

Abstract
Integrase (IN) from human immunodeficiency virus, type 1 (HIV-1) exerts pleiotropic effects in the viral replication cycle. Besides integration, IN mutations can impact nuclear import, viral maturation, and reverse transcription. IN and reverse transcriptase (RT) interact in vitro, and the IN C-terminal domain (CTD) is both necessary and sufficient for binding RT. We used nuclear magnetic resonance spectroscopy to identify a putative RT-binding surface on the IN CTD, and surface plasmon resonance to obtain kinetic parameters and the binding affinity for the IN-RT interaction. An IN K258A substitution that disrupts reverse transcription in infected cells is located at the putative RT-binding surface, and we found that this substitution substantially weakens IN CTD-RT interactions. We also identified two additional IN amino acid substitutions located at the putative RT-binding surface (W243E and V250E) that significantly impair viral replication in tissue culture. These results strengthen the notion that IN-RT interactions are biologically relevant during HIV-1 replication and also provide insights into this interaction at the molecular level.
A key step in the replication cycle of human immunodeficiency virus, type 1 (HIV-1)2 involves the synthesis of double-stranded DNA copies of the diploid, single-stranded RNA genome by viral reverse transcriptase (RT) within the cytoplasm of infected cells. This newly synthesized viral DNA becomes an integral component of a large nucleoprotein complex termed the preintegration complex (PIC). PIC-associated proteins include HIV-1 matrix, Vpr, RT, and integrase (IN) (1–4), as well as cellular components such as lens epithelium-derived growth factor p75 (5), barrier-to-autointegration factor (6), and the high mobility group I(Y) protein (7). The PIC traverses the nuclear envelope, and within the nucleus the viral DNA is subsequently inserted into the chromosomes of the infected host, an event specifically catalyzed by IN.
HIV-1 IN and RT (a heterodimeric enzyme of 66- and 51-kDa subunits) are initially synthesized as part of the Gag-Pol polyprotein, which is subsequently processed by HIV-1 protease during viral maturation to produce active IN and RT. IN (288 amino acids; 32 kDa) comprises three domains: an N-terminal zinc-binding domain (residues 1–50), a catalytic core domain (residues 51–212), and a C-terminal domain (CTD; residues 213–288) that binds DNA nonspecifically (reviewed in Ref. 8). IN has multiple effects throughout the viral life cycle, as perturbations within the IN coding sequence of the pol gene can impact not only integration but also PIC nuclear import, proper virion maturation, and reverse transcription (9–20).
Purified HIV-1 IN and RT have been found to physically interact in co-immunoprecipitation, glutathione S-transferase-based pulldown, and surface plasmon resonance (SPR) experiments (15, 18, 21, 22), and assays with IN deletion constructs showed the IN CTD is both necessary and sufficient for this association (18, 21). The significance of this IN-RT interaction in the context of viral replication has not yet been firmly established. In vitro studies with purified IN and RT, however, have shown that HIV-1 IN can stimulate both the initiation and elongation modes of RT-catalyzed reverse transcription by enhancing RT processivity (23).
To aid our ongoing studies of functional interactions between IN and RT during HIV-1 replication, we have characterized in detail the RT-binding surface on the IN CTD using nuclear magnetic resonance (NMR) spectroscopy, identifying particular IN CTD amino acids that form contacts with RT through chemical shift perturbation experiments. In addition, we have assessed kinetic parameters and binding constants for both IN CTD-RT and IN-RT interactions using SPR. This experimental approach allows us to define and quantify the IN-RT interaction to a level that is not possible through co-immunoprecipitation or other fusion protein pulldown techniques.
With the intention of perturbing IN-RT interactions, we introduced an alanine substitution for lysine at position 258 (K258A) within the IN CTD and subsequently used SPR to characterize the effect of this substitution upon IN·RT complex formation. Lys258 in IN lies at the putative RT-binding site identified here by NMR, and the IN K258A substitution has been previously shown to significantly impair reverse transcription in infected cells (20). We found that the K258A substitution substantially weakened IN CTD-RT interactions. Finally, we introduced various IN amino acid substitutions at the putative RT-binding surface into the NL4-3 viral clone, and replication of viruses containing these substitutions was significantly impaired in tissue culture assays. Collectively, the observations presented here suggest that IN-RT contacts are biologically relevant during HIV-1 replication and that an IN CTD·RT complex may provide a useful predictive model that can guide studies of IN-RT interactions in the viral life cycle.
EXPERIMENTAL PROCEDURES
Expression Plasmids and Protein Purification—Full-length HIV-1 IN with a 7 × His tag (MHHHHHHHIVPRGSHM) at the N terminus was produced using pT7-7 (His)H-IN, an expression plasmid bearing the IN coding sequence from HIV-1 NL4-3 (24). The expression plasmid encoding IN residues 220–270 (IN 220–270) with a 6 × His tag (MGSSHHHHHHSSGLVPRGSHM) at the N terminus was provided by Dr. Robert Craigie (National Institutes of Health). Mutagenic primers and a PCR-based site-directed mutagenesis protocol (25) were employed to introduce a specific mutation within the IN 220–270 expression plasmid to yield the IN 220–270 protein sequence bearing a K258A substitution (IN 220–270/K258A). All IN constructs were expressed in CodonPlus Escherichia coli cells (Stratagene) and were purified using nickel-nitrilotriacetic acid immobilized metal affinity chromatography. RT was expressed in E. coli cells (M15::pDMI.1 strain) using a plasmid (p6HRT-PROT) with dual inducible expression cassettes for (6 × His tag)-p66 and untagged HIV-1 protease. In this expression system, partial cleavage of the p66 product by protease generates the His-tagged p66/p51 RT heterodimer (26). RT was purified using Ni2+-nitrilotriacetic acid immobilized metal affinity chromatography followed by cation exchange chromatography, and SDS-PAGE analysis confirmed the presence of highly (>95%) pure RT heterodimer. Protein concentration was measured by Bradford assay (Bio-Rad) using bovine serum albumin (BSA) as a standard.
