

Abstract
Alzheimer’s disease (AD) is the most common type of dementia, comprising 60–80% of all reported cases, and currently affects 5.2 million Americans. AD is characterized pathologically by the accumulation of senile plaques (SPs), neurofibrillary tangles (NFTs), and synapse loss. The early stages of memory loss associated with AD have been studied in a condition known as amnestic mild cognitive impairment (MCI), arguably the earliest form of AD. In spite of extensive research across a variety of disciplines the cause of AD remains elusive. Proteomics techniques have helped to advance knowledge about AD by identifying irregularities in protein expression and post-translational modifications (PTMs) in AD brain. Glycosylation is a less studied PTM with regards to AD and MCI. This PTM is important to study because glycosylation is involved in proper protein folding, protein anchoring to cell membranes, and the delivery of proteins to organelles, and these processes are impaired in AD. Concanavalin-A (Con-A) binds to N-linked glycoproteins, but hydrophobic sites on non-glycoproteins are also known to bind Con-A. To our knowledge, the present study is the first to examine Con-A-associated brain proteins in MCI and AD with focus on the hippocampus and inferior parietal lobule (IPL) brain regions. Proteins found in AD hippocampus with altered levels are glutamate dehydrogenase (GDH), glial fibrillary acidic protein (GFAP), tropomyosin 3 (TPM3), Rab GDP-dissociation inhibitor XAP-4 (XAP4), and heat shock protein 90 (HSP90). Proteins found with altered levels in AD IPL are α-enolase, γ-enolase, and XAP-4. MCI hippocampal proteins with altered levels are dihydropyrimidase-2 (DRP2), glucose-regulated protein 78 (GRP-78), protein phosphatase related protein Sds-22 (Sds22), and GFAP and the only protein found with altered levels in MCI IPL was β-synuclein. These results are discussed with reference to biochemical and pathological alterations in and progression of AD.
Introduction
Alzheimer’s disease (AD) is the most common type of dementia, comprising 60–80% of all reported cases1, and currently affects 5.2 million Americans. Pathologically, AD is characterized by the accumulation of senile plaques (SPs), neurofibrillary tangles (NFTs), and loss of synapses. SPs are composed of a core of amyloid β-peptide (Aβ) and exist as either diffuse or neuritic variants.2–4 Diffuse plaques are believed to develop into the neuritic variant, which accumulate in the hippocampus, amygdala, association cortices of frontal, temporal, and parietal lobes, and in other brain areas that project to these regions in AD.4 Neuritic plaques have dense fibrillary Aβ cores surrounded by dystrophic axons and dendrites, activated microglia, and reactive astrocytes.5 NFTs are often associated with SPs.4 NFTs consist as paired helical filaments (PHFs) composed of hyperphosphorylated tau protein and occur in the cytoplasm of the soma and in neuritic projections.4, 6–11
Synapse loss is another pathological hallmark observed in AD in addition to the deposition of SPs and NFTs. The cortex of AD patients has a 25–35% reduction of total synaptic density and a 15–35% reduction of synapses per neuron.12 This finding suggests that communication within and among brain regions is limited in AD. The early stages of memory loss in arguably the earliest form of AD have been studied in a condition known as amnestic mild cognitive impairment (MCI) and have provided an important tool in the studies of the progression of AD.13
Amnestic MCI patients have observable deficits in memory or other essential cognitive abilities; however, these deficits have not developed extensively enough to interfere with activities of daily living and patients do not present dementia.1 The fate of persons who are living with MCI does not always lead to dementia and MCI patients can remain cognitively stable throughout the remainder of life. In some cases, patients revert back to a cognitively normal status.1 MCI provides an important tool for AD research because patient lifestyles and phenotypes can be analyzed to provide clues for normal brain aging or potential paths that lead to AD development.
In spite of extensive knowledge about AD and its potential precursor MCI,4, 14–17 the cause of disease remains elusive. However, proteomics techniques have helped to advance knowledge about AD by identifying irregularities in protein expression and post-translational modifications (PTMs) in affected AD brain regions.14, 18 These observations have helped formulate and support hypotheses for disease pathogenesis. For example, proteomics studies of nitration, 4-hydroxynonenalylation, and carbonylation of proteins have been studied extensively in Alzheimer’s disease (AD) and mild cognitive impairment (MCI).19–24 From such studies it is apparent that an increase in oxidative stress in brain occurs in these disorders relative to age-matched normal subjects.22–24
Glycosylation is a less studied PTM with regards to AD and MCI. This PTM becomes important to study because glycosylation is involved in proper protein folding, protein quality control, protein anchoring to cell membranes, and the delivery of proteins to organelles, and all of these processes are impaired in AD.4, 25 N-linked glycosylation is the linkage of carbohydrates to asparagine amino acids and occurs in the endoplasmic reticulum (ER), while O-linked glycosylation involves the addition of carbohydrates to serine or threonine residues and takes place in the Golgi apparatus. It is important to note that late stage N-linked glycosylation takes place in the Golgi, but the initial addition of oligosaccharide-lipid precursor occurs in the ER.26
Glycoproteomics studies have previously been conducted in culture, plasma, and cerebral spinal fluid (CSF) using a variety of chromatographic techniques such as immunoaffinity, lectin affinity, multiple affinity removal system (MARS) depletion, strong cation exchange, and reverse phase high-performance liquid chromatography (HPLC).27–35
To our knowledge, the present study is the first to examine Concanavalin-A (Con-A) associated proteins isolated from brain of MCI and AD subjects with focus on the hippocampus and inferior parietal lobule (IPL) brain regions. Hippocampal neurons project to the IPL and are critical for memory formation and are atrophied in AD.36 Our proteomics method employed Con-A lectin affinity columns coupled to 2D gel-electrophoresis, MALDI/TOF mass spectrometry, and MASCOT database searching methods for the separation and identification of glycoproteins. Con-A recognizes high-mannose, hybrid and complex bi-antennary glycoproteins, and terminal glucose carbohydrate moieties.26, 37–41 In addition to binding carbohydrate moieties, Con-A has been shown to have an independent hydrophobic binding domain separate from the carbohydrate-binding domain. Carbohydrate and hydrophobic substrate binding at their respective binding sites is neither competitive nor interactive with each other.42
Proteomics identified, Con-A-associated proteins in AD and MCI brain provide insight into impaired cellular functions observed in AD.
Results
Method overview
Figure 1 represents an overview of the experimental processes used to isolate Con-A-associated proteins. Hippocampus and inferior parietal lobule (IPL) sections from subjects with AD and MCI were homogenized and subjected to Con-A affinity chromatography. All brains were isolated from subjects at an average of 3 hours post mortem interval (PMI) to ensure minimal damage to the brain that is unrelated to disease. AD brain had an average Braak stage of 5.8. Braak staging indicates the severity of AD pathology (i.e., based on the number of senile plaques and neurofibrillary tangles) with the most severe stage being 6. MCI patients had an average PMI of 3 hours and an average Braak stage score of 3.8. (Refer to Tables 1 and 2 for individual details.) Age- matched controls for both AD and MCI had average PMI of less than 3 hours and an average Braak stage less than 2.
Figure 1.

Method Overview.
The use of Con-A lectin-affinity chromatography coupled to 2D electrophoresis allows the separation and the resolution of Con-A-associated proteins.
Alzheimer’s disease (AD) is the most common type of dementia, comprising 60–80% of all reported cases, and currently affects 5.2 million Americans. Mild cognitive impairment (MCI) arguably is the earliest for of AD. The present study identifies Con-A-fractionated brain proteins AD and MCI in two brain regions. The identity of proteins with altered levels in AD and MCI brain are consistent with biochemical and/or pathological alterations in both disorders.
Table 1.
AD subject profiles
| Age (years) | Sex | Brain Weight (g) | PMI (h) | Braak | |
|---|---|---|---|---|---|
| Control 1 | 77 | Male | 1310 | 3.50 | 1 |
| Control 2 | 83 | Male | 1275 | 2.00 | 3 |
| Control 3 | 87 | Male | 1150 | 2.00 | 2 |
| Control 4 | 72 | Male | 1150 | 3.75 | 1 |
| Control 5 | 85 | Female | 1020 | 2.50 | 3 |
| Control 6 | 81 | Male | 1410 | 2.00 | 2 |
| Average | 81 ± 5.5 | 1219 ± 139 | 2.60 ± 0.8 | 2.00 ± 0.9 | |
| AD 1 | 80 | Female | 1160 | 2.75 | 6 |
| AD 2 | 90 | Female | 1050 | 2.60 | 6 |
| AD 3 | 88 | Male | 1230 | 5.75 | 5 |
| AD 4 | 81 | Male | 1260 | 3.00 | 6 |
| AD 5 | 81 | Female | 835 | 3.00 | 6 |
| AD 6 | 92 | Female | 1090 | 2.00 | 6 |
| Average | 85 ± 5.3 | 1104 ± 154 | 3.2 ± 1.3 | 5.8 ± 0.4 |
PMI = post mortem interval
Braak = indicates severity of AD pathology (based on the number of senile plaques and neurofibrillary tangles in the brain.) The scale ranges from 1 to 6.
Table 2.
MCI subject profiles
| Age (years) | Sex | Brain Weight (g) | PMI (h) | Braak | |
|---|---|---|---|---|---|
| Control 1 | 93 | Female | 1080 | 2.75 | 2 |
| Control 2 | 74 | Male | 1140 | 4.00 | 1 |
| Control 3 | 86 | Female | 1150 | 1.75 | 1 |
| Control 4 | 76 | Female | 1315 | 2.00 | 1 |
| Control 5 | 79 | Male | 1240 | 1.75 | 2 |
| Control 6 | 86 | Female | 1300 | 3.75 | 1 |
| Average | 82 ± 7.2 | 1204 ± 95 | 2.67 ± 1.01 | 1.33 ± 0.52 | |
| MCI 1 | 99 | Female | 930 | 2.00 | 5 |
| MCI 2 | 88 | Female | 1080 | 2.25 | 5 |
| MCI 3 | 87 | Male | 1200 | 3.50 | 4 |
| MCI 4 | 87 | Male | 1170 | 2.25 | 3 |
| MCI 5 | 91 | Female | 1155 | 5.00 | 3 |
| MCI 6 | 82 | Female | 1075 | 3.00 | 3 |
| Average | 89 ± 5.7 | 1102 ± 98 | 3.0 ± 1.1 | 3.8 ± 1.0 |
PMI = post mortem interval
Braak = indicates severity of AD pathology (based on the number of senile plaques and neurofibrillary tangles in the brain.) The scale ranges from 1 to 6.
