

Abstract
Obstructive sleep apnea leads to chronic intermittent hypoxia (CIH) and is associated with atherosclerosis. We have previously shown that C57BL/6J mice exposed to CIH and a high-cholesterol diet develop dyslipidemia, atherosclerosis of the aorta, and upregulation of a hepatic enzyme of lipoprotein secretion, stearoyl coenzyme A desaturase 1 (SCD-1). We hypothesized that (1) SCD-1 deficiency will prevent dyslipidemia and atherosclerosis during CIH; and (2) human OSA is associated with dyslipidemia and upregulation of hepatic SCD. C57BL/6J mice were exposed to CIH or normoxia for 10 weeks while being treated with either SCD-1 or control antisense oligonucleotides. Obese human subjects underwent sleep study and bariatric surgery with intraoperative liver biopsy. In mice, hypoxia increased hepatic SCD-1 and plasma very-low-density lipoprotein cholesterol levels and induced atherosclerosis lesions in the ascending aorta (the cross-section area of 156514±57408 μm2), and descending aorta (7.0±1.2% of the total aortic surface). In mice exposed to CIH and treated with SCD-1 antisense oligonucleotides, dyslipidemia and atherosclerosis in the ascending aorta were abolished, whereas lesions in the descending aorta showed 56% reduction. None of the mice exposed to normoxia developed atherosclerosis. In human subjects, hepatic SCD mRNA levels correlated with the degree of nocturnal hypoxemia (r=0.68, P=0.001). Patients exhibiting oxyhemoglobin desaturations at night showed higher plasma triglyceride and low-density lipoprotein cholesterol levels, compared to subjects without hypoxemia. In conclusion, CIH is associated with dyslipidemia and overexpression of hepatic SCD in both humans and mice alike; SCD-1 deficiency attenuates CIH-induced dyslipidemia and atherosclerosis in mice.
Keywords: atherosclerosis, hypercholesterolemia, hypoxia, lipoproteins, obstructive sleep apnea
Obstructive sleep apnea (OSA) is characterized by chronic intermittent hypoxia (CIH) during sleep.1 OSA occurs in 9% of women and 24% of men in the United States, but the prevalence exceeds 30% to 50% in the obese population.2,3 OSA poses significant cardiovascular risk, which has been attributed to the high prevalence of atherosclerosis in patients with OSA.4–6 OSA is associated with dyslipidemia, lipid peroxidation, and vascular inflammation, all of which induce atherosclerosis.7–10 Recent studies have shown independent associations between the hypoxic stress of OSA and increased carotid artery intima–media thickness11,12 that is reversed by treatment with continuous positive airway pressure.13
Our group has previously explored relationships between CIH and atherosclerosis using a mouse model of CIH that mimics the oxygen profile in patients with severe OSA.14,15 We have shown that CIH induces atherosclerosis in C57BL/6J mice on a high-cholesterol diet.16 We have also found that the development of atherosclerosis during CIH was associated with increases in lipoprotein secretion, plasma very-low-density lipoprotein VLDL levels, and expression of hepatic stearoyl coenzyme A desaturase 1 (SCD-1).16–18
Four isoforms of SCD have been described in mice, of which SCD-1 is the best characterized.19 SCD converts saturated fatty acids, 18:0 and 16:0, into monounsaturated fatty acids (MUFAs), 18:1 and 16:1. Increased availability of MUFAs facilitates cholesterol ester and triglyceride biosynthesis and enhances lipoprotein secretion leading to hyperlipidemia.20,21 SCD-1 expression is regulated by sterol SREBP-1c, a key transcription factor of lipid biosynthesis.22 SCD-1 has been implicated in the pathogenesis of obesity, fatty liver, and insulin resistance in rodents,19,23–26 but the role of SCD-1 in atherosclerosis has not been explored. Two human SCD genes have been identified with high homology to the murine SCD-1.19 Expression of hepatic SCD in human subjects has not been evaluated.
In this study, we explored a hypothesis that SCD-1 is involved in the pathogenesis of dyslipidemia and atherosclerosis during CIH. We examined our hypothesis by administering antisense SCD-1 oligonucleotides (ASOs) or control ASOs to C57BL/6J mice fed a high-cholesterol diet and exposed to CIH and measuring plasma lipid levels and aortic atherosclerotic lesions. We also tested the relevance of our findings for human OSA by examining hepatic SCD and plasma lipid levels in obese patients who underwent bariatric surgery and correlating these measurements with anthropometric and polysomnographic parameters.
Materials and Methods
A total of 24 wild-type, 8 week-old male C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, Me). The study was approved by the Johns Hopkins University Animal Care and Use Committee. Mice were fed a high-cholesterol diet and subjected either to CIH during the 12-hour light phase with an FiO2 nadir of 5%, 60 times per hour, or intermittent air (IA) for 10 weeks. Animals from each exposure group were treated either with SCD-1 ASOs or control ASOs.
Nineteen consecutive patients (18 women and 1 man) without history of diabetes, cardiovascular or liver disease, or dyslipidemia were recruited from the Johns Hopkins Bayview Medical Center Bariatric Surgery Clinic. The protocol was approved by the Western Institutional Review Board and the subjects gave informed consent. A detailed description of the experimental methods is available in the online data supplement at http://circres.ahajournals.org.