Virus Preparation and Replication Kinetics—Generation of the HIV-1 NL4-3 molecular clones (27) bearing the IN single amino acid substitutions W243E and V250E were prepared by first introducing the desired substitutions into pSK-SC001, a pBluescript-derived subcloning vector containing an engineered AgeI site and the 2.3-kilobase pair AgeI-EcoRI fragment of NL4-3 (nucleotide positions 3486–5744), which encompasses the IN gene. Mutagenic primers and a PCR-based site-directed mutagenesis protocol (25) were used to introduce W243E and V250E substitutions into the IN coding sequence in pSK-SC001. The modified AgeI-EcoRI fragment was then excised from the subcloning vector and ligated into NL4-3 that had been sequentially digested with AgeI and then EcoRI. Top10 cells (Invitrogen) were transformed with the ligation product, plated on LB agar containing 80 μg/ml ampicillin, and incubated at 30 °C. The presence of the desired mutation within the IN gene was confirmed by sequencing.
Virus stocks were prepared by transfecting 293T cells grown at 37 °C in Dulbecco modified Eagle's medium containing l-glutamine, sodium pyruvate, and 4.5 g/liter glucose (Invitrogen) supplemented with 10% fetal bovine serum (HyClone), 100 IU/ml penicillin G, and 100 μg/ml streptomycin. Transfections were performed in 75-cm2 flasks with PolyFect Transfection Reagent (Qiagen) using 8 μg of plasmid DNA bearing the wild-type or mutant HIV-1 clone. Medium containing virus was collected 36–48 h later and gravity-filtered through 0.45-μm cellulose acetate filters, and the levels of p24 antigen contained in the viral supernatant were determined by enzyme-linked immunosorbent assay (PerkinElmer).
One hundred nanograms of p24 equivalent of viral stocks were added to 2 × 106 CEM cells grown at 37 °C in 2 ml of RPMI 1640 (Invitrogen) having 100 IU/ml penicillin G, 100 μg/ml streptomycin, and 10% fetal bovine serum. The medium was removed 12 h postinfection and replaced with 3 ml of fresh medium. Aliquots of the culture medium were then collected for p24 analysis every 2 days after the infection, with the cells being split 1:3 following these collections of viral supernatant. Mock infections were performed using wild-type virus that had been heat-inactivated by treatment at 95 °C for 4 min.
NMR Experiments with 15N-labeled IN 220–270 and RT—15N-labeled IN 220–270 was expressed in E. coli cells using M9 minimal medium supplemented with 15NH4Cl, and the 15N-labeled, His-tagged protein was purified using Ni2+-nitrilotriacetic acid immobilized metal affinity chromatography. The purified RT heterodimer was dialyzed at 4 °C in NMR buffer (50 mm sodium phosphate buffer, pH 6.5, 100 mm NaCl, and 0.5 mm EDTA), and the chemical shift perturbation experiment was performed by adding aliquots of an unlabeled RT heterodimer to 200 μm 15N-labeled IN 220–270 (concentration calculated assuming monomeric IN 220–270) in NMR buffer with 93% H2O, 7% D2O. Successive aliquots of RT were added to IN 220–270 to produce IN 220–270:RT molar ratios of 10:1 and 5:1, and 2:1. Molar ratios were calculated assuming monomeric amounts of IN 220–270. After each addition, a two-dimensional 15N-1H heteronuclear single quantum coherence (HSQC) spectrum (28) of the sample was recorded at 25 °C on a Bruker 800 MHz Avance spectrometer. The same buffer and temperature conditions used in the NMR structure determination of IN 220–270 (29) were also employed in our NMR experiments. Prior to performing titrations with RT, a preliminary spectrum of free 15N-labeled IN 220–270 was collected for the purpose of establishing peak assignments, which were made by comparison of peak positions with the NMR data archived at the Protein Data Bank (Protein Data Bank code 1IHV). Structural representations for IN 220–270 were produced using the program MacPyMOL (30).
SPR Experiments with IN and RT—SPR experiments using IN, IN 220–270, IN 220–270/K258A, and RT were performed on a Biacore T100 instrument. The carboxydextran surface of a CM5 chip (Biacore) was activated by injecting a 1:1 (by volume) mixture of 400 mm 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride: 100 mm N-hydroxysuccinimide over two flow cells, followed by immobilization of ∼1300 response units (RU) of recombinant RT on one of the flow cells. The remaining flow cell was left unmodified and used as a reference. The cross-linking reaction was quenched by injecting 1 m ethanolamine (pH 8.5) across both flow cells. Concentrations of 500, 250, 125, 62.5, 31.3, 15.6, 7.8, 3.9, and 0 nm (serial 2-fold dilutions) of the various IN constructs were injected across both flow cells. All of these experiments at a given concentration were performed in triplicate, and double-referencing (i.e. both solvent subtraction and correction for nonspecific binding to the unmodified reference flow cell) was employed to produce signals resulting only from IN-RT interactions. All of the SPR experiments were performed at 25 °C in a running buffer containing 20 mm Hepes (pH 7.5), 300 mm NaCl, 0.05% Tween 20, and 1 mm β-mercaptoethanol (SPR buffer) using a 50 μl/min flow rate. Regeneration of the chip surface between individual experiments was accomplished using a 120 s pulse of 10 mm glycine (pH 2.5) at a flow rate of 10 μl/min. In addition, the running buffers for experiments with IN and IN 220–270 contained 1% and 0.011% glycerol, respectively, to match residual glycerol levels in the samples that were injected across the chip surface. The association and dissociation phases of all of these experiments were 240 and 300 s, respectively. Similar experiments using 0, 0.125, 0.25, 0.5, and 1 μm BSA were performed in duplicate as negative controls. Assessment of the validity of 1:1 or two-state conformational exchange binding models, as well as the calculation of binding parameters, was performed using Biacore T100 Evaluation software (Biacore). Residual χ2 values are a measure of the average squared difference between experimental data and fitted models and were calculated using the formula χ2 =Σ(RUexp - RUfitted)2/n, where the summation takes place over n time points in the sensorgrams, and RUexp and RUfitted are the experimental and fitted response unit values, respectively, at a given time point.