After Con-A chromatography, fractions were concentrated and subjected to isoelectric focusing and SDS-PAGE electrophoresis. The resultant gels were stained with SYPRO ruby stain and scanned. Gel images were analyzed using PD-Quest software and spots from AD or MCI brain that were identified as significantly different from their age-matched controls were excised, treated with trypsin, and subjected to mass spectrometric analysis. Interrogation of protein databases led to identification of Con-A-associated proteins from AD and MCI brain. The proteins that were identified are listed in Table 3 and 4 and are enumerated below. All comparisons of gels were made between each disease state and their respective age-matched control (i.e., AD compared to AD age-matched controls and MCI compared to MCI age-matched controls.)
Table 3.
Summary of MALDI/TOF mass spectrometry-based characterizations and hippocampal protein identifications of Con-A-fractionated proteins from MCI and AD
| NCBI GI no. | NCBI accession no. | Protein | # of Peptides matched/searched | Percent coverage of the matched peptides | pI, Mr (kDa) | Mowse | Fold Increase/Decrease | P-Value | Disorder |
|---|---|---|---|---|---|---|---|---|---|
| 4503377 | NM_001386.4 | Dihydropyrimdase-2 (spot A) | 7/23 | 26% | 4.84, 41,653 | 99 | 1.72 decrease | 0.04 | MCI |
| 4503377 | NM_001386.4 | Dihydropyrimdase-2 (spot B) | 12/38 | 27% | 4.84, 41,653 | 166 | 3.82 decrease | 0.02 | MCI |
| 386758 | M19645.1 | Glucose-regulated protein-78 | 14/40 | 25% | 5.03, 72,185 | 117 | 1.51 decrease | 0.05 | MCI |
| 4506013 | NM_002712.1 | Protein phosphatase related protein SDS-22 | 7/23 | 26% | 4.84, 41,653 | 68 | 2.26 increase | 0.05 | MCI |
| 4503979 | NM_002055.3 | Glial fibrillary acidic protein | 20/42 | 39% | 5.84, 49,533 | 187 | 17.1 increase | 0.05 | MCI |
| 4885281 | NM_005271.2 | Glutamate Dehydrogenase | 15/33 | 32% | 7.66, 61,701 | 142 | 3.2 increase | 0.03 | AD |
| 4885281 | NM_005271.2 | Glutamate Dehydrogenase | 14/26 | 29% | 7.66, 61,701 | 147 | 2.6 increase | 0.03 | AD |
| 4503979 | NM_002055.3 | Glial fibrillary acidic protein | 13/18 | 32% | 5.84, 49,533 | 158 | 16.0 increase | 0.05 | AD |
| 109016549 | XM_001113666.1 | Tropomyosin-3 | 11/29 | 34% | 4.71, 29,019 | 106 | 2.0 increase | 0.02 | AD |
| 4503971 | NM_001493.2 | Rab GDP-dissociation inhibitor XAP-4 | 9/26 | 25% | 5.00, 51,177 | 83 | 6.0 decrease | 0.03 | AD |
| 4503971 | NM_001493.2 | Rab GDP-dissociation inhibitor XAP-4 | 14/35 | 34% | 5.00, 51,177 | 136 | 1.6 decrease | 0.03 | AD |
| 153792590 | NM_001017963.2 | Heat shock protein 90 | 18/40 | 31% | 5.11, 68,614 | 128 | 1.7 decrease | 0.04 | AD |
Table 4.
Summary of MALDI/TOF mass spectrometry-based characterizations and IPL protein identifications of Con-A-fractionated proteins from MCI and AD
| NCBI GI no. | NCBI accession no. | Protein | # of Peptides matched/searched | Percent coverage of the matched peptides | pI, Mr (kDa) | Mowse | Fold Increase/Decrease | P-Value | Disorder |
|---|---|---|---|---|---|---|---|---|---|
| 4507111 | NM_003085.3 | β-synuclein | 7/39 | 51% | 4.41, 14,279 | 85 | 1.45 decrease | 0.04 | MCI |
| 30583767 | BT007464.1 | α-enolase | 6/14 | 21% | 5.0, 51,177 | 66 | 3.14 increase | 0.05 | AD |
| 32880095 | BT009876.1 | γ-enolase | 19/47 | 57% | 6.99, 47,350 | 69 | 13.4 increase | 0.05 | AD |
| 4503971 | NM_001493.2 | Rab GDP-dissociation inhibitor XAP-4 | 7/19 | 18% | 4.91, 47,450 | 175 | 1.25 increase | 0.05 | AD |
Con-A associated proteins with altered levels in AD and MCI hippocampus
Comparing the Con-A associated protein levels in AD and MCI hippocampus to their respective age-matched controls resulted in identification of 6 proteins with altered expression in AD and 4 proteins in MCI. Proteins with altered expression in AD are glutamate dehydrogenase (GDH), glial fibrillary acidic protein (GFAP), tropomyosin 3 (TPM3), Rab GDP-dissociation inhibitor XAP-4 (XAP4), and heat shock protein 90 (HSP90). XAP4 and HSP90 have decreased levels, while GDH, GFAP, and TPM3 levels are increased. GDH and XAP4 occur as multiple spots (see Figure 2). The 4 proteins found in MCI are dihydropyrimidase-2 (DRP2), glucose-regulated protein 78 (GRP-78), protein phosphatase related protein Sds-22 (Sds22), and GFAP. GRP78 and DRP2 have decreased levels, with DRP2 occurring in multiple spots (see Figure 3). Sds-22 and GFAP levels are increased. Multiple spots for the same protein in these gels suggest post-translational modification heterogeneity of these particular proteins, affecting protein isoelectric point (pI). Representative 2D gels with protein identifications for AD and MCI hippocampus are shown in Figure 2 and Figure 3, respectively. Identification, statistical, and protein level data are summarized in Table 3.
Figure 2.

Sypro-stained 4–16% polyacrylamide representative 2D gels of Con-A-eluted proteins from AD hippocampus and age-matched control with statistically significant protein spot identities. For protein spot p-values please refer to Table 3.
Figure 3.

Sypro-stained 4–16% polyacrylamide representative 2D gels of Con-A-eluted proteins from MCI hippocampus and age-matched control with statistically significant protein spot identities. For protein spot p-values please refer to Table 3.
Con-A associated proteins with altered levels in AD and MCI inferior parietal lobule
Comparing the protein levels in AD and MCI inferior parietal lobule (IPL) to their respective age-matched controls resulted in 3 proteins found in AD and a single protein to have altered levels in MCI. In AD IPL the proteins were α-enolase, γ-enolase, and XAP-4. All of these proteins have increased levels in AD. The protein found in MCI was β-synuclein with a decreased level. Representative 2D gels with protein identifications for AD and MCI IPL are shown in Figures 4 and Figure 5, respectively. Identification, statistical, and protein level data are summarized in Table 4.
Figure 4.

Sypro-stained 4–16% polyacrylamide representative 2D gels of Con-A-eluted proteins from AD IPL and age-matched control with statistically significant protein spot identities. For protein spot p-values please refer to Table 4.
Figure 5.
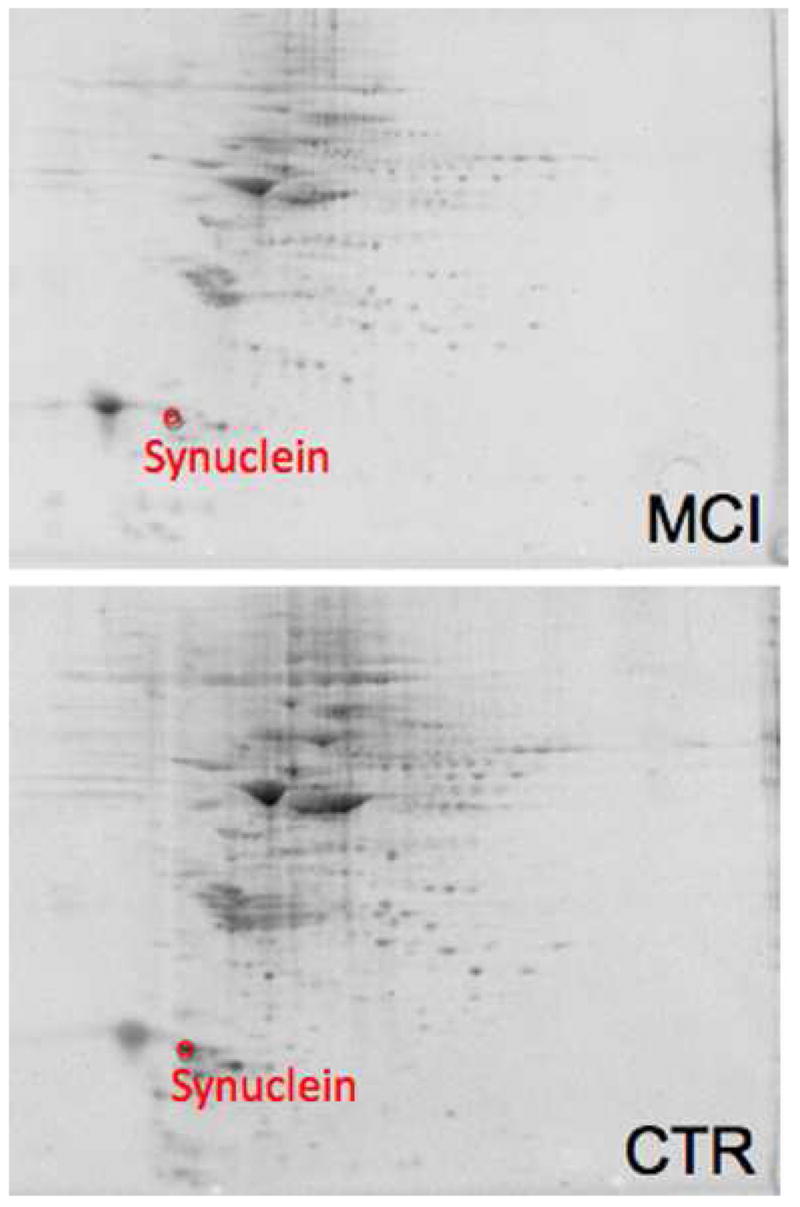
Sypro-stained 4–16% polyacrylamide representative 2D gels of Con-A-eluted proteins from MCI IPL and age-matched control with statistically significant protein spot identities. For protein spot p-values please refer to Table 4.
