-dependent and that the fusion of
lysosomes to phagosomes is required for phagosomal acidification and,
consequently, the bactericidal activity of macrophages. In contrast, Deretic
and co-workers
(23–25)
have reported that CFTR mutation results in hyperacidification of
cellubrevin-positive (recycling) endosomes and the Golgi, which produces
defects in protein processing, trafficking, and signaling. Hyperacidification
of organelles was proposed to result from CFTR-dependent dys-regulation of
(organellar) epithelial sodium channels (ENaC), which could be corrected by
cGMP-mediated ENaC inhibition
(24). However, several
independent laboratories have refuted these reports. Endosomal acidification
has been demonstrated to be CFTR-independent in transfected cell lines and
epithelial cells expressing endogenous CFTR
(26–29).
Some of these studies also reported that protein kinase A-mediated stimulation
of CFTR channel activity results in chloride efflux from endosomes (driven by
the electrochemical gradient), concluding that at baseline CFTR is inactive in
endosomes (26,
27). CFTR expression was also
reported to have no (29) or
very minor (∼0.2 pH units more acidic in CF
(30)) influence on the pH of
the trans-Golgi. In discrepant reports, CFTR dysfunction has been
found to hyperacidify (by 0.5 pH units
(25)) or mildly acidify (0.2
pH units (30)) Golgi pH in the
same cell types using similar GFP-based pH probes. Of direct relevance to this
study, CFTR activity was reported not to alter lysosomal pH in pancreatic
epithelial cells (28,
31). Finally, phagosomal and
lysosomal acidification in macrophages were found to be CFTR-independent
(16,
32).
), membrane potential, and
cytoplasmic ionic content. Presently, methods to measure each of these
parameters are not available; however, there is evidence using
chloride-sensitive fluorophores to indicate that chloride is the major ionic
species required for endosomal and Golgi acidification
(15,
21). Divergent roles for CFTR
in the regulation of organellar acidification, and consequent production of
the CF phenotype, have been proposed. In 1991, Al-Awqati and co-workers
(6) reported that CFTR
constitutes a prominent counterion conductance pathway required for organellar
acidification and that defective acidification in CF is responsible for
defective protein processing and consequent manifestations of CF disease.
Similarly, Nelson and co-workers
(22) reported that lysosomal
acidification in alveolar macrophages is CFTR-dependent and that the fusion of
lysosomes to phagosomes is required for phagosomal acidification and,
consequently, the bactericidal activity of macrophages. In contrast, Deretic
and co-workers
(23–25)
have reported that CFTR mutation results in hyperacidification of
cellubrevin-positive (recycling) endosomes and the Golgi, which produces
defects in protein processing, trafficking, and signaling. Hyperacidification
of organelles was proposed to result from CFTR-dependent dys-regulation of
(organellar) epithelial sodium channels (ENaC), which could be corrected by
cGMP-mediated ENaC inhibition
(24). However, several
independent laboratories have refuted these reports. Endosomal acidification
has been demonstrated to be CFTR-independent in transfected cell lines and
epithelial cells expressing endogenous CFTR
(26–29).
Some of these studies also reported that protein kinase A-mediated stimulation
of CFTR channel activity results in chloride efflux from endosomes (driven by
the electrochemical gradient), concluding that at baseline CFTR is inactive in
endosomes (26,
27). CFTR expression was also
reported to have no (29) or
very minor (∼0.2 pH units more acidic in CF
(30)) influence on the pH of
the trans-Golgi. In discrepant reports, CFTR dysfunction has been
found to hyperacidify (by 0.5 pH units
(25)) or mildly acidify (0.2
pH units (30)) Golgi pH in the
same cell types using similar GFP-based pH probes. Of direct relevance to this
study, CFTR activity was reported not to alter lysosomal pH in pancreatic
epithelial cells (28,
31). Finally, phagosomal and
lysosomal acidification in macrophages were found to be CFTR-independent
(16,
32).
There is good evidence that non-CFTR chloride channels are involved in organellar acidification. Using an elegant genetic screen, Maeda et al. (33) recently identified a novel, ubiquitous anion channel necessary for Golgi acidification (GPHR, Golgi pH regulator). Taken together with studies by Tsien and co-workers (34), which indicate that Golgi pH is predominantly controlled by H+-pump and leak pathways, and the aforementioned studies of Seksek et al. (29) and Chandy et al. (30), it seems unlikely that CFTR is a major determinant of Golgi pH. Using primary cells from knock-out mice and direct measurement of chloride and pH, endosomal acidification was found to require ClC-5 in proximal tubule cells and ClC-3 in hepatocytes (35, 36). ClC-3 was also found to be responsible for the acidification of neuronal synaptic vesicles (37). In lysosomes, biophysical evidence has indicated that ClC-7 is a major determinant of acidification in hepatocytes and Hela cells (7). However, data from knock-out mice have suggested that ClC-mediated organellar acidification may be cell type-specific. Lysosomal acidification in neurons and fibroblasts was not impaired in ClC-7 knock-out or gray-lethal mice (which lack Ostm1, the essential β-subunit of ClC-7, Refs. 38, 39).