Results
Effects of SCD-1 ASOs and CIH on Hepatic SCD-1 Levels and Activity in Mice
SCD-1 ASOs induced marked decreases in liver SCD-1 mRNA levels in both normoxic mice (ΔΔCt=2.9) and hypoxic mice (ΔΔ Ct=3.1) (Figure 1A). Given efficiencies of real-time PCR for SCD-1 and 18S (see the online data supplement), SCD-1 ASOs led to 84% to 87% depletion of SCD-1 mRNA in the liver (Figure 1A). SCD-1 ASOs decreased hepatic SCD-1 protein levels (Figure 1B and 1C), as well as 16:1/16:0 and 18:1/18:0 plasma fatty acid ratios (Figure I in the online data supplement), which indicated reduced enzymatic activity.
Figure 1.
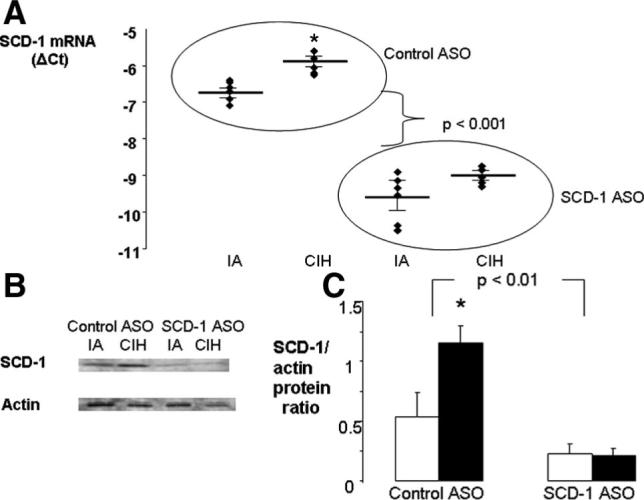
Analysis of SCD-1 in the livers of C57BL/6J mice receiving SCD-1 or control ASOs and exposed to CIH or IA for 10 weeks (n=6 per group). A, Hepatic SCD-1 mRNA levels by real-time RT-PCR; Ct indicates the critical threshold cycle; ΔCt is the difference between 18S and SCD Ct values. B and C, SCD-1 protein levels by immunoblot with total liver lysate. B, SCD-1 and α-actin bands in representative samples. C, Mean optical density of SCD-1 bands normalized to α-actin. Solid bars indicate CIH; open bars, IA. *P<0.05 for the difference between CIH and IA.
CIH led to a 71.6% elevation in SCD-1 mRNA and greater than a 2-fold increase in SCD-1 protein levels in the liver of mice receiving control ASOs (Figure 1). CIH also raised plasma 16:1/16:0 and 18:1/18:0 fatty acid ratios by 17% and 22%, respectively (supplemental Figure I). In contrast, animals injected with SCD-1 ASOs did not exhibit significant changes in SCD-1 mRNA, protein levels, or activity in response to CIH (Figure 1 and supplemental Figure I).
Effects of SCD-1 ASOs and CIH on Body Weight and Plasma Lipids in Mice
CIH caused significant weight loss by day 30 (Table), which was consistent with our previous observations.17 Mice treated with control ASOs from day 30 to day 70 of CIH regained weight by day 70, whereas SCD-1 ASO–treated mice did not. As a result, 10 weeks of CIH caused weight loss only in the SCD-1 ASO–treated mice but not in mice receiving control ASOs. SCD-1 ASOs did not affect liver weight, fasting blood glucose, or insulin levels but induced a significant loss of epididymal fat and declines in hepatic cholesterol, triglyceride, and free fatty acid content (Table). CIH had no impact on any of these parameters.
Table.