Column affinity experiments
To address non-glycoprotein, hydrophobic binding of brain proteins by Con-A, α-enolase, a protein identified in this study, underwent oxidation with the Fe2+/H2O2 Fenton reaction and PNGase F treatments in vitro. Protein oxidation makes hydrophobic regions more surface accessible43 and PNGase F removes oligosaccharides from N-linked glycoproteins. The resultant enolase was then loaded onto a 1-D SDS-PAGE gel as described. The protein in the gel was transferred onto a Western blot and probed with Con-A/biotin conjugate followed by streptavidin/horse radish peroxidase treatment (Figure 6). The results of this experiment show that Con-A binds to oxidized and un-oxidized enolase. However, consistent with the notion that increased hydrophobic residue exposure exists in oxidized proteins,43 apparent increased Con-A binding occurs with oxidized enolase. Both the oxidized and un-oxidized forms of enolase treated with PNGase F are also bound by Con-A. This experiment shows that enolase is a substrate for Con-A binding independent of oxidation state or the presence of any potential N-linked glycans, but Con-A has an apparently higher affinity for oxidized enolase.
Figure 6.

Enolase 1-D Western blot probed with Con-A/biotin conjugate. Equal amonts of un-oxidized enolase, oxidized enolase, PNGase F-treated un-oxidized enolase, and PNGase F-treated oxidized enolase were probed with Con-A/biotin.
To further investigate whether the brain protein fractions isolated from the Con-A affinity columns bind this lectin due to carbohydrate or hydrophobic interactions, brain samples were prepared in buffers made with the Con-A competitive haptens, α-methyl D-glucopyranoside and α-methyl D-mannopyranoside, and the hydrophobic interaction disrupting non-ionic detergent NP-40. Samples were then processed through Con-A columns as described and loaded onto a 1-D SDS-PAGE gel. The proteins were then transferred and the subsequent Western blot was probed with Con-A/biotin conjugate followed by streptavidin/horse radish peroxidase. When hydrophobic interactions are disrupted with NP-40 treatment Con-A column resolution is diminished. In contrast, protein bands are still visualized when samples are treated with methylated sugars acting as competitive inhibitors. This result suggests that Con-A still binds some proteins without the presence of glycans. This is consistent with the existence of a hydrophobic binding region of Con-A that binds brain proteins independently of the presence of carbohydrate residues.
Discussion
The proteins identified in AD and MCI hippocampus and IPL with altered levels compared to control brain (Tables 3 and 4) have not been previously reported as N-linked glycoproteins. The proteins α- and γ-enolase, DRP-2, Sds22, GDH, and HSP 90 have one or more Asn-X-Ser/Thr sequences that potentially can be N-glycosylated, but lack a hydrophobic amino acid signal sequence at the N-terminus for canonical ER localization. Alternatively, proteins devoid of glycans may bind Con-A as a result of interactions with this lectin’s hydrophobic binding domain. Experiments with α-enolase suggest that binding of some proteins found in this study is due to some mechanism other than carbohydrate binding. However, binding due to hydrophobic interactions has significance. An increased hydrophobicity of proteins would drive most of these normally cytosolic or lumen-associated proteins to the various membranes of the cell or to aggregate with other hydrophobic proteins in the cell’s aqueous environments. The bulk of the remainder of the discussion describes the normal role of the identified proteins and possible implications of their impairment.
Glutamate dehydrogenase (GDH) is an enzyme located in the mitochondrial matrix that can act in either a metabolic or a catabolic direction. In the biosynthetic direction, GDH catalyzes the reductive amination of α-ketoglutarate with NADPH to yield glutamate. Alternatively, GDH can catalyze the formation of α-ketoglutarate from glutamate with NAD+ and ammonium ion. The catabolic activity of GDH is particularly important for the elimination of excitotoxic glutamate. Excess glutamate can stimulate NMDA receptors leading to an increase in Ca2+ influx and altered calcium homeostasis, which would lead to alteration in long-term potentiation (LTP) and consequently, learning and memory deficits as seen in AD.44 In our study we found a significant increase of Con-A-associated GDH in AD hippocampus compared to control. This increase might be related with an increased presence of astrocytes, which can use glutamate as a substrate for energy metabolism,45 particularly when the extracellular glutamate levels increase.46, 47 In addition, the GDH pathway is thought be mostly active when glucose levels are low,48 and it is known from both positron emisson tomography (PET) studies and proteomics analysis that glucose utilization is markedly depressed in AD brain. Our results confirm numerous studies that report an increase in the total amount of GDH in AD.49–52 These studies support the possible compensatory role of GDH in the pathogenesis of AD, potentially reflecting decreased glucose energy metabolism.53
Glial fibrillary acidic protein (GFAP) is the building block of glial intermediate filaments and is the major cytoskeletal structure in mature reactive astrocytes. Increased expression of GFAP is a characteristic feature of reactive astrocytes during chronic inflammation that could be associated with the presence of tangles, neuritic plaques and Aβ pathology, and represent the maturation of the astrocytes.54 The levels of GFAP dynamically respond to aging as well as to neurodegenerative lesions,55,56 and many studies have shown that the amount of GFAP generally increases in neurodegenerative diseases such as AD.57 We report in this work an increased expression of Con-A- associated GFAP in MCI and AD hippocampus, supporting the hypothesis that neuro-inflammation starts early and is maintained throughout AD progression.
Tropomyosin 3 (TPM3) and tropomyosin proteins in general are implicated in the differentiation and maturation of neurons as well as polarization of developing neurons, growth of dendrites and axons, and growth cone size and formation. Neuronal cells express a limited spectrum of TPMs, some of which are specific to this cell type and show both developmental and spatial patterns of expression.58 TPM3 is a brain specific TPM found in all regions of the brain that appear late in development and increase in concentration as the brain matures.59, 60 TPM3 is concentrated in axons and synaptic glomeruli60 and is thought to play a role in synaptic function as well as neuronal maturation. Our experimental results demonstrate an increased level of Con-A-associated TPM3 in AD hippocampus. These data, coupled with previous studies that report an up-regulation and oxidation of TPM3 in the olfactory system of aged mice61 and in gerbil synaptosomes treated with the lipid peroxidation product acrolein,62 are consistent with the notion of an impairment of neuron maturation that may lead to dysfunction in axonal transport and dendrite formation occurs in AD. Consistent with this notion, axonal transport is severely disrupted in AD.63
Rab-GDP dissociation inhibitor XAP-4 (XAP4) is a member of the GDP dissociation inhibitor (GDI) family that controls the recycling of the Rab GTPases involved in membrane trafficking.64, 65 GDIs retrieve the GDP-bound form of Rab from the membrane to form a heterodimeric complex, which maintains a cytosolic reservoir for the reuse of inactive Rab during multiple rounds of vesicle budding and fusion.66, 67 In the brain, Rab3 proteins play an important role in neurotransmitter release and are substrates for XAP4,68–70 demonstrating the importance of this particular GDI in brain plasticity. A decrease of Con-A-associated GDI levels in AD hippocampus suggests a possible impairment in synaptic vesicle exocytosis and recycling during AD progression, potentially contributing to the known impairment in neurotransmission in AD.71
The 90-kDa heat shock protein (HSP90) is a highly conserved molecular chaperone widely expressed in cells of various species that plays a key role in protein refolding under stress conditions and in signal transduction under non-stress conditions.72,73 HSP90 can prevent the aggregation of unfolded proteins and cooperate with the HSP70/HSP40 chaperone system in the ATP-dependent refolding of unfolded model proteins.74,75 In eukaryotes, cytoplasmic HSP90s act as specific chaperones for a wide range of client proteins such as the cytoplasmic receptor steroid hormone.76 In mammalian cells, HSP90 association with Raf, MEK, and Src family proteins is required for signaling.72 HSP90 has also been shown to facilitate G-protein coupled pathways,77 but whether HSP90 directly regulates G protein-coupled receptors is unknown. In AD and other neurodegenerative diseases HSP90 has been shown to modulate tau association with microtubules and help determine microtubule integrity.78,79 In addition, HSP 90 in AD may act to attenuate the cytotoxicity of Aβ by preventing processes such as oxidation, oligomerization, misfolding, and aggregation therefore inhibiting the formation of Aβ toxic variants.80,81 Further, HSP 90 could act via the NF-KB and p38 MAPK pathways to induce microglial activation and facilitate the phagocytosis and clearance of Aβ.82, 83 Our data show a decrease in the levels of Con-A-associated HSP 90. This condition in the AD hippocampus conceivably could contribute to both the well-recognized deposition of Aβ and the formation of NFTs.
Dihydropyrimidase-2 (DRP-2), a protein member of the DRP gene family, is a path-finding and guidance protein for axonal outgrowth during the formation of neuronal connections and for the maintenance of neuronal communication. DRP-2 exploits its function through the transmission and modulation of extracellular signals like collapsin, a protein responsible for the elongation and guidance of dendrites. DRP-2 is normally expressed during development, however it was found to be present in adult brain, indicating its involvement in repairing and maintaining the plasticity of neuronal connections in aged brains.23, 84 Dysfunction of the DRP-2 repairing activity in brain suggests that depletion of DRP-2 may result in neuronal abnormalities, thus accelerating the neuritic degeneration in many neurodegenerative disorders.84 Proteomic data from our laboratory showed that DRP-2 is significantly oxidatively modified in AD brain20 and has been shown to have decreased expression in AD brain.85 The present study, showing a decreased level of Con-A-associated DRP-2 confirms a decrement in hippocampus of MCI subjects and is consistent with the known shortened dendritic length in AD.86 We speculate that this loss of ability to form neuronal connections could be important in a memory disorder and may be a possible mechanism involved in the onset of AD.