In summary, our measurements do not support the conclusions of Teichgräber et al. (5) that lysosomal acidification in airway epithelial cells is CFTR-dependent, impaired in CF, and responsible for ceramide accumulation. Determination of the mechanism responsible for ceramide accumulation in CF airway tissue is important to the development of rational approaches for CF therapies. Investigation into the role of ceramide in other tissues that manifest inflammatory responses in CF is warranted. Finally, given the complex nature of CFTR regulation and the effects of CFTR channel activity on alternative conductance pathways, we emphasize the importance of studying appropriate cell types and the application of rigorously validated methods to determine the role of CFTR in organellar pH regulation.
Acknowledgments
We thank Panjamaporn Sungwang for isolation of murine trachea, Dr. D. W. Nielson for providing human epithelial cells, and Dr. W. E. Finkbeiner for culture of human epithelial cells.
This work was supported, in whole or in part, by National Institutes of Health Grants EB00415, DK35124, HL73856, HL59198, EY13574, DK72517, and DK81355. This work was also supported by the Cystic Fibrosis Foundation (Research Development Program and Drug Discovery grants) and from the American Heart Association (0765070Y). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: CF, cystic fibrosis; CFTR, cystic fibrosis transmembrane conductance regulator protein; PBS, phosphate-buffered saline; TMR, tetramethylrhodamine; GFP, green fluorescent protein.
References
- 1.Rowe, S. M., Miller, S., and Sorscher, E. (2005) N. Engl. J. Med. 352 1992-2001 [DOI] [PubMed] [Google Scholar]
- 2.Rommens, J. M., Iannuzzi, M. C., Kerem, B., Drumm, M. L., Melmer, G., Dean, M., Rozmahel, R., Cole, J. L., Kennedy, D., Hidaka, N., Zsiga, M., Buchwald, M., Riordan, J. R., Tsui, L. C., and Collins, F. S. (1989) Science 245 1059-1065 [DOI] [PubMed] [Google Scholar]
- 3.Verkman, A. S., Song, Y., and Thiagarajah, J. R. (2003) Am. J. Physiol. 284 C2-C15 [DOI] [PubMed] [Google Scholar]
- 4.Boucher, R. C. (2007) Trends Mol. Med. 13 231-240 [DOI] [PubMed] [Google Scholar]
- 5.Teichgräber, V., Ulrich, M., Endlich, N., Reithmüller, J., Wilker, B., Conceição Ce Olivereira-Munding, C., van Heeckeren, A. M., Barr, M. L., von Kürthy, G., Schmid, K. W., Weller, M., Tümmler, B., Lang, F., Grassme, H., Döring, G., and Gulbins, E. (2008) Nat. Med. 14 382-391 [DOI] [PubMed] [Google Scholar]
- 6.Barasch, J., Kiss, B., Prince, A., Saiman, L., Gruenert, D., and Al-Awqati, Q. (1991) Nature 352 70-73 [DOI] [PubMed] [Google Scholar]
- 7.Graves, A. R., Curran, P. K., Smith, C. L., and Mindell, J. A. (2008) Nature 453 788-792 [DOI] [PubMed] [Google Scholar]
- 8.Ohkuma, S. (1989) Methods Enzymol. 174 131-154 [DOI] [PubMed] [Google Scholar]
- 9.Mohamed, A., Ferguson, D., Seibert, F. S., Cai, H. M., Kartner, N., Grinstein, S., Riordan, J. R., and Lukacs, G. L. (1997) Biochem. J. 322 259-265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamaya, M., Finkbeiner, W. E., Chun, S. Y., and Widdicombe, J. H. (1992) Am. J. Physiol. 262 L713-L724 [DOI] [PubMed] [Google Scholar]
- 11.Fulcher, M. L., Gabriel, S., Burns, K. A., Yankaskas, J. R., and Randell, S. H. (2005) Methods Mol. Med. 107 183-206 [DOI] [PubMed] [Google Scholar]
- 12.von Bonsdorff, C. H., Fuller, S. D., and Simons, K. (1985) EMBO J. 4 2781-2792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caci, E., Caputo, A., Hinzpeter, A., Arous, N., Fanen, P., Sonawane, N., Verkman, A. S., Ravazzolo, R., Zegarra-Moran, O., and Galietta, L. J. (2008) Biochem. J. 413 135-142 [DOI] [PubMed] [Google Scholar]
- 14.Zen, K., Biwersi, J., Periasamy, N., and Verkman, A. S. (1992) J. Cell Biol. 119 99-110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sonawane, N., Thiagarajah, J. R., and Verkman, A. S. (2002) J. Biol. Chem. 277 5506-5513 [DOI] [PubMed] [Google Scholar]
- 16.Haggie, P. M., and Verkman, A. S. (2007) J. Biol. Chem. 282 31422-31428 [DOI] [PubMed] [Google Scholar]