Effects of CIH and SCD-1 ASOs on Metabolic Characteristics in C57BL/6J Mice
| Characteristic | Control ASO |
SCD-1 ASO |
|||
|---|---|---|---|---|---|
| IA | CIH | IA | CIH | Effect of SCD-1 (P) | |
| N | 6 | 6 | 6 | 6 | N/A |
| Starting age, wk | 8 | 8 | 8 | 8 | N/A |
| Body weight, g | |||||
| Day 0 | 23.1±0.3 | 22.9±1.9 | 23.1±0.2 | 23.5±0.7 | >0.05 |
| Day 30 | 23.5±0.4 | 20.8±1.9** | 24.0±0.6 | 21.8±0.7*** | >0.05 |
| Day 70 | 24.2±0.5 | 23.7±0.7* | 24.3±0.4 | 22.2±0.7 | >0.05 |
| Daily food intake, g | 2.2±0.1 | 2.3±0.1 | 2.4±0.3 | 2.6±0.1 | >0.05 |
| Liver weight, g | 2.11±0.07 | 1.89±0.08 | 1.90±0.11 | 1.80±0.10 | >0.05 |
| Liver weight/body wt, % | 8.85±0.49 | 8.01±0.34 | 7.85±0.52 | 8.12±0.89 | >0.05 |
| Epididymal fat, g | 0.28±0.02 | 0.29±0.03 | 0.20±0.01 | 0.14±0.01 | <0.001 |
| Epididymal fat/body wt, % | 1.15±0.07 | 1.22±0.14 | 0.83±0.06 | 0.62±0.02 | <0.001 |
| Fasting blood glucose, mg/dL | 167±14 | 163±14 | 165±18 | 172±6 | >0.05 |
| Fasting plasma insulin, ng/mL | 0.39±0.05 | 0.42±0.04 | 0.51±0.10 | 0.55±0.12 | >0.05 |
| Fasting plasma total cholesterol, mg/dL | 224±7 | 270±12† | 202±9 | 218±10 | <0.05 |
| Fasting plasma triglycerides, mg/dL | 14.4±3.6 | 13.2±5.8 | 16.7±5.3 | 13.9±2.4 | >0.05 |
| Fasting plasma free fatty acids, mmol/L | 0.31±0.06 | 0.28±0.08 | 0.41±0.04 | 0.36±0.06 | >0.05 |
| Liver cholesterol, mg/g | 9.60±0.11 | 9.75±0.27 | 7.80±1.17 | 9.36±0.24 | <0.05 |
| Liver triglyceride, mg/g | 13.4±0.8 | 13.5±2.0 | 7.91±1.88 | 9.84±1.63 | <0.01 |
| Liver free fatty acids, μmol/g | 7.20±1.46 | 8.49±2.73 | 5.66±1.61 | 5.90±1.36 | 0.05 |
P<0.05 between day 30 and day 70
P<0.01 between day 0 and day 30
P<0.001 between day 0 and day 30
P<0.01 between IA and CIH.
SCD-1 ASO administration resulted in a significant decrease in fasting plasma total cholesterol levels, especially during exposure to CIH (Table). High-performance liquid chromatography (HPLC) revealed that cholesterol-lowering effect of SCD-1 ASOs occurred in the VLDL cholesterol (VLDL-C) fraction (Figure 2). SCD-1 ASOs induced a 38% decrease in VLDL-C at normoxic conditions, from 116.8±3.6 mg/dL in mice receiving control ASOs to 71.9±7.1 mg/dL (P=0.001), and a 36% decrease in VLDL-C at hypoxic conditions, from 135.4±3.6 mg/dL to 86.5±7.7 mg/dL (P=0.001), which was consistent with the previously described role of SCD-1 in secretion of VLDL particles by the liver.19,21 SCD-1 ASOs also increased plasma levels of high-density lipoprotein cholesterol (HDL-C), especially at normoxic conditions, from 62.0±2.0 mg/dL in mice receiving control ASOs to 74.7±2.1 mg/dL (P<0.01) (Figure 2). CIH resulted in a significant increase in fasting plasma total cholesterol levels in mice treated with control ASOs (Table) with the majority of the increase occurring in the VLDL fraction (Figure 2). In mice receiving control ASOs, CIH reduced HDL-C levels. SCD-1 ASO deficiency essentially abolished detrimental effects of CIH on the cholesterol profile (Figure 2). Neither SCD-1 ASOs nor CIH impacted plasma triglycerides and free fatty acid levels.
Figure 2.
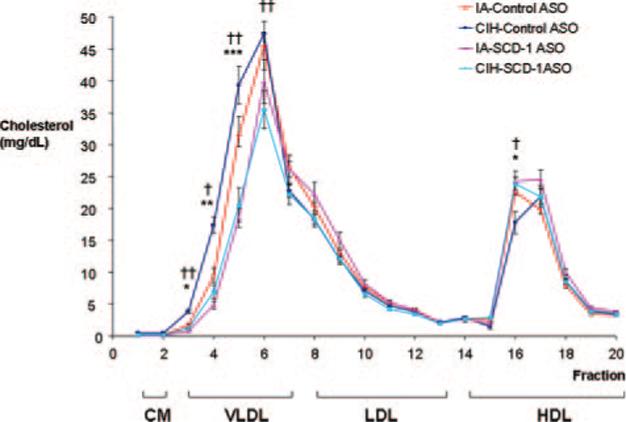
The HPLC profile of plasma cholesterol in C57BL/6J mice on a high-cholesterol diet after exposure to CIH or IA while receiving SCD-1 or control ASOs. †P<0.01, ††P<0.001 for the effect of CIH; *P<0.05, **P<0.01, ***P<0.001 for the effect of SCD-1 ASOs.
Effects of SCD-1 ASOs and CIH on the Aorta in Mice
In the absence of the hypoxic stimulus, male C56BL/6J mice did not exhibit any evidence of atherosclerosis in the aortic origin, regardless of the type of ASO treatment (Figure 3A and 3C). In contrast, all mice exposed to CIH and treated with control ASOs developed extensive atherosclerosis in the aortic origin. The aortic lesions had the appearance of mature atherosclerotic plaques, with extensive lipid depositions in the intima and media, thickening of the aortic wall, mononuclear infiltration, and a necrotic acellular core (Figure 3B). The plaques varied in size from 26 726 to 560 836 μm2, with an average cross-section area of 156 514±57 408 μm2. Interestingly, none of the mice exposed to CIH and SCD-1 ASOs exhibited atherosclerotic plaques in the aortic origin, suggesting that SCD-1 ASOs opposed atherogenic effects of CIH. Two of 6 animals in this group revealed a small amount of oil red O–positive subintimal lipid deposits (Figure 3D), whereas in 4 others, the proximal aorta was intact and similar in appearance to mice exposed to IA.