Glucose-regulated protein 78 (GRP78), also known as BiP or HSPA5, is a member of the heat shock protein 70 (Hsp70) family of proteins, which function as molecular chaperones by binding transiently to proteins traversing through the ER and facilitating their folding, assembly, and transport.87 GRP78 is the main constituent involved in the unfolded protein response (UPR). GRP78 recognizes unfolded polypeptides, inhibits intra- and intermolecular aggregation, and promotes oligomerization and proper folding. During ER stress response, GRP78 binds misfolded proteins and translocates them through ER membranes for their proteosomal degradation. GRP78 interacts with APP and reduces the levels of APP, Aβ40, and Aβ42 secretion.87 This transient interaction may impair access of APP to β-/γ-secretases within the ER/Golgi or may influence APP metabolism by facilitating its correct folding.87 GRP78 has also been implicated in the cellular response to altered intracellular Ca2+ homeostasis.88 Some studies verified that the protein levels of BiP/GRP78 decreased in the temporal cerebral cortex brain region of sporadic AD as well as familial AD depending on the mutations in the PS1 gene that affect the regulation of expression of GRP78 mRNA.89 In contrast, other investigators have found that overall protein levels of GRP78 are increased in AD temporal cortex and hippocampus compared to non-demented control cases confirming a marked ER stress during AD progression.90 The present study demonstrates a decreased level of Con-A associated GRP78 in MCI hippocampus. Because there are conflicting reports in the literature about GRP78 levels in AD brain, further studies are needed to elucidate the role GRP-78 in AD pathogenesis.
The cellular role of protein phosphatase related protein Sds-22 (Sds22) is still under debate. Sds22 has been shown to be essential for chromosome disjunction during the metaphase/anaphase transition.91 In addition to its role in the cell cycle, Sds22 may facilitate protein phosphatase 1 (PP-1) mediated dephosphorylation, although it does not itself display protein phosphatase activity. Two mechanisms has been proposed for the interaction of Sds22 with PP-1: direct interaction with the PP-1 catalytic site increases PP-1 nuclear activity, or a chaperone-like role for the nuclear PP-1 catalytic site preventing aggregation of the free PP1c subunit and/or helping to retain it in the nucleus.92 Human Sds22 homologs are expressed in a variety of tissues and are present in both the cytoplasm and the nucleus.91 In the current study, we demonstrate a decrease in the levels of Con-A-associated Sds22 in MCI hippocampus, however its involvement in MCI progression to AD, if any, needs further investigation.
The proteins α-enolase and γ-enolase catalyze the reversible glycolytic reaction of 2-phosphoglycerate to phosphoenolpyruvate. The α-enolase isoform is ubiquitously expressed in tissue, while γ-enolase is brain specific, and in the present investigation both show increased Con-A-associated levels in AD IPL. It is well documented that the AD brain is metabolically challenged, and increased production in enolase isoforms may be a response to combat the compromise in energy production.93, 94 The increased levels of Con-A-associated enolase, coupled with previous studies that show enolase as oxidized and glutathionylated in AD brain,20, 95 suggests that enolase might be newly synthesized, oxidatively damaged and recruited back to the ER for repair and subsequently kept in the ER due to impaired ER-Golgi trafficking. This would render enolase ineffective in overcoming metabolic deficits observed in AD. Enolase has multiple functions in addition to glycolysis, and investigations of this oxidatively modified protein for other potential roles in AD brain is ongoing in our laboratory.
α-Synuclein is well known as the primary component of pathological Lewy bodies that occur in Parkinson’s disease (PD) and in diffuse Lewy body disease. The primary function of synuclein is not known, but it is typically a component of pre-synaptic neurons.96 β-synuclein is homologous to α-synuclein and binds to Aβ.97 The role of synuclein in the pathogenesis of PD has been investigated and these insights may be relevant to AD. Cooper et al. showed that ER-Golgi trafficking is blocked by synuclein and rescued by Rab1 protein.96 β-synuclein is largely localized to synaptic membranes, and Aβ (1–42) leads to oxidation of this protein in vivo.98 The present study reports that Con-A-associated β-synuclein level is decreased in MCI IPL, which conceivably could contribute to altered vesicular trafficking and synaptic function in AD. The current study also reports that XAP-4 has an increased expression in the IPL and a decreased expression in AD hippocampus. This seemly confounded result may be due to a loss in communication between the two brain regions. We speculate that less β-synuclein and more XAP-4 in the IPL may alter synaptic trafficking in an attempt to re-establish communication between the two brain regions. The decreased expression in XAP-4 in hippocampus suggests decreased neurotransmission and supports the idea that communication is impaired between the hippocampus and IPL.
All the aforementioned proteins were isolated using Con-A lectin. Con-A recognizes α-linked mannose and terminal glucose carbohydrate moieties that are characteristic of the initial sugars added to N-linked glycoproteins and hybrid and complex bi-antennary glycoproteins. As noted above, Con-A also has an independent hydrophobic binding domain.42 The results of experiments using α-enolase probed with Con-A/biotin conjugate described here suggest that the brain proteins identified in the current study to have altered levels in AD and MCI could be a result of hydrophobic binding to Con-A. This observation does not rule out the possibility that the proteins were not glycosylated at some point in their history or are not currently glycoproteins. Controversial studies have reported that glycosylation occurs in the nucleus and cytoplasm and is not limited to the canonical pathway through the cell’s endomembrane system.99 However, non-canonical pathways remain to be generally accepted by the glycobiology community because hypotheses to explain the existence of nucleoplasmic and cytoplasmic glycoproteins, either through direct glycosylation in these compartments or glycoprotein transport into these environments, have yet to be elucidated.99 Therefore, the proteins that were found in this study with potential N-linked glycosylation sites may be glycosylated in a non-canonical pathway. An alternative explanation of these results is the hydrophobicity of proteins from AD and MCI brain compared to age-matched controls is altered. In addition, a number of proteins in this study, such as HSP90 and GRP78, are chaperone proteins. Therefore, the possibility exists that proteins that formed conjugates with these chaperones may have been included in the protein fraction eluted from the Con-A column and did not necessarily bind to hydrophobic or carbohydrate binding domains of Con-A. The reverse scenario of proteins bound to Con-A-associated proteins being identified as having altered levels in AD and MCI brain is another possibility, i.e., chaperone proteins conceivably are binding partners to Con-A fractionated proteins.
As noted above, one way to increase hydrophobic region availability of brain proteins is by oxidative modification of proteins43, a phenomenon known in AD and MCI.18, 100 The exact nature of the binding of these proteins to Con-A remains to be determined, but the identity of proteins of altered levels in AD and MCI brain are consistent with biochemical and/or pathological alterations in both disorders.
Conclusions
The present study is the first to report Con-A-fractionated proteins in the brains from subjects with AD and MCI and identified a number of proteins with varying levels compared to the respective control brains. These proteins encompass a wide assortment of functions including cellular trafficking, metabolism, signal transduction, and neurotransmission, all known to be disrupted in AD. In the discussion of these findings we posit about the implications of these results in AD pathogenesis as well as aberrant cellular processes in the disease. Future studies coupling Con-A and other lectins with higher substrate specificity may provide more information on the pathogenesis and potential treatment of AD.
Materials and Methods
Sample preparation
Hippocampus and inferior parietal lobule samples (n=6 each) from well- characterized subjects with AD and MCI and respective age-matched controls were obtained from the University of Kentucky Alzheimer’s Disease Clinical Center Neuropathology Core. Samples were homogenized on ice with sucrose isolation buffer (0.32 M sucrose with protease inhibitors, 4 ug/mL leupeptin, 4 ug/mL pepstatin A, 0.125 M Tris pH 8.0, 5 ug/mL aprotinin, 0.2 mM phenylmethylsulfonylfluoride (PMSF), and 0.6 mM MgCl2). Table 1 and Table 2 represent demographic data of the AD and MCI subjects, respectively. Note the very short post mortem interval of these samples.
Protein Estimation and Concanavalin A lectin affinity columns
Protein concentrations were determined using the BCA method and aliquots (1500 μg) were diluted with 5× Binding/Wash buffer in a 4:1 volumetric ratio and loaded into Con-A affinity columns. Con-A lectin columns, binding/wash, and elution buffers were prepared as described by the manufacturer’s instructions (Pierce Biotechnology, Rockford IL). The samples were loaded onto columns and incubated (10 min) with end-over-end mixing using an elliptical rotor. The samples were spun at 1000 × g (1 min) and the effluent was reloaded onto the columns and the process was repeated. The effluent was discarded after this step. Binding/Wash buffer (400 μl) was added to the Con-A lectin columns, and the columns then were centrifuged at 1000 × g (1 min) and the process was repeated. The columns were loaded with elution buffer (200 μl) and incubated (10 min) with end-over-end mixing using an elliptical rotor. The samples were spun at 1000 × g for 1 min and the effluent was retained. This process was repeated and the elution buffer containing the Con-A-associated proteins were combined (400 μl) and concentrated (to ~25 μl) using 30 kDa cut-off filters (Millipore, Billerica MA). Protein concentration was re-determined using the BCA method and the remaining aliquots were suspended in 200 μl of rehydration buffer composed of a 1:1 ratio (v/v) of the Zwittergent solubilization buffer (7M urea, 2M thiourea, 2% Chaps, 65 mM DTT, 1% Zwittergent 0.8% 3–10 ampholytes and bromophenol blue) and ASB-14 solubilization buffer (7M urea, 2M thiourea 5 mM TCEP, 1% (w/v) ASB-14, 1% (v/v) Triton X-100, 0.5% Chaps, 0.5% 3–10 ampholytes) for 2 h.
First dimension electrophoresis
For the first-dimension electrophoresis, approximately 50 μL of sample solution was applied to 110-mm pH 3–10 ReadyStrip™ IPG strips (Bio-Rad, Hercules CA). The strips were then actively rehydrated in the protean isoelectric focusing (IEF) cell (Bio-Rad) at 50 V for 18 h. The isoelectric focusing was performed in increasing voltages as follows; 300 V for 1 h, then linear gradient to 8000 V for 5 h and finally 20 000 V/h. Strips were then stored at −80 °C until the second dimension electrophoresis was to be performed.