Analytical Ultracentrifugation—The oligomeric state of IN 220–270 in both NMR and SPR buffers was assessed by sedimentation equilibrium analysis with a Beckman XL-A analytical ultracentrifuge using absorption optics at 280 nm. IN 220–270 in NMR buffer was analyzed at a concentration of 150 μm at 20 °C in a 3-mm-path length double sector cell and at a concentration of 93 μm at 25 °C in a 12-mm path length six-sector cell. Absorbance values in this data set were corrected for differences in cell path length, and partial specific volumes for ensuing calculations were derived from the IN 220–270 amino acid composition and corrected for temperature (v̄25 °C = 0.734 ml/g and v̄20 °C = 0.732 ml/g) (31, 32). IN 220–270 was analyzed in SPR buffer at concentrations of 150, 93, and 31 μm at 25 °C in a 12-mm-path length six-sector cell. Sedimentation equilibrium profiles for IN 220–270 in both NMR and SPR buffers were measured at speeds of 20,000, 24,000, and 30,000 rpm. For the two buffer conditions, the absorbance profiles at each speed were initially fitted by a nonlinear least squares algorithm within Beckman Origin-based software (version 3.01) to an exponential curve corresponding to a single ideal species, yielding weight-averaged molecular weight values for the IN 220–270 sample. The data set encompassing the entire collection of scans corresponding to different rotor speeds and protein loading concentrations were globally fitted to a monomer-dimer equilibrium model (using the “multifit” option) to obtain a dimer dissociation constant for each buffer condition. Higher order association models ultimately resulted in poorer fits and thus were not analyzed further.
RESULTS
NMR Spectroscopy to Map the RT-binding Surface on the IN CTD—To obtain a detailed map of the RT-binding surface on the IN CTD, we performed NMR chemical shift perturbation experiments with 15N-labeled IN 220–270 and unlabeled RT. A preliminary HSQC spectrum of 15N-labeled IN 220–270 indicated excellent agreement between observed peak chemical shifts and NMR chemical shift data archived at the Protein Data Bank (Protein Data Bank code 1IHV), allowing straightforward peak assignment. For mapping experiments, a HSQC spectrum of the free 15N-labeled IN 220–270 was collected, and successive aliquots of unlabeled RT were subsequently added to produce IN 220–270:RT molar ratios of 10:1, 5:1, and 2:1, with a 15N-1H HSQC spectrum being collected after each titration (Fig. 1). The peaks showed no changes in chemical shift as the titration progressed, and most IN 220–270 signals showed relatively gradual reductions in peak intensity during the course of the titration. At a 2:1 IN 220–270:RT molar ratio, most of the peaks in the acquired spectrum became broadened to the noise level (data not shown), and so no further titrations were performed.
FIGURE 1.

HSQC spectra of free IN 220–270 (blue) at a concentration of 200 μm, and of IN 220–270 mixed with RT at IN 220–270:RT molar ratios of 10:1 (red) and 5:1 (green). A representative subset of IN 220–270 signals is shown in the expanded view. Residues whose amide group signals show a significant change in intensity are labeled in red.
Amide backbone peaks associated with nine residues showed significant decreases in intensity during the titration, as compared with most other peaks in the spectra: Arg231, Leu242, Trp243, Gly247, Ala248, Val250, Ile251, Gln252, and Lys258 (Fig. 1). With the exception of Arg231, these residues mapped to one side of the IN 220–270 structure, thus identifying a putative RT-binding surface (Fig. 2). Arg231 is located within a stretch of amino acids connecting strands β1 and β2 in the NMR structure (29) and is positioned close to the putative RT-binding surface. The signal decreases observed during the titration suggest that these nine residues experience intermediate exchange on the NMR timescale (i.e. chemical exchange occurs at a frequency that is on the order of the difference between free and bound amide group chemical shift) (33, 34). Finally, the selective broadening of the peaks for the above nine amino acids indicates that binding of IN 220–270 to RT is specific, as nonspecific binding would produce uniform changes across all of the peaks in the spectra.
FIGURE 2.

A, ribbon diagrams illustrating two views of the putative IN 220–270 binding surface (shown in “red”) that interacts with RT. B, space-filling model with orientation similar to the leftmost representation in A. The positions of amino acids whose peak signals decreased significantly in intensity in the presence of RT are indicated.
SPR Studies to Characterize IN-RT Interactions—Previous co-immunoprecipitation and glutathione S-transferase pulldown studies of IN-RT interactions provided relatively qualitative measures of IN-RT affinity. To provide a quantitative measurement of IN-RT binding affinity and kinetics, SPR experiments using RT and either full-length IN or IN 220–270 were performed. RT was immobilized via lysine residues to a CM5 chip using amine coupling chemistry. The crystal structure for RT (Protein Data Bank code 1HMV) shows that lysines lie throughout the RT surface; thus the cross-linking method is anticipated to attach RT to the chip surface in a random orientation. As such, any putative IN-binding site on RT is expected to be accessible for some percentage of the immobilized RT molecules. The p66/p51 heterodimer exhibits long term stability under the conditions employed for SPR experiments (35), and therefore the RT heterodimer species (rather than isolated p66 and p51 subunits) are anticipated to be immobilized on the biosensor chip surface.
Purified IN or IN 220–270 was passed over the immobilized RT at concentrations of 0–500 nm, and sensorgrams were recorded. The sensorgrams clearly show that both IN and IN 220–270 bound immobilized RT under these experimental conditions (Fig. 3). Analogous experiments using BSA as a negative control indicated no binding to RT (Fig. 3), and BSA present in 10-fold molar excess of IN 220–270 did not influence IN 220–270 binding to RT (data not shown), indicating that IN·RT complex formation is not due to nonspecific protein-protein interactions.
FIGURE 3.

Sensorgrams showing binding events for IN 220–270, IN, and IN 220–270/K258A, as well as negative control experiments using BSA. The concentrations employed were 0–500 nm IN 220–270, IN, or IN 220–270/K258A, or 0–1 μm BSA. The red lines are experimental data, whereas the black lines are simulated binding curves employing a two-state conformational exchange model.