Figure 3.
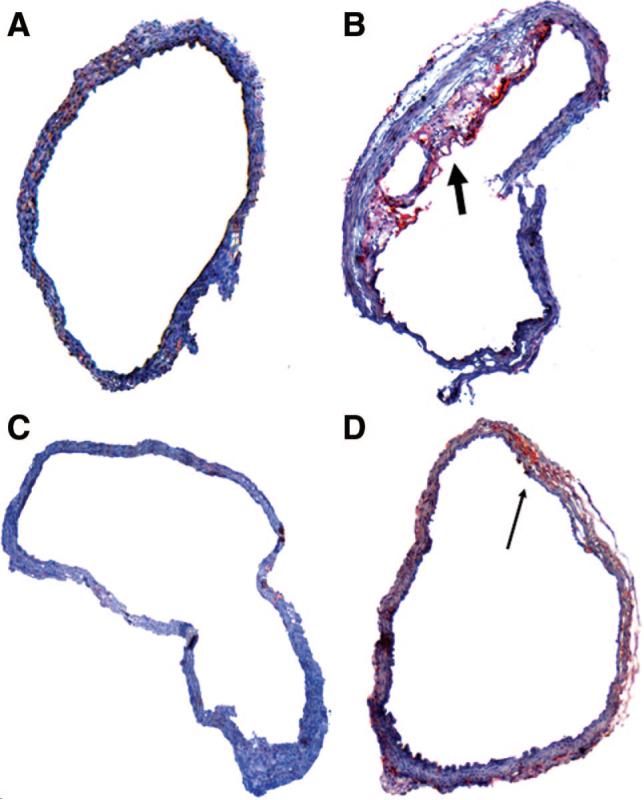
Representative cross-sections of the ascending aorta (sinus of Valsalva) in C57BL/6J mice exposed to IA and control ASOs injections (A), CIH and control ASOs (B), IA and SCD-1 ASOs (C), or CIH and SCD-1 ASOs (D). Transverse frozen sections of the aorta were stained with oil red O and hematoxylin. Original magnification, ×100. The thick arrow points to the distinct atherosclerotic plaque in a mouse exposed to CIH and control ASOs. The thin arrow points to the small amount of lipids in the aorta stained in red in a mouse exposed to CIH and SCD-1 ASOs.
The en face preparation showed no lipid accumulation in the thoracic and abdominal aorta of mice exposed to IA (Figure 4A and 4C). In contrast, all mice exposed to CIH exhibited bright red Sudan IV–positive areas in the intima. Consistent with findings in the aortic origin, hypoxic mice treated with SCD-1 ASOs exhibited a 56% decrease in the size of atherosclerotic lesions, compared to hypoxic animals treated with control ASOs (Figure 4B, 4D, and 4E).
Figure 4.
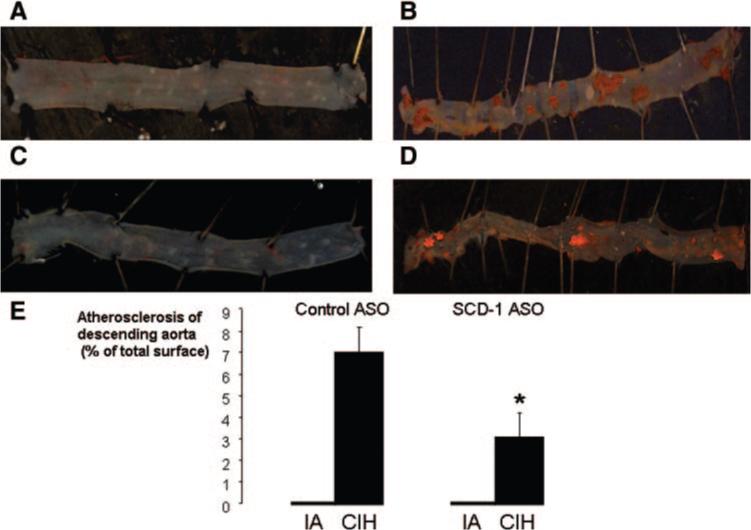
Representative images of the thoracic (aortic arch and descending aorta) and abdominal aorta by the en face method in C57BL/6J mice exposed to IA and control ASO injections (A), CIH and control ASOs (B), IA and SCD-1 ASOs (C), or CIH and SCD-1 ASOs (D); Sudan IV staining; original magnification, ×10, water immersion. E, Mean area of the total aortic surface covered by atherosclerotic lesions. *P<0.05 for the difference between control ASOs and SCD-1 ASOs.
Dihydroethidium staining revealed that CIH approximately doubled superoxide production in the aortic wall (supplement Figure II). SCD-1 deficiency did not have a significant effect on ROS generation in the vasculature.