Second dimension electrophoresis
For the second dimension, the IPG® Strips, pH 3–10, were thawed and equilibrated for 10 min in 50 mM Tris–HCl (pH 6.8) containing 6 M urea, 1% (w/v) sodium dodecyl sulfate (SDS), 30% (v/v) glycerol, and 0.5% dithiothreitol, and then re-equilibrated for 15 min in the same buffer containing 4.5% iodacetamide instead of dithiothreitol. Linear gradient precast criterion Tris–HCl gels (8–16%) (Bio-Rad) were used to perform second dimension electrophoresis. Precision Protein™ Standards (Bio-Rad, CA) were run along with the samples at 200 V for 65 min.
SYPRO ruby staining
After the second dimension electrophoresis, the gels were incubated in fixing solution (7% acetic acid, 10% methanol) for 20 min and stained overnight at room temperature with 50ml SYPRO Ruby gel stain (Bio-Rad). The SYPRO ruby gel stain was then removed and gels stored in deionized water.
Con-A affinity experiments
Enolase (Sigma) was oxidized by hydroxyl free radicals as previously described.101 Aliquots of un-oxidized and oxidized enolase were treated with PNGase F (Sigma) as described by manufacturer’s directions. Samples were loaded onto linear gradient 18-well precast criterion Tris–HCl gels (8–16%) (Bio-Rad), which were used to perform 1-dimensional electrophoresis. Precision Protein™ Standards (Bio-Rad, CA) were run along with the sample at 120 V for 120 min. Gels were transferred onto nitrocellulose membranes (Bio-Rad) at 20 V for 2 h. Blots were blocked with blocking reagent (Licor) for 1 h then probed with ConA/Biotin conjugate (0.001 %) for an additional 1 h. Blots were washed (3x) with PBST. A solution of streptavidin/horse radish peroxidase conjugate (0.2 μg/mL in PBST) was added and incubated for 1 h. Blots were visualized using a STORM phosphoimager (Ex. 470 nm, Em. 618 nm, Molecular Dynamics, Sunnyvale, CA, USA).
MCI age-matched control hippocampus samples were homogenized on ice with sucrose isolation buffer, (0.32 M sucrose with protease inhibitors, 4 ug/mL leupeptin, 4 ug/mL pepstatin A, 0.125 M Tris pH 8.0, 5 ug/mL aprotinin, 0.2 mM phenylmethylsulfonylfluoride (PMSF), and 0.6 mM MgCl2), sucrose isolation buffer plus NP-40, sucrose isolation buffer plus 0.1 M α-methyl D-glucopyranoside and 0.5 M α-methyl D-mannopyranoside, and sucrose isolation buffer plus 0.1 M α-methyl D-glucopyranoside, 0.5 M α-methyl D-mannopyranoside, and NP40. These samples underwent Con-A column chromatography as described above. Protein fractions were loaded onto linear gradient 18-well precast criterion Tris–HCl gels (8–16%) (Bio-Rad) were used to perform 1-dimensional electrophoresis. Precision Protein™ Standards (Bio-Rad, CA) were run along with the sample at 120 V for 120 min. Gels were transferred onto nitrocellulose membranes (Bio-Rad) at 20 V for 2 h. Blots were blocked with blocking reagent (Licor) for 1 h then probed with ConA/Biotin conjugate (0.001 %) for an additional h. Blots were washed (3x) with PBST. A solution of streptavidin/horse radish peroxidase conjugate (0.2 μg/mL in PBST) was added and incubated for 1 h. Blots were visualized using a STORM phosphoimager as described above.
Image analysis
SYPRO ruby-stained gel images were obtained using a STORM phosphoimager as indicated above and saved in TIFF format. Gel imaging was software-aided using PD-Quest (Bio-Rad) imaging software. Briefly, a master gel was selected followed by normalization of all gels (Control and AD or Control and MCI) according to the total spot density. Gel to gel analysis was then initiated in two parts. First, manual matching of common spots that could be visualized among the differential 2D gels was performed. After obtaining a significant number of spots the automated matching of all spots was then initiated. Automated matching is based on user-defined parameters for spot detection. These parameters are based on the faintest spot, the largest spot, and the largest spot cluster that occur in the master gel and are defined by the user. Based on these parameters the software defines spot centers for the gel. If the software “misses” spots that are obvious to the naked eye that manual and automated matching fails to identify, the user can manually assign a spot center. This process generates a large pool of data, approximately 350 spots.
Only proteins showing computer-determined significant differential levels between the two groups being analyzed were considered for identification. To determine significant differential levels of proteins, analysis sets were created using the analysis set manger software incorporated into the PD-Quest software. The number of pixels that occur in a protein spot were computed by the software corresponding to an increase/decrease in protein level. A quantitative analysis set was created that recognized matched spots that had increases or decreases of 1.5-fold. A statistical analysis set was created that compared matched spots using a Student’s t-test at 95% confidence. Spots with p-values of p < 0.05 were considered significant. A Boolean analysis set was created that identified overlapping spots from the aforementioned quantitative and statistical sets. These spots were selected for subsequent mass spectrometric analysis.
In-gel trypsin digestion
Protein spots statistically different than controls were digested in-gel by trypsin using protocols previously described and modified by Thongboonkerd et al.102 Spots were taken from indivdual gels and not pooled for mass spectrometric analysis. The amount of protein from one gel-spot was sufficient for identification. Briefly, spots of interest were excised using a clean blade and placed in Eppendorf tubes, which were then washed with 0.1 M ammonium bicarbonate (NH4HCO3) at room temperature for 15 min. Acetonitrile was then added to the gel pieces and incubated at room temperature for 15 min. This solvent mixture was then removed and gel pieces dried. The protein spots were then incubated with 20 μL of 20 mM DTT in 0.1 M NH4HCO3 at 56 °C for 45 min. The DTT solution was removed and replaced with 20 μL of 55 mM iodoacetamide in 0.1 M NH4HCO3. The solution was then incubated at room temperature for 30 min. The iodoacetamide was removed and replaced with 0.2 mL of 50 mM NH4HCO3 and incubated at room temperature for 15 min. Acetonitrile (200 μL) was added. After 15 min incubation, the solvent was removed, and the gel spots were dried in a flow hood for 30 min. The gel pieces were rehydrated with 20 ng/μL-modified trypsin (Promega, Madison, WI) in 50 mM NH4HCO3 with the minimal volume enough to cover the gel pieces. The gel pieces were incubated overnight at 37 °C in a shaking incubator.
Mass spectrometry
All mass spectrometry results reported in this study were obtained in collaboration with the Department of Pharmacology in the University of Louisville Mass Spectrometry Facility. A Bruker Autoflex matrix assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometer in the reflectron mode was used to generate peptide mass fingerprints (Bruker Daltonic, Billerica, MA). Peptides resulting from in-gel digestion with trypsin were analyzed on a 384 position, 600 μm AnchorChip™ Target (Bruker Daltonics, Bremen, Germany) and prepared according to AnchorChip recommendations (AnchorChip Technology, Rev. 2, Bruker Daltonics, Bremen, Germany). Briefly, 1 μL of digestate was mixed with 1 μL of alpha-cyano-4-hydroxycinnamic acid (0.3 mg/mL in ethanol: acetone, 2:1 ratio) directly on the target and allowed to dry at room temperature. The sample spot was washed with 1 μL of a 1% TFA solution for approximately 60 seconds. The TFA droplet was gently blown off the sample spot with compressed air. The resulting diffuse sample spot was recrystallized (refocused) using 1 μL of a solution of ethanol: acetone: 0. 1 % TFA (6:3:1 ratio). Reported spectra are a summation of 100 laser shots. External calibration of the mass axis was used for acquisition and internal calibration using either trypsin autolysis ions or matrix clusters and was applied post acquisition for accurate mass determination. The raw data were exported as a text file and subsequently generated peak lists were interrogated using the online Mascot database.
Analysis of peptide sequences
Peptide mass fingerprinting was used to identify proteins from tryptic peptide fragments by utilizing the MASCOT search engine (MASCOT Server Version 2.2 http://www.matrixscience.com ) based on the entire NCBI and SwissProt protein databases (Release 31 containing 5,859,648 proteins from 5,513 species http://www.ncbi.nlm.nih.gov/RefSeq/). Database searches were conducted allowing for up to one missed trypsin cleavage and using the assumption that the peptides were monoisotopic, oxidized at methionine residues, and carbamidomethylated at cysteine residues. Mass tolerance of 150 ppm, 0.1 Da peptide tolerance and 0.2 Da fragmentation tolerance was the window of error allowed for matching the peptide mass values 103. Probability-based MOWSE scores were estimated by comparison of search results against estimated random match population and were reported as -10*log10 (p), where p is the probability that the identification of the protein is a random event. MOWSE scores greater than 63 were considered to be significant (p < 0.05). All protein identifications were in the expected size and isoelectric point (pI) range based on the position in the gel. A Supplemental Material file to this paper contains MS peak lists, sequences of matched peaks, and complete sequences for all identified proteins.
Statistical analysis
Statistical analysis of protein levels matched with spots on 2D-gels from AD and MCI hippocampus and inferior parietal lobule compared to age-matched controls were carried out using Student’s t-tests. A value of p < 0.05 was considered statistically significant. (Please see the Image Analysis section above for generation of analysis sets.) Only proteins that were considered significantly different by Student’s t-test were subjected to in-gel trypsin digestion and subsequent proteomic analysis. According to Maurer and Peters, generalized statistical tests that apply to proteomics studies are unavailable.104 Due to small numbers of proteins that are typically identified in proteomics studies compared to microarray data, statistical methods applied to thousands of hits in microarray studies are not applicable for proteomics studies with relatively small numbers of identified proteins.94,95 Hence, Student’s t-test was used for analysis.104,105 A summary of the methods employed in this study is shown in Figure 1.
Supplementary Material
MS and protein database information associated with each Con-A fractionated protein identified was provided by the University of Louisville Mass Spectrometry Facility for this study. This information is available free of charge via the Internet at http://pubs.acs.org.
Figure 7.

Con-A/biotin binding of Con-A column fractions in the presence or absence of competitive haptens and the non-ionic detergent NP-40. C = Control H = Hapten (0.1 M α-methyl glucopyranoside, 0.5 M α-methyl mannopyranoside) NP40 = Non-ionic detergent.