A “two-state conformational exchange” binding model, where an initial bimolecular encounter to form a nascent species is followed by a conformational transition to a stable complex, has been recently described for the IN-RT interaction (22), and this model was fitted to the sensorgram data to extract binding parameters. The quality of the fit for the two-state conformational exchange model was assessed by a residual χ2 value, a parameter that measures the difference between the fitted model and the experimental data. A simple bimolecular “1:1 binding” model was also fitted to the sensorgram data for comparison purposes. Experiments with both IN and IN 220–270 resulted in suboptimum fits of the 1:1 binding model, with relatively high χ2 values (6.67 and 22.3 for IN and IN 220–270, respectively). 2–3-fold decreases in χ2 were achieved when fitting the two-state conformational exchange model to the IN and IN 220–270 data sets (3.55 and 8.82, respectively; Fig. 3 and Table 1),
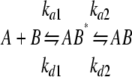 |
REACTION 1 |
and the resulting dissociation constants (KD) for IN and IN 220–270 binding to RT were 61.2 and 87.3 nm, respectively (Fig. 4 and Table 1). Both the kinetic parameters and the dissociation constant for the full-length IN-RT interaction were in good agreement with previously published measurements (22).
TABLE 1.
Fitted binding parameters for two-state conformational exchange reaction See Reaction 1, which shows the two-state reaction with conformational exchange.
| ka1 | kd1 | ka2 | kd2 | KD | X2 | |
|---|---|---|---|---|---|---|
| m–1 s–1 | s–1 | s–1 | s–1 | nm | ||
| IN 220–270 | 5.45 (± 0.10) × 105 | 1.19 (± 0.02) × 10–1 | 4.11 (± 0.02) × 10–3 | 2.75 (± 0.01) × 10–3 | 87.3 ± 2.2 | 8.82 |
| IN | 4.82 (± 0.04) × 104 | 1.45 (± 0.02) × 10–2 | 6.23 (± 0.10) × 10–3 | 1.59 (± 0.03) × 10–3 | 61.2 ± 1.6 | 3.55 |
| IN 220–270/K258A | 1.55 (± 0.01) × 104 | 3.25 (± 0.03) × 10–2 | 4.64 (± 0.03) × 10–3 | 2.69 (± 0.02) × 10–3 | 769.5 ± 9.9 | 0.97 |
FIGURE 4.

Kinetic parameters (ka1, kd1, ka2, and kd2) and dissociation constants (KD) obtained by employing a two-state conformational exchange binding model for IN 220–270, IN, and IN 220–270/K258A.
Although the dissociation constants for IN and IN 220–270 binding to RT were similar, two of the kinetic parameters found for these two systems differed: both the initial on-rate and off-rate values (ka1 and kd1, respectively) for IN binding to RT were ∼10-fold lower than those found for IN 220–270 (Fig. 4 and Table 1). The reduction in ka1 for IN·RT complex formation, compared with that for IN 220–270 binding to RT, might be attributed to steric hindrance from the IN N-terminal zinc-binding domain or catalytic core domain, with these domains possibly preventing the IN CTD from gaining clear access to the binding site on RT. In such a situation, the smaller IN 220–270 construct might gain access to the binding site more freely. The reduced kd1 found for IN·RT complex formation, as compared with that found in experiments involving IN 220–270, suggests increased stabilization of the IN·RT complex at this particular step in the binding mechanism. The added stability could possibly arise from IN N-terminal zinc-binding domain and catalytic core domain residues, which may contribute to RT binding. Altogether, the differences in individual kinetic parameters suggest that the mechanistic details for IN and IN 220–270 binding to RT are not entirely the same, even though the overall dissociation constants for these two systems are comparable.
Analytical Ultracentrifugation Analysis of IN 220–270—A previous study has reported that 1 mm IN 220–270 in NMR buffer is predominantly dimeric (29). The current NMR experiments with IN 220–270 were performed at concentrations appreciably lower than 1 mm (100–200 μm), and the current SPR experiments were performed at much lower concentrations (3.9–500 nm) and in the presence of nondenaturing detergent (0.05% Tween 20). As such, some proportion of the dimeric IN 220–270 may have dissociated to its monomeric form under our NMR and SPR experimental conditions.
To measure the extent of IN 220–270 self-association in our NMR and SPR experiments, IN 220–270 in both NMR and SPR buffers was analyzed by sedimentation equilibrium techniques. IN 220–270 sedimentation equilibrium profiles at 150 and 93 μm in NMR buffer were initially fitted to a single ideal species model. This fit showed a distinct decrease in apparent molecular mass (from 16,305 to 14,269 g/mol) with decreasing concentration, an observation that is consistent with an equilibrium shift from dimeric IN 220–270 (molecular mass, 16,580.8 g/mol) to an increased proportion of monomeric IN 220–270 (molecular mass, 8290.4 g/mol). The absorbance profiles for all IN 220–270 concentrations and rotor speeds were then globally fitted to a monomer-dimer equilibrium model, yielding an IN 220–270 dimer dissociation constant of 2.3 μm in NMR buffer. Such a dissociation constant suggests that ∼10–14% of the total IN 220–270 exists as a monomer under the conditions of our NMR experiment.
Sedimentation equilibrium experiments using 150, 93, and 31 μm IN 220–270 were also performed in SPR buffer. As with the corresponding experiments in NMR buffer, the absorbance profiles associated with all IN 220–270 loading concentrations and rotor speeds were globally fitted to a monomer-dimer equilibrium model. The fit yielded an IN 220–270 dimer dissociation constant of 28 μm in SPR buffer, assuming an insignificant effect of bound Tween 20 detergent upon IN 220–270 buoyant density. A dissociation constant of this magnitude suggests that 98.3% of the total IN 220–270 is monomeric at the highest concentration employed (500 nm) in the SPR experiments. The dissociation constant measured in SPR buffer was ∼12-fold higher than the corresponding dissociation constant measured in NMR buffer.