- 17.Sharma, M., Pampinella, F., Nemes, C., Benharouga, M., So, J., Du, K., Bache, K. G., Papsin, B., Zerangue, N., Stenmark, H., and Lukacs, G. L. (2004) J. Cell Biol. 164 923-933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar, K. G., Barriere, H., Carbone, C. J., Liu, J., Swaminathan, G., Xu, P., Li, Y., Baker, D. P., Peng, J., Lukacs, G. L., and Fuchs, S. Y. (2007) J. Cell Biol. 179 935-950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verghese, B., Barriere, H., Carbone, C. J., Banerjee, A., Swaminathan, G., Plotnikov, A., Xu, P., Peng, J., Goffin, V., Lukacs, G. L., and Fuchs, S. Y. (2008) Mol. Cell. Biol. 28 5275-5287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ostedgaard, L. S., Rogers, C. S., Dong, Q., Randak, C. O., Vermeer, D. W., Rokhlina, T., Karp, P. H., and Welsh, M. J. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 15370-15375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sonawane, N. D., and Verkman, A. S. (2003) J. Cell Biol. 160 1129-1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di, A., Brown, M. E., Deriy, L. V., Li, C., Szeto, F. L., Chen, Y., Huang, P., Tong, J., Naren, A. P., Bindokas, V., Palfrey, H. C., and Nelson, D. J. (2006) Nat. Cell Biol. 8 933-944 [DOI] [PubMed] [Google Scholar]
- 23.Poschet, J. F., Skidmore, J., Boucher, J. C., Firoved, A. M., Van Dyke, R. W., and Deretic, V. (2002) J. Biol. Chem. 277 13959-13965 [DOI] [PubMed] [Google Scholar]
- 24.Poschet, J. F., Fazio, J. A., Timmins, G. S., Ornatowski, W., Perkett, E., Delgado, M., and Deretic, V. (2006) EMBO Rep. 7 553-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poschet, J. F., Boucher, J. C., Tatterson, L., Skidmore, J., Van Dyke, R. W., and Deretic, V. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 13972-13977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lukacs, G. L., Chang, X.-B., Kartner, N., Rotstein, O. D., Riordan, J. R., and Grinstein, S. (1992) J. Biol. Chem. 267 14568-14572 [PubMed] [Google Scholar]
- 27.Biwersi, J., and Verkman, A. S. (1994) Am. J. Physiol. 266 C149-C156 [DOI] [PubMed] [Google Scholar]
- 28.Dunn, K. W., Park, J., Semrad, C. E., Gelman, D. L., Shevell, T., and McGraw, T. E. (1994) J. Biol. Chem. 269 5336-5345 [PubMed] [Google Scholar]
- 29.Seksek, O., Biwersi, J., and Verkman, A. S. (1996) J. Biol. Chem. 271 15542-15548 [DOI] [PubMed] [Google Scholar]
- 30.Chandy, G., Grabe, M., Moore, H. P., and Machen, T. E. (2001) Am. J. Physiol. 281 C908-C921 [DOI] [PubMed] [Google Scholar]
- 31.Van Dyke, R. W., Root, K. V., Schreiber, J. H., and Wilson, J. M. (1992) Biochem. Biophys. Res. Commun. 184 300-305 [DOI] [PubMed] [Google Scholar]
- 32.Lamonthe, J., and Valvano, M. A. (2008) Microbiology 154 3825-3834 [DOI] [PubMed] [Google Scholar]
- 33.Maeda, Y., Ide, T., Koike, M., Uchiyama, Y., and Kinoshita, T. (2008) Nat. Cell Biol. 10 1135-1145 [DOI] [PubMed] [Google Scholar]
- 34.Wu, M. M., Llopis, J., Adams, S., McCaffery, J. M., Kulomaa, M. S., Machen, T. E., Moore, H.-P. H., and Tsien, R. Y. (2000) Chem. Biol. 7 197-209 [DOI] [PubMed] [Google Scholar]
- 35.Hara-Chikuma, M., Yang, B., Sonawane, N., Sasaki, S., Uchida, S., and Verkman, A. S. (2005) J. Biol. Chem. 280 1241-1247 [DOI] [PubMed] [Google Scholar]
- 36.Hara-Chikuma, M., Wang, Y., Guggino, S. E., Guggino, W. B., and Verkman, A. S. (2005) Biochem. Biophys. Res. Commun. 329 941-946 [DOI] [PubMed] [Google Scholar]
- 37.Stobrawa, S. M., Breiderhoff, T., Takamori, S., Engel, D., Schweizer, M., Zdebik, A. A., Bosl, M. R., Ruether, K., Jahn, H., Draguhn, A., Jahn, R., and Jentsch, T. J. (2001) Neuron 29 185-196 [DOI] [PubMed] [Google Scholar]
- 38.Kasper, D., Planells-Cases, R., Fuhrmann, J. C., Scheel, O., Zeitz, O., Ruether, K., Schmitt, A., Poët, M., Steinfeld, R., Schweizer, M., Kornak, U., and Jentsch, T. J. (2005) EMBO J. 24 1079-1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lange, P. F., Wartosch, L., Jentsch, T. J., and Fuhrmann, J. C. (2006) Nature 440 220-223 [DOI] [PubMed] [Google Scholar]