Acknowledgments
This research was supported in part by grants from NIH to DAB [AG-05119; AG-10836; AG-029839].
References
- 1.2008 Alzheimer’s disease facts and figures. Alzheimers Dement. 2008;4(2):110–33. doi: 10.1016/j.jalz.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 2.Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120(3):885–90. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 3.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985;82(12):4245–9. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Selkoe DJ. Cell biology of protein misfolding: the examples of Alzheimer’s and Parkinson’s diseases. Nat Cell Biol. 2004;6(11):1054–61. doi: 10.1038/ncb1104-1054. [DOI] [PubMed] [Google Scholar]
- 5.Selkoe DJ. Cell biology of the amyloid beta-protein precursor and the mechanism of Alzheimer’s disease. Annu Rev Cell Biol. 1994;10:373–403. doi: 10.1146/annurev.cb.10.110194.002105. [DOI] [PubMed] [Google Scholar]
- 6.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83(13):4913–7. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kosik KS, Joachim CL, Selkoe DJ. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U S A. 1986;83(11):4044–8. doi: 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee VM, Balin BJ, Otvos L, Jr, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science. 1991;251(4994):675–8. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- 9.Nukina N, Ihara Y. Proteolytic fragments of Alzheimer’s paired helical filaments. J Biochem. 1985;98(6):1715–8. doi: 10.1093/oxfordjournals.jbchem.a135443. [DOI] [PubMed] [Google Scholar]
- 10.Selkoe DJ, Ihara Y, Salazar FJ. Alzheimer’s disease: insolubility of partially purified paired helical filaments in sodium dodecyl sulfate and urea. Science. 1982;215(4537):1243–5. doi: 10.1126/science.6120571. [DOI] [PubMed] [Google Scholar]
- 11.Wischik CM, Novak M, Thogersen HC, Edwards PC, Runswick MJ, Jakes R, Walker JE, Milstein C, Roth M, Klug A. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A. 1988;85(12):4506–10. doi: 10.1073/pnas.85.12.4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies CA, Mann DM, Sumpter PQ, Yates PO. A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer’s disease. J Neurol Sci. 1987;78(2):151–64. doi: 10.1016/0022-510x(87)90057-8. [DOI] [PubMed] [Google Scholar]
- 13.Petersen RC, Negash S. Mild cognitive impairment: an overview. CNS Spectr. 2008;13(1):45–53. doi: 10.1017/s1092852900016151. [DOI] [PubMed] [Google Scholar]
- 14.Butterfield DA, Sultana R. Redox proteomics identification of oxidatively modified brain proteins in Alzheimer’s disease and mild cognitive impairment: insights into the progression of this dementing disorder. J Alzheimers Dis. 2007;12(1):61–72. doi: 10.3233/jad-2007-12107. [DOI] [PubMed] [Google Scholar]
- 15.Meguro K. Clinical features of mild cognitive impairment and dementia in a community: an update of the Osaki-Tajiri Project. Tohoku J Exp Med. 2008;215(2):125–31. doi: 10.1620/tjem.215.125. [DOI] [PubMed] [Google Scholar]
- 16.Selkoe DJ. Aging, amyloid, and Alzheimer’s disease: a perspective in honor of Carl Cotman. Neurochem Res. 2003;28(11):1705–13. doi: 10.1023/a:1026065122854. [DOI] [PubMed] [Google Scholar]
- 17.Wu CK. Mild cognitive impairment, healthy aging and Alzheimer’s disease. Med Health R I. 2008;91(5):132–3. [PubMed] [Google Scholar]
- 18.Butterfield DA, Reed T, Newman SF, Sultana R. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic Biol Med. 2007;43(5):658–77. doi: 10.1016/j.freeradbiomed.2007.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med. 2002;33(4):562–71. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- 20.Castegna A, Aksenov M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part II: dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J Neurochem. 2002;82(6):1524–32. doi: 10.1046/j.1471-4159.2002.01103.x. [DOI] [PubMed] [Google Scholar]
- 21.Castegna A, Thongboonkerd V, Klein JB, Lynn B, Markesbery WR, Butterfield DA. Proteomic identification of nitrated proteins in Alzheimer’s disease brain. J Neurochem. 2003;85(6):1394–401. doi: 10.1046/j.1471-4159.2003.01786.x. [DOI] [PubMed] [Google Scholar]
- 22.Reed T, Perluigi M, Sultana R, Pierce WM, Klein JB, Turner DM, Coccia R, Markesbery WR, Butterfield DA. Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiol Dis. 2008;30(1):107–20. doi: 10.1016/j.nbd.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 23.Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Merchant M, Markesbery WR, Butterfield DA. Redox proteomics identification of oxidized proteins in Alzheimer’s disease hippocampus and cerebellum: an approach to understand pathological and biochemical alterations in AD. Neurobiol Aging. 2006;27(11):1564–76. doi: 10.1016/j.neurobiolaging.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 24.Sultana R, Reed T, Perluigi M, Coccia R, Pierce WM, Butterfield DA. Proteomic identification of nitrated brain proteins in amnestic mild cognitive impairment: a regional study. J Cell Mol Med. 2007;11(4):839–51. doi: 10.1111/j.1582-4934.2007.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki T, Araki Y, Yamamoto T, Nakaya T. Trafficking of Alzheimer’s disease-related membrane proteins and its participation in disease pathogenesis. J Biochem. 2006;139(6):949–55. doi: 10.1093/jb/mvj121. [DOI] [PubMed] [Google Scholar]
- 26.Spiro RG. Glucose residues as key determinants in the biosynthesis and quality control of glycoproteins with N-linked oligosaccharides. J Biol Chem. 2000;275(46):35657–60. doi: 10.1074/jbc.R000022200. [DOI] [PubMed] [Google Scholar]
- 27.Kullolli M, Hancock WS, Hincapie M. Preparation of a high-performance multi-lectin affinity chromatography (HP-M-LAC) adsorbent for the analysis of human plasma glycoproteins. J Sep Sci. 2008;31(14):2733–2739. doi: 10.1002/jssc.200800233. [DOI] [PubMed] [Google Scholar]
- 28.Orazine CI, Hincapie M, Hancock WS, Hattersley M, Hanke JH. A proteomic analysis of the plasma glycoproteins of a MCF-7 mouse xenograft: a model system for the detection of tumor markers. J Proteome Res. 2008;7(4):1542–54. doi: 10.1021/pr7008516. [DOI] [PubMed] [Google Scholar]
- 29.Yang Z, Harris LE, Palmer-Toy DE, Hancock WS. Multilectin affinity chromatography for characterization of multiple glycoprotein biomarker candidates in serum from breast cancer patients. Clin Chem. 2006;52(10):1897–905. doi: 10.1373/clinchem.2005.065862. [DOI] [PubMed] [Google Scholar]
- 30.Ogata Y, Charlesworth MC, Muddiman DC. Evaluation of protein depletion methods for the analysis of total-, phospho- and glycoproteins in lumbar cerebrospinal fluid. J Proteome Res. 2005;4(3):837–45. doi: 10.1021/pr049750o. [DOI] [PubMed] [Google Scholar]
- 31.Liu T, Qian WJ, Gritsenko MA, Camp DG, 2nd, Monroe ME, Moore RJ, Smith RD. Human plasma N-glycoproteome analysis by immunoaffinity subtraction, hydrazide chemistry, and mass spectrometry. J Proteome Res. 2005;4(6):2070–80. doi: 10.1021/pr0502065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qiu Y, Patwa TH, Xu L, Shedden K, Misek DE, Tuck M, Jin G, Ruffin MT, Turgeon DK, Synal S, Bresalier R, Marcon N, Brenner DE, Lubman DM. Plasma glycoprotein profiling for colorectal cancer biomarker identification by lectin glycoarray and lectin blot. J Proteome Res. 2008;7(4):1693–703. doi: 10.1021/pr700706s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vercoutter-Edouart AS, Slomianny MC, Dekeyzer-Beseme O, Haeuw JF, Michalski JC. Glycoproteomics and glycomics investigation of membrane N-glycosylproteins from human colon carcinoma cells. Proteomics. 2008;8(16):3236–3256. doi: 10.1002/pmic.200800151. [DOI] [PubMed] [Google Scholar]
- 34.Yang Z, Hancock WS, Chew TR, Bonilla L. A study of glycoproteins in human serum and plasma reference standards (HUPO) using multilectin affinity chromatography coupled with RPLC-MS/MS. Proteomics. 2005;5(13):3353–66. doi: 10.1002/pmic.200401190. [DOI] [PubMed] [Google Scholar]
- 35.Zhao J, Simeone DM, Heidt D, Anderson MA, Lubman DM. Comparative serum glycoproteomics using lectin selected sialic acid glycoproteins with mass spectrometric analysis: application to pancreatic cancer serum. J Proteome Res. 2006;5(7):1792–802. doi: 10.1021/pr060034r. [DOI] [PubMed] [Google Scholar]
- 36.Clower DM, West RA, Lynch JC, Strick PL. The inferior parietal lobule is the target of output from the superior colliculus, hippocampus, and cerebellum. J Neurosci. 2001;21(16):6283–91. doi: 10.1523/JNEUROSCI.21-16-06283.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goldstein IJ. Studies on the combining sites of concanavalin A. Adv Exp Med Biol. 1975;55:35–53. doi: 10.1007/978-1-4684-0949-9_3. [DOI] [PubMed] [Google Scholar]
- 38.Kornfeld R, Ferris C. Interaction of immunoglobulin glycopeptides with concanavalin A. J Biol Chem. 1975;250(7):2614–9. [PubMed] [Google Scholar]