Metabolic and Sleep Characteristics and Hepatic SCD Expression in Patients With Severe Obesity
The patients were divided in 2 groups according to the median values for body mass index (BMI) and the severity of obstructive sleep apnea, which was measured during routine polysomnography as the apnea–hypopnea index (AHI) and the magnitude of oxygen desaturation (ΔSaO2; supplemental Table I). BMI, AHI, and indices of nocturnal hypoxemia were not associated with free fatty acid levels. The AHI and nocturnal oxyhemoglobin desaturation were similar in individuals with higher and lower BMI (supplemental Table I), whereas more severe obesity was associated with a trend to higher SCD gene expression (P=0.053). Nonetheless, a correlation between BMI and SCD mRNA levels was not significant. The AHI had no relationship with SCD levels. In contrast, patients showing severe intermittent hypoxemia (ΔSaO2, ≥5%) exhibited significantly greater levels of hepatic SCD gene expression than patients experiencing mild intermittent hypoxemia (ΔSaO2 <5%; supplemental Table I). Furthermore, there was a strong positive correlation between the magnitude of nocturnal oxygen desaturation and SCD mRNA levels in the liver (r=0.68, P<0.001, Figure 5A). Consequently, individuals with severe nocturnal oxygen desaturation showed higher SCD protein levels in the liver (Figure 5 B). The patients with severe nocturnal hypoxemia also exhibited a 17% increase in total plasma cholesterol and an 89% increase in plasma triglyceride levels compared to the nonhypoxemic patients (supplemental Table I). The increase in plasma triglycerides occurred predominantly in the VLDL fraction and to a lesser extent in the LDL fraction, whereas the increase in plasma cholesterol levels was entirely attributable to LDL-C (Figure 6).
Figure 5.
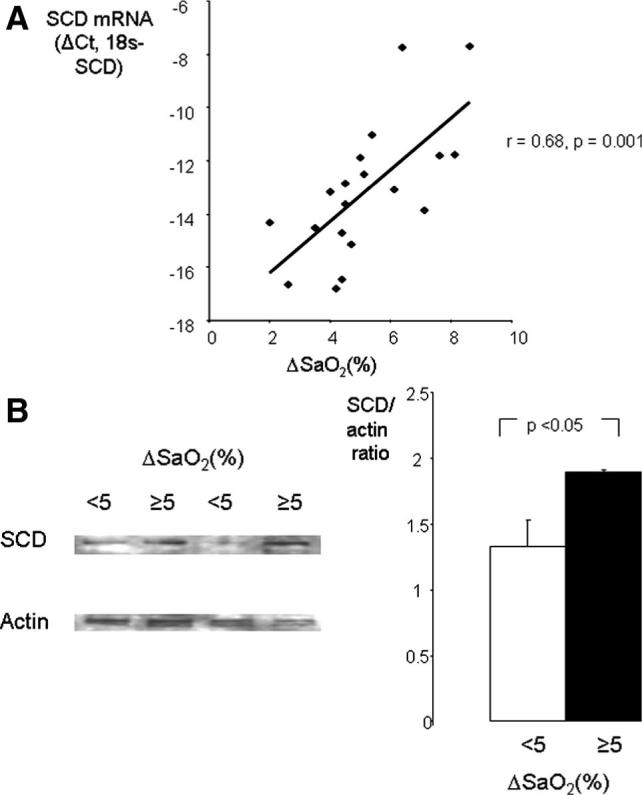
Relationships between nocturnal oxyhemoglobin desaturation (ΔSaO2) and hepatic levels of SCD in patients undergoing bariatric surgery; ΔSaO2 is a difference between baseline SaO2 and an average nocturnal SaO2 nadir during disordered breathing events (average low SaO2). A, mRNA levels measured by real-time RT-PCR; Ct indicates the critical threshold cycle; ΔCt, is the difference between 18S and SCD Ct values. Each data point represents 1 patient. B, SCD protein by immunoblot. Left, SCD and α-actin bands in representative samples of patients with mild (<5%) and severe (≥5%) oxyhemoglobin desaturation. Right, Mean optical density of SCD-1 bands normalized to α-actin. Solid bars indicate ΔSaO2 ≥5%; open bars, ΔSaO2 <5%.
Figure 6.
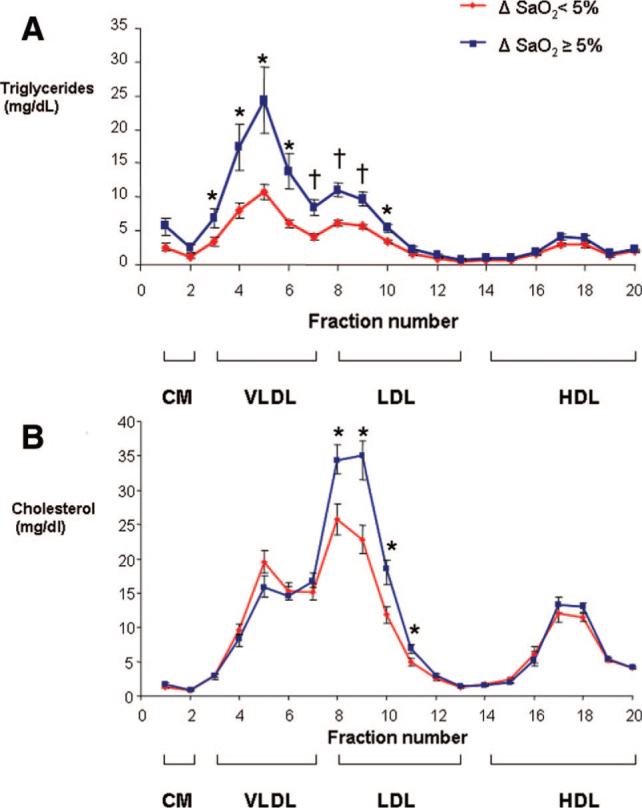
The HPLC profile of serum triglycerides (A) and cholesterol (B) in patients undergoing bariatric surgery separated in 2 groups based on the median value of ΔSaO2, which is a difference between baseline nocturnal SaO2 and average low SaO2 during disordered breathing events. *P<0.05 and †P<0.01 for the difference between the groups.