An IN Substitution at the Putative IN-RT Binding Interface Weakens RT Binding—Lys258 in IN lies at the putative RT-binding site and is a well conserved residue with 99.7% sequence identity among 325 HIV-1 and SIVcpz strains (20). Furthermore, a K258A substitution has been previously shown to significantly impair reverse transcription in infected cells (20). A K258A substitution was introduced into IN 220–270 (IN 220–270/K258A), and the binding parameters for IN 220–270/K258A·RT complex formation were quantitated by SPR. Using the two-state conformational exchange model, the K258A substitution in IN 220–270 raises the KD for complex formation nearly 9-fold over that found for the unmodified IN 220–270 construct (769.5 nm for IN 220–270/K258A versus 87.3 nm for IN 220–270). The difference in binding affinities stems mostly from changes in ka1 (Fig. 4 and Table 1). As such, initial association of IN 220–270 with RT may be mediated by electrostatic attraction, because removal of positive charge from the putative RT-binding surface adversely affected the on-rate for complex formation. The binding data show that the same IN K258A substitution that impairs reverse transcription (20) also results in significantly weaker IN-RT binding interactions, indicating that productive IN-RT interactions may be important for efficient reverse transcription during HIV-1 replication.
NL4-3 Viral Clones Bearing IN W243E and V250E Substitutions Are Replication-defective—Mutant viral clones encoding IN amino acid substitutions W243E and V250E were constructed to test whether such substitutions at the putative RT-binding surface significantly impair viral replication in tissue culture. Viruses prepared by transient transfection in 293T cells were used to infect CEM cells, and the infectivity was monitored by measuring levels of p24 antigen in culture media every 2 days following infection. Both of the IN substitutions resulted in impaired HIV-1 replication (Fig. 5). The p24 levels for wild-type virus peaked after 6 days (∼600 ng/ml), whereas p24 levels for the mutant viruses and the heat-inactivated wild-type virus never exceeded 40 ng/ml during the course of the experiment. These observations show that nonconservative substitutions at the putative IN-RT interface are not well tolerated by replicating virus and suggest that such substitutions may inhibit viral replication by disrupting important IN-RT contacts.
FIGURE 5.

Replication kinetics of wild-type and mutant HIV-1 viruses. CEM cells were infected by wild type (♦) or mutant HIV-1 bearing IN substitutions W243E (▴) or V250E (×). The infections with each mutant virus were repeated to confirm the replication-defective phenotype (W243E, n = 2; V250E, n = 3), and representative data for each viral clone are shown. Mock infections were performed using heat-inactivated wild-type virus (□).
DISCUSSION
Although the IN CTD has been long known to bind to DNA (36, 37), additional studies indicate that this domain also promotes binding to various cellular proteins. Such proteins include the survival of motor neuron complex component Gemin2 (38), the nuclear import receptor importin 7 (39), the cytidine deaminase APOBEC3G (40), the human homolog of the mouse EED (embryonic ectoderm development) gene product (41), and the histone acetyl transferase p300 (42). The diverse array of IN CTD binding partners suggests that the IN CTD is a versatile protein component that can mediate a variety of physiological processes during HIV-1 replication.
Previous studies have also shown that the IN CTD is both necessary and sufficient for binding to RT (18, 21), and both the NMR and SPR data presented in the current study further confirm these reports. In addition, Hehl et al. (21) identified regions within RT involved in forming IN contacts, demonstrating that either the p66 or the p51 subunit from HIV-1 RT is sufficient for binding HIV-1 IN. Additional experiments with RT deletion constructs narrowed the IN-interacting residues on RT to a bipartite region encompassing residues 1–242 in the fingers-palm domain and residues 387–422 in the connection domain. In vitro IN-RT interactions in murine leukemia virus have also been identified (43) but are less well characterized than in HIV-1.
The role of IN-RT interactions during viral replication is currently unknown. Nonetheless, diverse reports collectively suggest that IN and RT may be functionally intertwined, not only in HIV-1 but in other retroviruses. For example, both RT and RT-derived peptides can inhibit the 3′-end processing and strand transfer activity of IN in vitro (44–46). In vitro assays also show that HIV-2 IN is inhibited by HIV-2 RT and that HIV-1 IN is inhibited by heterologous RTs from HIV-2 or murine leukemia virus (45). As such, the possibility that RT may serve as a viral cofactor that prevents suicidal autointegration events in retroviruses has been proposed (45, 46). Furthermore, in avian sarcoma leukosis virus IN exhibits activity not only in its free form as a 32-kDa phosphoprotein, but also as part of a larger β subunit that includes RT (47). The overt tethering of IN with RT in this case suggests that a tight IN-RT linkage may be an important feature for retroviral replication.
In addition, genetic studies have indicated that IN is important for reverse transcription during both HIV-1 and murine leukemia virus replication (15, 48) and have identified a number of HIV-1 IN mutant viruses that are impaired in viral cDNA synthesis (9, 14–20). Furthermore, continuous passage of a replication-impaired chimeric HIV-1 viral clone bearing the HIV-2 IN gene ultimately yielded compensating mutations within the IN and RT genes, resulting in increased synthesis of viral DNA and improved viral fitness (49). A recent study from our laboratory has shown that IN directly stimulates both the initiation and elongation modes of reverse transcription in vitro and that the processivity of RT in the presence of IN is enhanced. Such findings suggest that direct physical interactions between IN and RT may facilitate reverse transcription in vivo (23).
In all, the above studies suggest that IN and RT may exert mutual effects upon one another's activity during viral replication. Whether such effects may always proceed through direct IN-RT interactions during infection is currently unclear. For example, some IN mutations that impact reverse transcription in infected cells could conceivably act indirectly by perturbing other steps in the viral life cycle prior to reverse transcription (e.g. uncoating). IN mutations may also affect reverse transcription indirectly through altered interactions with cellular factors. One such cellular factor may be Gemin2, a host cell protein that binds IN and that also appears to play a role in reverse transcription (38).
Nonetheless, one straightforward explanation for the various findings described above is that IN and RT influence each other's activity through direct IN-RT contacts. The overall aim of this study is to investigate whether physical interactions between IN and RT are functionally relevant. A necessary step in achieving this aim is pinpointing the particular amino acids involved in the IN-RT interaction. IN-RT binding interactions were therefore characterized in detail through mapping the RT-binding surface on IN 220–270 by NMR spectroscopy. Although IN 220–270 does not encompass the full set of IN CTD amino acids (residues 213–288), the availability of both the IN 220–270 NMR structure and assignment data for peaks in the IN 220–270 NMR spectra (29) strongly motivated the use of IN 220–270 in NMR mapping experiments. In addition, the binding data gathered by both NMR and SPR confirm that the IN 220–270 subdomain represents a useful construct for investigating IN CTD-RT interactions.