- 39.Ogata S, Muramatsu T, Kobata A. Fractionation of glycopeptides by affinity column chromatography on concanavalin A-sepharose. J Biochem. 1975;78(4):687–96. doi: 10.1093/oxfordjournals.jbchem.a130956. [DOI] [PubMed] [Google Scholar]
- 40.Narasimhan S, Wilson JR, Martin E, Schachter H. A structural basis for four distinct elution profiles on concanavalin A--Sepharose affinity chromatography of glycopeptides. Can J Biochem. 1979;57(1):83–96. doi: 10.1139/o79-011. [DOI] [PubMed] [Google Scholar]
- 41.Brewer CF, Bhattacharyya L. Specificity of concanavalin A binding to asparagine-linked glycopeptides. A nuclear magnetic relaxation dispersion study. J Biol Chem. 1986;261(16):7306–10. [PubMed] [Google Scholar]
- 42.Edelman GM, Wang JL. Binding and functional properties of concanavalin A and its derivatives. III. Interactions with indoleacetic acid and other hydrophobic ligands. J Biol Chem. 1978;253(9):3016–22. [PubMed] [Google Scholar]
- 43.Butterfield DA, Stadtman ER. Protein Oxidation Processes in Aging Brain. In: Mattson M, Geddes J, editors. Adv Cell Aging Gerontol. 1997. pp. 161–191. [Google Scholar]
- 44.Boyd-Kimball D, Castegna A, Sultana R, Poon HF, Petroze R, Lynn BC, Klein JB, Butterfield DA. Proteomic identification of proteins oxidized by Abeta(1–42) in synaptosomes: implications for Alzheimer’s disease. Brain Res. 2005;1044(2):206–15. doi: 10.1016/j.brainres.2005.02.086. [DOI] [PubMed] [Google Scholar]
- 45.Yu AC, Schousboe A, Hertz L. Metabolic fate of 14C-labeled glutamate in astrocytes in primary cultures. J Neurochem. 1982;39(4):954–60. doi: 10.1111/j.1471-4159.1982.tb11482.x. [DOI] [PubMed] [Google Scholar]
- 46.Malthankar-Phatak GH, de Lanerolle N, Eid T, Spencer DD, Behar KL, Spencer SS, Kim JH, Lai JC. Differential glutamate dehydrogenase (GDH) activity profile in patients with temporal lobe epilepsy. Epilepsia. 2006;47(8):1292–9. doi: 10.1111/j.1528-1167.2006.00543.x. [DOI] [PubMed] [Google Scholar]
- 47.McKenna MC, Sonnewald U, Huang X, Stevenson J, Zielke HR. Exogenous glutamate concentration regulates the metabolic fate of glutamate in astrocytes. J Neurochem. 1996;66(1):386–93. doi: 10.1046/j.1471-4159.1996.66010386.x. [DOI] [PubMed] [Google Scholar]
- 48.Erecinska M, Nelson D. Activation of glutamate dehydrogenase by leucine and its nonmetabolizable analogue in rat brain synaptosomes. J Neurochem. 1990;54(4):1335–43. doi: 10.1111/j.1471-4159.1990.tb01967.x. [DOI] [PubMed] [Google Scholar]
- 49.Burbaeva G, Boksha IS, Tereshkina EB, Savushkina OK, Starodubtseva LI, Turishcheva MS. Glutamate metabolizing enzymes in prefrontal cortex of Alzheimer’s disease patients. Neurochem Res. 2005;30(11):1443–51. doi: 10.1007/s11064-005-8654-x. [DOI] [PubMed] [Google Scholar]
- 50.Butterworth RF, Besnard AM. Thiamine-dependent enzyme changes in temporal cortex of patients with Alzheimer’s disease. Metab Brain Dis. 1990;5(4):179–84. doi: 10.1007/BF00997071. [DOI] [PubMed] [Google Scholar]
- 51.Miulli DE, Norwell DY, Schwartz FN. Plasma concentrations of glutamate and its metabolites in patients with Alzheimer’s disease. J Am Osteopath Assoc. 1993;93(6):670–6. [PubMed] [Google Scholar]
- 52.Op den Velde W, Stam FC. Some cerebral proteins and enzyme systems in Alzheimer’s presenile and senile dementia. J Am Geriatr Soc. 1976;24(1):12–6. doi: 10.1111/j.1532-5415.1976.tb03247.x. [DOI] [PubMed] [Google Scholar]
- 53.Gibson GE, Park LC, Zhang H, Sorbi S, Calingasan NY. Oxidative stress and a key metabolic enzyme in Alzheimer brains, cultured cells, and an animal model of chronic oxidative deficits. Ann N Y Acad Sci. 1999;893:79–94. doi: 10.1111/j.1749-6632.1999.tb07819.x. [DOI] [PubMed] [Google Scholar]
- 54.Korolainen MA, Auriola S, Nyman TA, Alafuzoff I, Pirttila T. Proteomic analysis of glial fibrillary acidic protein in Alzheimer’s disease and aging brain. Neurobiol Dis. 2005;20(3):858–70. doi: 10.1016/j.nbd.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 55.Eddleston M, Mucke L. Molecular profile of reactive astrocytes--implications for their role in neurologic disease. Neuroscience. 1993;54(1):15–36. doi: 10.1016/0306-4522(93)90380-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gomes FC, Paulin D, Moura Neto V. Glial fibrillary acidic protein (GFAP): modulation by growth factors and its implication in astrocyte differentiation. Braz J Med Biol Res. 1999;32(5):619–31. doi: 10.1590/s0100-879x1999000500016. [DOI] [PubMed] [Google Scholar]
- 57.Beach TG, Walker R, McGeer EG. Patterns of gliosis in Alzheimer’s disease and aging cerebrum. Glia. 1989;2(6):420–36. doi: 10.1002/glia.440020605. [DOI] [PubMed] [Google Scholar]
- 58.Gunning P, O’Neill G, Hardeman E. Tropomyosin-based regulation of the actin cytoskeleton in time and space. Physiol Rev. 2008;88(1):1–35. doi: 10.1152/physrev.00001.2007. [DOI] [PubMed] [Google Scholar]
- 59.Stamm S, Casper D, Lees-Miller JP, Helfman DM. Brain-specific tropomyosins TMBr-1 and TMBr-3 have distinct patterns of expression during development and in adult brain. Proc Natl Acad Sci U S A. 1993;90(21):9857–61. doi: 10.1073/pnas.90.21.9857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vrhovski B, Schevzov G, Dingle S, Lessard JL, Gunning P, Weinberger RP. Tropomyosin isoforms from the gamma gene differing at the C-terminus are spatially and developmentally regulated in the brain. J Neurosci Res. 2003;72(3):373–83. doi: 10.1002/jnr.10586. [DOI] [PubMed] [Google Scholar]
- 61.Poon HF, Vaishnav RA, Butterfield DA, Getchell ML, Getchell TV. Proteomic identification of differentially expressed proteins in the aging murine olfactory system and transcriptional analysis of the associated genes. J Neurochem. 2005;94(2):380–92. doi: 10.1111/j.1471-4159.2005.03215.x. [DOI] [PubMed] [Google Scholar]
- 62.Mello CF, Sultana R, Piroddi M, Cai J, Pierce WM, Klein JB, Butterfield DA. Acrolein induces selective protein carbonylation in synaptosomes. Neuroscience. 2007;147(3):674–9. doi: 10.1016/j.neuroscience.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Konzack S, Thies E, Marx A, Mandelkow EM, Mandelkow E. Swimming against the tide: mobility of the microtubule-associated protein tau in neurons. J Neurosci. 2007;27(37):9916–27. doi: 10.1523/JNEUROSCI.0927-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sedlacek Z, Konecki DS, Korn B, Klauck SM, Poustka A. Evolutionary conservation and genomic organization of XAP-4, an Xq28 located gene coding for a human rab GDP-dissociation inhibitor (GDI) Mamm Genome. 1994;5(10):633–9. doi: 10.1007/BF00411459. [DOI] [PubMed] [Google Scholar]
- 65.Wu SK, Zeng K, Wilson IA, Balch WE. Structural insights into the function of the Rab GDI superfamily. Trends Biochem Sci. 1996;21(12):472–6. doi: 10.1016/s0968-0004(96)10062-1. [DOI] [PubMed] [Google Scholar]
- 66.Luan P, Balch WE, Emr SD, Burd CG. Molecular dissection of guanine nucleotide dissociation inhibitor function in vivo. Rab-independent binding to membranes and role of Rab recycling factors. J Biol Chem. 1999;274(21):14806–17. doi: 10.1074/jbc.274.21.14806. [DOI] [PubMed] [Google Scholar]
- 67.Sasaki T, Kikuchi A, Araki S, Hata Y, Isomura M, Kuroda S, Takai Y. Purification and characterization from bovine brain cytosol of a protein that inhibits the dissociation of GDP from and the subsequent binding of GTP to smg p25A, a ras p21-like GTP-binding protein. J Biol Chem. 1990;265(4):2333–7. [PubMed] [Google Scholar]
- 68.Fischer von Mollard G, Stahl B, Khokhlatchev A, Sudhof TC, Jahn R. Rab3C is a synaptic vesicle protein that dissociates from synaptic vesicles after stimulation of exocytosis. J Biol Chem. 1994;269(15):10971–4. [PubMed] [Google Scholar]
- 69.Geppert M, Bolshakov VY, Siegelbaum SA, Takei K, De Camilli P, Hammer RE, Sudhof TC. The role of Rab3A in neurotransmitter release. Nature. 1994;369(6480):493–7. doi: 10.1038/369493a0. [DOI] [PubMed] [Google Scholar]
- 70.Wang Y, Okamoto M, Schmitz F, Hofmann K, Sudhof TC. Rim is a putative Rab3 effector in regulating synaptic-vesicle fusion. Nature. 1997;388(6642):593–8. doi: 10.1038/41580. [DOI] [PubMed] [Google Scholar]
- 71.Wenk GL. Neuropathologic changes in Alzheimer’s disease: potential targets for treatment. J Clin Psychiatry. 2006;67(Suppl 3):3–7. quiz 23. [PubMed] [Google Scholar]
- 72.Pratt WB. The role of the hsp90-based chaperone system in signal transduction by nuclear receptors and receptors signaling via MAP kinase. Annu Rev Pharmacol Toxicol. 1997;37:297–326. doi: 10.1146/annurev.pharmtox.37.1.297. [DOI] [PubMed] [Google Scholar]
- 73.Rutherford SL, Zuker CS. Protein folding and the regulation of signaling pathways. Cell. 1994;79(7):1129–32. doi: 10.1016/0092-8674(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 74.Freeman BC, Morimoto RI. The human cytosolic molecular chaperones hsp90, hsp70 (hsc70) and hdj-1 have distinct roles in recognition of a non-native protein and protein refolding. EMBO J. 1996;15(12):2969–79. [PMC free article] [PubMed] [Google Scholar]