Discussion
We have previously shown that CIH induces atherosclerosis in the aorta of C57BL/6J mice on a high-cholesterol diet in association with hypercholesterolemia, increased lipoprotein secretion, and upregulation of a key hepatic enzyme of lipoprotein secretion, SCD-1.16 The purpose of this study was to explore the role of SCD-1 in CIH-induced dyslipidemia and atherosclerosis. Several novel findings resulted from the study. Our main finding was that CIH-induced atherosclerosis was completely abolished by SCD-1 ASOs in the ascending aorta and decreased by 56% in the descending aorta. Secondly, SCD-1 ASOs abolished CIH-induced increases in hepatic SCD-1 mRNA and protein levels, as well as an increase in plasma 16:1 and 18:1 MUFAs. Third, SCD-1 ASOs reversed a CIH-induced increase in plasma VLDL-C. Finally, we demonstrated that patients with OSA exhibit an increase in hepatic SCD expression in direct proportion to the severity of nocturnal intermittent hypoxemia and that SCD overexpression in human subjects with OSA was associated with marked increases in plasma triglyceride and LDL-C levels. In the discussion below, we explore relationships between CIH, SCD-1, and atherosclerosis and elaborate on the clinical implications of our work.
CIH Increases SCD Expression in the Liver
We have demonstrated that, in mice, CIH upregulated hepatic SCD-1 mRNA and protein levels, consistent with our earlier reports.16–18,27 Other investigators found that hypoxia augments SCD expression in cell culture of human macrophages in association with increased intracellular lipid droplets.28 A novel finding of the present study was that SCD expression in the liver significantly correlated with the depth of oxyhemoglobin desaturation in human subjects with OSA, suggesting that SCD is upregulated by intermittent hypoxia (IH) in humans. We have previously reported that IH enhances expression of SREBP-1c and SREBP cleavage–activating protein (SCAP),18,27,29 which mediates posttranscriptional modifications of SREBP from the immature form to the active nuclear form.30,31 Because SREBP-1c regulates SCD-1 expression,22 it is conceivable that IH affects SCD-1 through the SREBP-1 pathway, either directly or through hypoxia inducible factors (HIFs).32–36 HIF-1, HIF-2, and HIF-3 consist of O2-regulated HIF-1α, HIF-2α, or HIF-3α subunits and a constitutively expressed HIF-1β subunit.33,35,37–40 IH up-regulates HIF-1α both in vitro and in vivo,33,35,41,42 whereas effects of IH on HIF-2α and HIF-3 α are unknown. HIF-1α activation has a direct proinflammatory effect and may promote atherogenesis by increasing macrophagal infiltration, angiogenesis, and lipid content in the atherosclerotic plaque.43,44 Transgenic mice with partial deficiency of HIF-1α exhibit significantly attenuated increases in serum lipids, hepatic SREBP-1, SCAP, and SCD-1 during IH.36 Based on these findings, we have previously formulated a hypothesis that IH can induce hepatic SCD-1 and dyslipidemia via sequential upregulation of HIF-1 and SREBP-1 (see Figure 7 in the report by Li et al36).