Our NMR studies show strong peak changes for a set of IN residues that mapped mostly (with the exception of Arg231) to one side of the IN 220–270 structure, pinpointing specific IN amino acids involved in contacting RT. As noted above, Arg231 is close to the putative IN-RT interface. Binding to RT could possibly induce a conformational change within IN 220–270 that subsequently positions Arg231 alongside the proposed RT-binding surface. Alternatively, the RT heterodimer is conceivably both large enough and flexible enough (see below) to engage both the putative RT-binding surface on IN 220–270 and the more distal Arg231 residue without requiring a significant conformational adjustment on the part of IN 220–270.
IN 220–270 was modeled as a homodimer in the NMR structure (29). Complementation studies indicate that IN functions as a multimer (50–52), and various models describing the IN oligomeric state have been proposed (53–58). However, the precise nature of the particular protein-protein interactions among IN monomers has not been fully characterized to date, and in contrast to the dimeric model for IN 220–270, a crystal structure of a dimeric two-domain IN construct encompassing the IN catalytic core domain and CTD shows that the CTDs are separated by about 55 Å (59). As such, the biological significance of a dimeric form of IN 220–270 for IN activity has not yet been established.
Our sedimentation equilibrium experiments show that IN 220–270 is 86–90% dimeric at the concentrations used in our NMR study (∼100–200 μm). Most residues identified by our mapping experiments (with the exception of R231) also comprise the IN 220–270 homodimer interface. This observation suggests that the IN 220–270 dimer must first dissociate before binding to RT can take place, because the putative RT-binding surface is removed from solvent in the IN 220–270 dimeric species. A sequence of events consistent with these observations is that IN 220–270 undergoes monomer-dimer exchange in solution (as dictated by a micromolar dimerization constant), with RT recognizing and sequestering the monomeric form of IN 220–270 with nanomolar affinity at what was previously the IN 220–270 homodimer interface.
An alternative (though less probable) binding model is that RT binds dimeric IN 220–270 at some region other than the IN 220–270 homodimer interface, inducing dissociation of the IN 220–270 homodimer to its monomeric form. Such an event might also change the chemical environment at the IN 220–270 homodimer interface, thus producing the observed NMR spectra. However, evidence supporting such a scenario is scant; aside from Arg231, no other RT binding sites on IN 220–270 (other than the homodimer interface) were identified by the NMR experiments. Therefore, in this alternative binding model, a single amino acid (Arg231) must mediate the entire interaction with the 117-kDa RT heterodimer. Although formally possible, such an event seems highly unlikely to provide sufficient binding energy to form a stable protein·protein complex. Altogether, the NMR data and structural analysis favor a sequence of events where (i) dimeric IN 220–270 first dissociates into monomers, and then (ii) RT forms a complex with the monomeric form of IN 220–270 at the homodimer interface, further shifting the IN 220–270 monomer-dimer equilibrium toward the monomeric species. Whether a comparable mechanism describes the interaction between RT and full-length IN is unclear, because dimerization of the IN CTD in the context of full-length IN has not yet been established.
Our SPR data indicate that both IN and IN 220–270 bind to RT with nanomolar affinity and are consistent with a two-state reaction process involving a conformational change step. Indeed, conformational flexibility is a prominent feature of RT recognition and binding of other substrates. Comparison of the x-ray crystal structures of unliganded RT (60) and complexes containing either nevirapine (61) or duplex DNA (62, 63) show significant structural differences between free and substrate-bound enzyme. In particular, the thumb subdomain within p66 undergoes a substantial (>30°) rotation upon complex formation, and the p66 fingers and palm subdomains reorient as well. Shifts in position of the p51 palm subdomain are also noted between DNA-bound and unliganded RT. Molecular dynamics simulations of RT also indicate that the thumb and fingers subdomains in the p66 subunit, as well as the p51 thumb subdomain, show considerable mobility (64). Such mobility could conceivably confer on RT the conformational adaptability needed to recognize IN through a two-step, conformational exchange mechanism.
The sedimentation equilibrium data indicate that the experimental conditions employed in the SPR binding assays strongly favor the monomeric form of IN 220–270, and previously published biophysical studies indicate that the SPR conditions should favor the monomeric form of full-length IN as well. IN has been previously shown to be monomeric at submicromolar concentrations in the presence of nondenaturing detergents at 25 °C (65). Given the concentrations of IN (3–500 nm), temperature (25 °C), and the levels of detergent (0.05% Tween 20) used in our SPR assays, we hypothesize that both IN 220–270 and IN bind RT as a monomer in these SPR experiments.
IN can directly stimulate both RT-mediated synthesis of early viral cDNA products as well as RT processivity in vitro (23). The current study further suggests that direct binding between IN and RT may have functional consequences during viral replication. Previous infectivity studies indicated that HIV-1 harboring a K258A substitution within IN resulted in substantially reduced levels of late reverse transcription products (<10% that of wild-type virus) and no detectable virus replication in a CD4+ T-cell line (20). Our NMR data indicate that Lys258 forms contacts with RT, and our SPR data indicate that introducing a K258A substitution into IN 220–270 had a disruptive impact on the dissociation constant by primarily affecting the on-rate for IN·RT complex formation. Thus, the IN K258A substitution results in both impaired reverse transcription (20) and significantly weaker IN-RT binding interactions (this study). In addition, our NMR data indicate that IN Val250 and Trp243 are involved in RT contacts, and introduction of nonconservative substitutions at these IN positions dramatically reduces viral infectivity. Collectively, the data presented here and by Lu et al. (20) suggest that IN CTD-RT interactions are biologically relevant during HIV-1 replication. Furthermore, our results indicate that the IN CTD·RT complex can be a useful predictive model for exploring mechanisms by which IN-RT interactions impact the viral life cycle. Further studies to learn whether other IN substitutions elsewhere within the putative IN-RT interface can affect viral replication and to identify whether such IN substitutions impact early or late replication events are currently ongoing.
HIV-1 RT and IN are responsible for catalyzing the essential steps of reverse transcription and integration, respectively, during the early stages of the retroviral life cycle. The physical interactions between RT and IN may serve to ensure that the PIC is assembled properly and can function efficiently. Characterization of the RT-IN interaction and determination of its biological significance may reveal new functional roles for IN, provide a mechanistic basis for the phenotypes observed with certain IN mutants, and identify new potential targets for anti-HIV therapy.