- 75.Young JC, Schneider C, Hartl FU. In vitro evidence that hsp90 contains two independent chaperone sites. FEBS Lett. 1997;418(1–2):139–43. doi: 10.1016/s0014-5793(97)01363-x. [DOI] [PubMed] [Google Scholar]
- 76.Joab I, Radanyi C, Renoir M, Buchou T, Catelli MG, Binart N, Mester J, Baulieu EE. Common non-hormone binding component in non-transformed chick oviduct receptors of four steroid hormones. Nature. 1984;308(5962):850–3. doi: 10.1038/308850a0. [DOI] [PubMed] [Google Scholar]
- 77.Garcia-Cardena G, Fan R, Shah V, Sorrentino R, Cirino G, Papapetropoulos A, Sessa WC. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature. 1998;392(6678):821–4. doi: 10.1038/33934. [DOI] [PubMed] [Google Scholar]
- 78.Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, Gouras GK, Greengard P, Xu H. Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci U S A. 2003;100(2):721–6. doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yokota T, Mishra M, Akatsu H, Tani Y, Miyauchi T, Yamamoto T, Kosaka K, Nagai Y, Sawada T, Heese K. Brain site-specific gene expression analysis in Alzheimer’s disease patients. Eur J Clin Invest. 2006;36(11):820–30. doi: 10.1111/j.1365-2362.2006.01722.x. [DOI] [PubMed] [Google Scholar]
- 80.Evans CG, Wisen S, Gestwicki JE. Heat shock proteins 70 and 90 inhibit early stages of amyloid beta-(1–42) aggregation in vitro. J Biol Chem. 2006;281(44):33182–91. doi: 10.1074/jbc.M606192200. [DOI] [PubMed] [Google Scholar]
- 81.Veereshwarayya V, Kumar P, Rosen KM, Mestril R, Querfurth HW. Differential effects of mitochondrial heat shock protein 60 and related molecular chaperones to prevent intracellular beta-amyloid-induced inhibition of complex IV and limit apoptosis. J Biol Chem. 2006;281(40):29468–78. doi: 10.1074/jbc.M602533200. [DOI] [PubMed] [Google Scholar]
- 82.Kakimura J, Kitamura Y, Takata K, Umeki M, Suzuki S, Shibagaki K, Taniguchi T, Nomura Y, Gebicke-Haerter PJ, Smith MA, Perry G, Shimohama S. Microglial activation and amyloid-beta clearance induced by exogenous heat-shock proteins. FASEB J. 2002;16(6):601–3. doi: 10.1096/fj.01-0530fje. [DOI] [PubMed] [Google Scholar]
- 83.Takata K, Kitamura Y, Tsuchiya D, Kawasaki T, Taniguchi T, Shimohama S. Heat shock protein-90-induced microglial clearance of exogenous amyloid-beta1-42 in rat hippocampus in vivo. Neurosci Lett. 2003;344(2):87–90. doi: 10.1016/s0304-3940(03)00447-6. [DOI] [PubMed] [Google Scholar]
- 84.Perluigi M, Fai Poon H, Hensley K, Pierce WM, Klein JB, Calabrese V, De Marco C, Butterfield DA. Proteomic analysis of 4-hydroxy-2-nonenal-modified proteins in G93A-SOD1 transgenic mice--a model of familial amyotrophic lateral sclerosis. Free Radic Biol Med. 2005;38(7):960–8. doi: 10.1016/j.freeradbiomed.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 85.Lubec G, Nonaka M, Krapfenbauer K, Gratzer M, Cairns N, Fountoulakis M. Expression of the dihydropyrimidinase related protein 2 (DRP-2) in Down syndrome and Alzheimer’s disease brain is downregulated at the mRNA and dysregulated at the protein level. J Neural Transm Suppl. 1999;57:161–77. doi: 10.1007/978-3-7091-6380-1_10. [DOI] [PubMed] [Google Scholar]
- 86.Coleman PD, Flood DG. Neuron numbers and dendritic extent in normal aging and Alzheimer’s disease. Neurobiol Aging. 1987;8:521–45. doi: 10.1016/0197-4580(87)90127-8. [DOI] [PubMed] [Google Scholar]
- 87.Yang Y, Turner RS, Gaut JR. The chaperone BiP/GRP78 binds to amyloid precursor protein and decreases Abeta40 and Abeta42 secretion. J Biol Chem. 1998;273(40):25552–5. doi: 10.1074/jbc.273.40.25552. [DOI] [PubMed] [Google Scholar]
- 88.Falahatpisheh H, Nanez A, Montoya-Durango D, Qian Y, Tiffany-Castiglioni E, Ramos KS. Activation profiles of HSPA5 during the glomerular mesangial cell stress response to chemical injury. Cell Stress Chaperones. 2007;12(3):209–18. doi: 10.1379/CSC-259.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Katayama T, Imaizumi K, Sato N, Miyoshi K, Kudo T, Hitomi J, Morihara T, Yoneda T, Gomi F, Mori Y, Nakano Y, Takeda J, Tsuda T, Itoyama Y, Murayama O, Takashima A, St George-Hyslop P, Takeda M, Tohyama M. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat Cell Biol. 1999;1(8):479–85. doi: 10.1038/70265. [DOI] [PubMed] [Google Scholar]
- 90.Hoozemans JJ, Veerhuis R, Van Haastert ES, Rozemuller JM, Baas F, Eikelenboom P, Scheper W. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 2005;110(2):165–72. doi: 10.1007/s00401-005-1038-0. [DOI] [PubMed] [Google Scholar]
- 91.Renouf S, Beullens M, Wera S, Van Eynde A, Sikela J, Stalmans W, Bollen M. Molecular cloning of a human polypeptide related to yeast sds22, a regulator of protein phosphatase-1. FEBS Lett. 1995;375(1–2):75–8. doi: 10.1016/0014-5793(95)01180-m. [DOI] [PubMed] [Google Scholar]
- 92.Peggie MW, MacKelvie SH, Bloecher A, Knatko EV, Tatchell K, Stark MJ. Essential functions of Sds22p in chromosome stability and nuclear localization of PP1. J Cell Sci. 2002;115(Pt 1):195–206. doi: 10.1242/jcs.115.1.195. [DOI] [PubMed] [Google Scholar]
- 93.Simpson IA, Chundu KR, Davies-Hill T, Honer WG, Davies P. Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann Neurol. 1994;35(5):546–51. doi: 10.1002/ana.410350507. [DOI] [PubMed] [Google Scholar]
- 94.Yamaguchi S, Meguro K, Itoh M, Hayasaka C, Shimada M, Yamazaki H, Yamadori A. Decreased cortical glucose metabolism correlates with hippocampal atrophy in Alzheimer’s disease as shown by MRI and PET. J Neurol Neurosurg Psychiatry. 1997;62(6):596–600. doi: 10.1136/jnnp.62.6.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Newman SF, Sultana R, Perluigi M, Coccia R, Cai J, Pierce WM, Klein JB, Turner DM, Butterfield DA. An increase in S-glutathionylated proteins in the Alzheimer’s disease inferior parietal lobule, a proteomics approach. J Neurosci Res. 2007;85(7):1506–14. doi: 10.1002/jnr.21275. [DOI] [PubMed] [Google Scholar]
- 96.Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, Lindquist S. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science. 2006;313(5785):324–8. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jensen PH, Hojrup P, Hager H, Nielsen MS, Jacobsen L, Olesen OF, Gliemann J, Jakes R. Binding of Abeta to alpha- and beta-synucleins: identification of segments in alpha-synuclein/NAC precursor that bind Abeta and NAC. Biochem J. 1997;323(Pt 2):539–46. doi: 10.1042/bj3230539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Boyd-Kimball D, Sultana R, Poon HF, Lynn BC, Casamenti F, Pepeu G, Klein JB, Butterfield DA. Proteomic identification of proteins specifically oxidized by intracerebral injection of amyloid beta-peptide (1–42) into rat brain: implications for Alzheimer’s disease. Neuroscience. 2005;132(2):313–24. doi: 10.1016/j.neuroscience.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 99.Hart GW, Haltiwanger RS, Holt GD, Kelly WG. Glycosylation in the nucleus and cytoplasm. Annu Rev Biochem. 1989;58:841–74. doi: 10.1146/annurev.bi.58.070189.004205. [DOI] [PubMed] [Google Scholar]
- 100.Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid beta-peptide. Trends Mol Med. 2001;7(12):548–54. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- 101.Joshi G, Perluigi M, Sultana R, Agrippino R, Calabrese V, Butterfield DA. In vivo protection of synaptosomes by ferulic acid ethyl ester (FAEE) from oxidative stress mediated by 2,2-azobis(2-amidino-propane)dihydrochloride (AAPH) or Fe(2+)/H(2)O(2): insight into mechanisms of neuroprotection and relevance to oxidative stress-related neurodegenerative disorders. Neurochem Int. 2006;48(4):318–27. doi: 10.1016/j.neuint.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 102.Thongboonkerd V, McLeish KR, Arthur JM, Klein JB. Proteomic analysis of normal human urinary proteins isolated by acetone precipitation or ultracentrifugation. Kidney Int. 2002;62(4):1461–9. doi: 10.1111/j.1523-1755.2002.kid565.x. [DOI] [PubMed] [Google Scholar]
- 103.Butterfield DA, Castegna A. Proteomics for the identification of specifically oxidized proteins in brain: technology and application to the study of neurodegenerative disorders. Amino Acids. 2003;25(34):419–25. doi: 10.1007/s00726-003-0027-7. [DOI] [PubMed] [Google Scholar]
- 104.Maurer HH, Peters FT. Toward high-throughput drug screening using mass spectrometry. Ther Drug Monit. 2005;27(6):686–8. doi: 10.1097/01.ftd.0000180224.19384.f0. [DOI] [PubMed] [Google Scholar]
- 105.Boguski MS, McIntosh MW. Biomedical informatics for proteomics. Nature. 2003;422(6928):233–7. doi: 10.1038/nature01515. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MS and protein database information associated with each Con-A fractionated protein identified was provided by the University of Louisville Mass Spectrometry Facility for this study. This information is available free of charge via the Internet at http://pubs.acs.org.