SCD-1 ASOs Reverse Dyslipidemia During CIH
SCD-1 ASOs prevented hypoxia-induced increases in 16:1/16:0 and 18:1/18:0 fatty acid ratios (supplemental Figure I). Given that all experimental animals consumed diet with the same MUFA content and that SCD-1 is the main mechanism of MUFA biosynthesis in the liver,19,45 our data provide solid evidence that CIH raises palmitoleate and oleate levels via the SCD-1. Relative abundance of palmitoleate and oleate induces biosynthesis of triglyceride and cholesterol esters in the liver, augmenting lipoprotein secretion and leading to hypercholesterolemia and hypertriglyceridemia.20,23,46 SCD-1 deficiency results in low plasma levels of cholesterol and triglycerides in Asebia mice and SCD-1−/− transgenic mice fed regular chow.26,47,48 Our murine data showed that the CIH-induced increase in plasma total cholesterol levels occurred exclusively in the VLDL fraction and was entirely abolished by SCD-1 ASOs. This increase in VLDL cholesterol is likely attributable to cholesterol esters, the synthesis of which is regulated by SCD.21,48 CIH slightly decreased HDL-C levels and SCD-1 ASOs abolished this decrease (Figure 2). The latter is consistent with the reports that SCD-1 inhibits reverse cholesterol transport, destabilizing ATP-binding cassette transporter A1,49 and that SCD-1 deficiency increases plasma HDL-C.45 Surprisingly, in our study, neither CIH nor SCD-1 ASOs affected plasma triglyceride levels in mice that could be ascribed to low baseline levels after a prolonged fast. In contrast, SCD-1 ASOs significantly decreased hepatic lipid content and the amount of epididymal fat, which is consistent with the previous observations in SCD-1–deficient mice24,50 and attributable to downregulation of lipid biosynthesis and upregulation of fatty acid oxidation.19,51 Human studies showed that hypoxic upregulation of SCD in the liver was associated with a 2-fold increase in plasma triglycerides, predominantly in the VLDL fraction, implying that intermittent hypoxemia could augment lipoprotein secretion via the SCD mechanism as it occurs in mice. Summarizing all of the above, our data demonstrate that CIH causes dyslipidemia with elevation of VLDL, which was attenuated by SCD-1 ASOs.
SCD-1 ASOs Attenuate Atherosclerosis During CIH
We have reproduced our recently reported results16 and have again shown that a combination of CIH with a high-cholesterol diet leads to atherosclerosis in C57BL/6J mice, whereas mice exposed to the dietary fat alone did not exhibit atherosclerotic lesions. The main finding of the present study is that atherosclerosis in the mouse aorta was associated with upregulation of hepatic SCD-1 and was attenuated by SCD-1 ASOs. Thus, our data clearly demonstrate that SCD-1 inhibition has a therapeutic effect for atherosclerotic lesions induced by CIH.
SCD-1 upregulation in CIH may be proatherogenic. What are the mechanisms by which SCD-1 can lead to atherosclerosis? The most obvious pathway would be dyslipidemia resulting from upregulation of VLDL secretion and down-regulation of reverse cholesterol transport.20,21,23,46,49 However, CIH led only to modest changes in VLDL-C and HDL-C levels (Figure 2), which are not likely to cause such a dramatic effect on atherosclerotic lesions, unless other pathways are involved. One of the potential mechanisms is SCD-1 upregulation in macrophages of the aortic intima, which may result in accelerated foam cell formation, similar to that previously described in human macrophages exposed to hypoxia in vitro.28 Accelerated lipid accumulation in the vascular wall may also activate the nuclear factor κB pathway and induce local production of proinflammatory cytokines, leading to vascular inflammation, necrosis, and fibrosis.52 Finally, CIH leads to systemic oxidative stress with serum lipid peroxidation and high levels of oxidized LDL.10,16 Lipid peroxidation may exacerbate proatherogenic effects of SCD-1–induced dyslipidemia.53 We hypothesize that CIH-induced upregulation of SCD-1 may accelerate atherogenesis by a number of mechanisms, including dyslipidemia, oxidative stress, and vascular inflammation.
Conclusions, Clinical Implications, and Limitations of the Study
Our study has shown that, in C57BL/6J mice, CIH upregulates a key hepatic enzyme of lipoprotein secretion, SCD-1, and induces dyslipidemia and atherosclerosis, which are markedly attenuated by SCD-1 depletion. The major limitation of the study is that ASOs inhibited SCD-1 expression in CIH-exposed mice to the levels, which were significantly lower than in control animals. Thus, our study has shown that SCD-1 depletion has an antiatherogenic effect during CIH but does not necessarily suggest that SCD-1 is responsible for CIH-induced atherosclerosis. We have also shown that, in human OSA, oxyhemoglobin desaturation is associated with upregulation of hepatic SCD and dyslipidemia, consistent with augmented lipoprotein secretion. Our data imply that SCD-1 ASOs could be a potential candidate for therapeutic use in patients with OSA, dyslipidemia, and atherosclerosis.
Supplementary Material
Acknowledgments
Sources of Funding
This work was supported by National Heart, Lung, and Blood Institute grants HL68715 and HL80105 and American Heart Association Mid-Atlantic Affiliate Grant 0765293U (to V.Y.P.); National Heart, Lung, and Blood Institute grant HL050381 (A.R.S.); National Heart, Lung, and Blood Institute grant HL077137 (to S.P.P.); Pilot and Feasibility grant DK72488 of the Clinical Nutrition Research Unit of Maryland (to V.S.); and American Heart Association Mid-Atlantic Affiliate Postdoctoral Fellowship 0625514U (to J.L.). This work was also supported in part by the Johns Hopkins Bayview General Clinical Research Center (grant M01-RR-02719).
Footnotes
This manuscript was sent to Peter Libby, Consulting Editor, for review by expert referees, editorial decision, and final disposition.
Disclosures S.B. is an employee of Isis Pharmaceuticals and has a commercial interest in the development of SCD-1 anti-sense oligonucleotides.