Acknowledgments
We thank Jian Ye, Charles Dobard, and Marisa Briones for technical advice and assistance rendered.
This work was supported, in whole or in part, by National Institutes of Health Grants CA68859 and AI077386 (to S. A. C.). This work was also supported by the UCLA AIDS Institute, UCLA Center for AIDS Research (AI28697 to T. A. W.), State of California Universitywide AIDS Research Program Grants CC95 LA 137 (to T. A. W.) and 05-LA-021 (to S. A. C.), and by the Intramural Research Program of the Center for Cancer Research, NCI, National Institutes of Health (to S. F. J. L.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: HIV-1, human immunodeficiency virus, type 1; IN, integrase; RT, reverse transcriptase; CTD, C-terminal domain; PIC, preintegration complex; SPR, surface plasmon resonance; BSA, bovine serum albumin; HSQC, heteronuclear single quantum coherence; RU, response unit(s); NMR, nuclear magnetic resonance.
References
- 1.Farnet, C. M., and Haseltine, W. A. (1991) J. Virol. 65 1910-1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heinzinger, N. K., Bukinsky, M. I., Haggerty, S. A., Ragland, A. M., Kewalramani, V., Lee, M. A., Gendelman, H. E., Ratner, L., Stevenson, M., and Emerman, M. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 7311-7315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller, M. D., Farnet, C. M., and Bushman, F. D. (1997) J. Virol. 71 5382-5390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bukrinsky, M. I., Sharova, N., McDonald, T. L., Pushkarskaya, T., Tarpley, W. G., and Stevenson, M. (1993) Proc. Natl. Acad. Sci. U. S. A. 90 6125-6129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Llano, M., Vanegas, M., Fregoso, O., Saenz, D., Chung, S., Peretz, M., and Poeschla, E. M. (2004) J. Virol. 78 9524-9537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin, C. W., and Engelman, A. (2003) J. Virol. 77 5030-5036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farnet, C. M., and Bushman, F. D. (1997) Cell 88 483-492 [DOI] [PubMed] [Google Scholar]
- 8.Holmes-Son, M. L., Appa, R. S., and Chow, S. A. (2001) Adv. Genet. 43 33-69 [DOI] [PubMed] [Google Scholar]
- 9.Tsurutani, N., Kubo, M., Maeda, Y., Ohashi, T., Yamamoto, N., Kannagi, M., and Masuda, T. (2000) J. Virol. 74 4795-4806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gallay, P., Hope, T., Chin, D., and Trono, D. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 9825-9830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quillent, C., Borman, A. M., Paulous, S., Dauguet, C., and Clavel, F. (1996) Virology 219 29-36 [DOI] [PubMed] [Google Scholar]
- 12.Bukovsky, A., and Gottlinger, H. (1996) J. Virol. 70 6820-6825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engelman, A., Englund, G., Orenstein, J. M., Martin, M. A., and Craigie, R. (1995) J. Virol. 69 2729-2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leavitt, A. D., Robles, G., Alesandro, N., and Varmus, H. E. (1996) J. Virol. 70 721-728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu, X., Liu, H., Xiao, H., Conway, J. A., Hehl, E., Kalpana, G. V., Prasad, V., and Kappes, J. C. (1999) J. Virol. 73 2126-2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Masuda, T., Planelles, V., Krogstad, P., and Chen, I. S. (1995) J. Virol. 69 6687-6696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shin, C. G., Taddeo, B., Haseltine, W. A., and Farnet, C. M. (1994) J. Virol. 68 1633-1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu, K., Dobard, C., and Chow, S. A. (2004) J. Virol. 78 5045-5055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu, R., Limon, A., Ghory, H. Z., and Engelman, A. (2005) J. Virol. 79 2493-2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu, R., Ghory, H. Z., and Engelman, A. (2005) J. Virol. 79 10356-10368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hehl, E. A., Joshi, P., Kalpana, G. V., and Prasad, V. R. (2004) J. Virol. 78 5056-5067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herschhorn, A., Oz-Gleenberg, I., and Hizi, A. (2008) Biochem. J. 412 163-170 [DOI] [PubMed] [Google Scholar]
- 23.Dobard, C. W., Briones, M. S., and Chow, S. A. (2007) J. Virol. 81 10037-10046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Appa, R. S., Shin, C. G., Lee, P., and Chow, S. A. (2001) J. Biol. Chem. 276 45848-45855 [DOI] [PubMed] [Google Scholar]
- 25.Fisher, C. L., and Pei, G. K. (1997) BioTechniques 23 570-571, 574 [DOI] [PubMed] [Google Scholar]
- 26.Le Grice, S. F., and Gruninger-Leitch, F. (1990) Eur. J. Biochem. 187 307-314 [DOI] [PubMed] [Google Scholar]
- 27.Adachi, A., Gendelman, H. E., Koenig, S., Folks, T., Willey, R., Rabson, A., and Martin, M. A. (1986) J. Virol. 59 284-291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kay, L. E., Keifer, P., and Saarinen, T. (1992) J. Am. Chem. Soc. 114 10663-10665 [Google Scholar]
- 29.Lodi, P. J., Ernst, J. A., Kuszewski, J., Hickman, A. B., Engelman, A., Craigie, R., Clore, G. M., and Gronenborn, A. M. (1995) Biochemistry 34 9826-9833 [DOI] [PubMed] [Google Scholar]
- 30.DeLano, W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific, Palo Alto, CA
- 31.Cohn, E. J., and Edsall, J. T. (1943) in Proteins, Amino Acids and Peptides as Ions and Dipolar Ions (Cohn, E. J., and Edsall, J. T., eds) pp. 370-381, Reinhold Publishing Corporation, New York
- 32.Laue, T. M., Shah, B. D., Ridgeway, T. M., and Pelletier, S. L. (1992) in Analytical Ultracentrifugation in Biochemistry and Polymer Science (Harding, S. E., Rowe, A. J., and Horton, J. C., eds) pp. 90-125, The Royal Society of Chemistry, Cambridge, UK
- 33.Lian, L.-Y., and Roberts, G. C. K. (1993) in NMR of Macromolecules: A Practical Approach (Roberts, G. C. K., ed) pp. 153-182, Oxford University Press Inc., New York
- 34.Palmer, A. G., III, Kroenke, C. D., and Loria, J. P. (2001) Methods Enzymol. 339 204-238 [DOI] [PubMed] [Google Scholar]
- 35.Divita, G., Rittinger, K., Restle, T., Immendorfer, U., and Goody, R. S. (1995) Biochemistry 34 16337-16346 [DOI] [PubMed] [Google Scholar]
- 36.Lutzke, R. A., Vink, C., and Plasterk, R. H. (1994) Nucleic Acids Res. 22 4125-4131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vink, C., Oude Groeneger, A. M., and Plasterk, R. H. (1993) Nucleic Acids Res. 21 1419-1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamamoto, S., Nishitsuji, H., Amagasa, T., Kannagi, M., and Masuda, T. (2006) J. Virol. 80 5670-5677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ao, Z., Huang, G., Yao, H., Xu, Z., Labine, M., Cochrane, A. W., and Yao, X. (2007) J. Biol. Chem. 282 13456-13467 [DOI] [PubMed] [Google Scholar]
- 40.Luo, K., Wang, T., Liu, B., Tian, C., Xiao, Z., Kappes, J., and Yu, X. F. (2007) J. Virol. 81 7238-7248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Violot, S., Hong, S. S., Rakotobe, D., Petit, C., Gay, B., Moreau, K., Billaud, G., Priet, S., Sire, J., Schwartz, O., Mouscadet, J. F., and Boulanger, P. (2003) J. Virol. 77 12507-12522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cereseto, A., Manganaro, L., Gutierrez, M. I., Terreni, M., Fittipaldi, A., Lusic, M., Marcello, A., and Giacca, M. (2005) EMBO J. 24 3070-3081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu, S. C., Court, D. L., Zweig, M., and Levin, J. G. (1986) J. Virol. 60 267-274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tasara, T., Maga, G., Hottiger, M. O., and Hubscher, U. (2000) FEBS Lett. 507 39-44 [DOI] [PubMed] [Google Scholar]
- 45.Oz, I., Avidan, O., and Hizi, A. (2002) Biochem. J. 361 557-566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oz Gleenberg, I., Avidan, O., Goldgur, Y., Herschhorn, A., and Hizi, A. (2005) J. Biol. Chem. 280 21987-21996 [DOI] [PubMed] [Google Scholar]
- 47.Brown, P. O. (1997) in Retroviruses (Coffin, J. M., Hughes, S. H., and Varmus, H. E., eds.) pp. 161-203, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [PubMed]
- 48.Lai, L., Liu, H., Wu, X., and Kappes, J. C. (2001) J. Virol. 75 11365-11372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Padow, M., Lai, L., Deivanayagam, C., DeLucas, L. J., Weiss, R. B., Dunn, D. M., Wu, X., and Kappes, J. C. (2003) J. Virol. 77 11050-11059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Gent, D. C., Vink, C., Groeneger, A. A., and Plasterk, R. H. (1993) EMBO J. 12 3261-3267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Engelman, A., Bushman, F. D., and Craigie, R. (1993) EMBO J. 12 3269-3275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ellison, V., Gerton, J., Vincent, K. A., and Brown, P. O. (1995) J. Biol. Chem. 270 3320-3326 [DOI] [PubMed] [Google Scholar]
- 53.Podtelezhnikov, A. A., Gao, K., Bushman, F. D., and McCammon, J. A. (2003) Biopolymers 68 110-120 [DOI] [PubMed] [Google Scholar]
- 54.Gao, K., Butler, S. L., and Bushman, F. (2001) EMBO J. 20 3565-3576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang, J. Y., Ling, H., Yang, W., and Craigie, R. (2001) EMBO J. 20 7333-7343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De Luca, L., Pedretti, A., Vistoli, G., Barreca, M. L., Villa, L., Monforte, P., and Chimirri, A. (2003) Biochem. Biophys. Res. Commun. 310 1083-1088 [DOI] [PubMed] [Google Scholar]
- 57.Heuer, T. S., and Brown, P. O. (1998) Biochemistry 37 6667-6678 [DOI] [PubMed] [Google Scholar]
- 58.Faure, A., Calmels, C., Desjobert, C., Castroviejo, M., Caumont-Sarcos, A., Tarrago-Litvak, L., Litvak, S., and Parissi, V. (2005) Nucleic Acids Res. 33 977-986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen, J. C., Krucinski, J., Miercke, L. J., Finer-Moore, J. S., Tang, A. H., Leavitt, A. D., and Stroud, R. M. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 8233-8238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rodgers, D. W., Gamblin, S. J., Harris, B. A., Ray, S., Culp, J. S., Hellmig, B., Woolf, D. J., Debouck, C., and Harrison, S. C. (1995) Proc. Natl. Acad. Sci. U. S. A. 92 1222-1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kohlstaedt, L. A., Wang, J., Friedman, J. M., Rice, P. A., and Steitz, T. A. (1992) Science 256 1783-1790 [DOI] [PubMed] [Google Scholar]
- 62.Jacobo-Molina, A., Ding, J., Nanni, R. G., Clark, A. D., Jr., Lu, X., Tantillo, C., Williams, R. L., Kamer, G., Ferris, A. L., Clark, P., Hizi, A., Hughes, S. H., and Arnold, A. (1993) Proc. Natl. Acad. Sci. U. S. A. 90 6320-6324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ding, J., Das, K., Hsiou, Y., Sarafianos, S. G., Clark, A. D., Jr., Jacobo-Molina, A., Tantillo, C., Hughes, S. H., and Arnold, E. (1998) J. Mol. Biol. 284 1095-1111 [DOI] [PubMed] [Google Scholar]
- 64.Temiz, N. A., and Bahar, I. (2002) Proteins 49 61-70 [DOI] [PubMed] [Google Scholar]
- 65.Deprez, E., Tauc, P., Leh, H., Mouscadet, J. F., Auclair, C., and Brochon, J. C. (2000) Biochemistry 39 9275-9284 [DOI] [PubMed] [Google Scholar]