References
- 1.Gastaut H, Tassinari CA, Duron B. Polygraphic study of the episodic diurnal and nocturnal (hypnic and respiratory) manifestations of the Pickwick syndrome. Brain Res. 1966;1:167–186. doi: 10.1016/0006-8993(66)90117-x. [DOI] [PubMed] [Google Scholar]
- 2.Punjabi NM, Sorkin JD, Katzel LI, Goldberg AP, Schwartz AR, Smith PL. Sleep-disordered breathing and insulin resistance in middle-aged and overweight men. Am J Respir Crit Care Med. 2002;165:677–682. doi: 10.1164/ajrccm.165.5.2104087. [DOI] [PubMed] [Google Scholar]
- 3.Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med. 1993;328:1230–1235. doi: 10.1056/NEJM199304293281704. [DOI] [PubMed] [Google Scholar]
- 4.Marin JM, Carrizo SJ, Vicente E, Agusti AG. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet. 2005;365:1046–1053. doi: 10.1016/S0140-6736(05)71141-7. [DOI] [PubMed] [Google Scholar]
- 5.Shahar E, Whitney CW, Redline S, Lee ET, Newman AB, Javier NF, O'Connor GT, Boland LL, Schwartz JE, Samet JM. Sleep-disordered breathing and cardiovascular disease: cross-sectional results of the Sleep Heart Health Study. Am J Respir Crit Care Med. 2001;163:19–25. doi: 10.1164/ajrccm.163.1.2001008. [DOI] [PubMed] [Google Scholar]
- 6.Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med. 2005;353:2034–2041. doi: 10.1056/NEJMoa043104. [DOI] [PubMed] [Google Scholar]
- 7.McArdle N, Hillman D, Beilin L, Watts G. Metabolic risk factors for vascular disease in obstructive sleep apnea: a matched controlled study. Am J Respir Crit Care Med. 2007;175:190–195. doi: 10.1164/rccm.200602-270OC. [DOI] [PubMed] [Google Scholar]
- 8.Chin K, Nakamura T, Shimizu K, Mishima M, Nakamura T, Miyasaka M, Ohi M. Effects of nasal continuous positive airway pressure on soluble cell adhesion molecules in patients with obstructive sleep apnea syndrome. Am J Med. 2000;109:562–567. doi: 10.1016/s0002-9343(00)00580-5. [DOI] [PubMed] [Google Scholar]
- 9.Robinson GV, Pepperell JC, Segal HC, Davies RJ, Stradling JR. Circulating cardiovascular risk factors in obstructive sleep apnoea: data from randomised controlled trials. Thorax. 2004;59:777–782. doi: 10.1136/thx.2003.018739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lavie L, Vishnevsky A, Lavie P. Evidence for lipid peroxidation in obstructive sleep apnea. Sleep. 2004;27:123–128. [PubMed] [Google Scholar]
- 11.Drager LF, Bortolotto LA, Lorenzi MC, Figueiredo AC, Krieger EM, Lorenzi-Filho G. Early signs of atherosclerosis in obstructive sleep apnea. Am J Respir Crit Care Med. 2005;172:613–618. doi: 10.1164/rccm.200503-340OC. [DOI] [PubMed] [Google Scholar]
- 12.Minoguchi K, Yokoe T, Tazaki T, Minoguchi H, Tanaka A, Oda N, Okada S, Ohta S, Naito H, Adachi M. Increased carotid intima-media thickness and serum inflammatory markers in obstructive sleep apnea. Am J Respir Crit Care Med. 2005;172:625–630. doi: 10.1164/rccm.200412-1652OC. [DOI] [PubMed] [Google Scholar]
- 13.Drager LF, Bortolotto LA, Figueiredo AC, Krieger EM, Lorenzi-Filho G. Effects of CPAP on early signs of atherosclerosis in obstructive sleep apnea. Am J Respir Crit Care Med. 2007;176:706–712. doi: 10.1164/rccm.200703-500OC. [DOI] [PubMed] [Google Scholar]
- 14.Polotsky VY, Rubin AE, Balbir A, Dean T, Smith PL, Schwartz AR, O'Donnell CP. Intermittent hypoxia causes REM sleep deficits and decreases EEG delta power in NREM sleep in the C57BL/6J mouse. Sleep Med. 2006;7:7–16. doi: 10.1016/j.sleep.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 15.Savransky V, Bevans S, Nanayakkara A, Li J, Smith PL, Torbenson MS, Polotsky VY. Chronic intermittent hypoxia causes hepatitis in a mouse model of diet-induced fatty liver. Am J Physiol Gastrointest Liver Physiol. 2007;293:G871–G877. doi: 10.1152/ajpgi.00145.2007. [DOI] [PubMed] [Google Scholar]
- 16.Savransky V, Nanayakkara A, Li J, Bevans S, Smith PL, Rodriguez A, Polotsky VY. Chronic intermittent hypoxia induces atherosclerosis. Am J Respir Crit Care Med. 2007;175:1290–1297. doi: 10.1164/rccm.200612-1771OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J, Savransky V, Nanayakkara A, Smith PL, O'Donnell CP, Polotsky VY. Hyperlipidemia and lipid peroxidation are dependent on the severity of chronic intermittent hypoxia. J Appl Physiol. 2007;102:557–563. doi: 10.1152/japplphysiol.01081.2006. [DOI] [PubMed] [Google Scholar]
- 18.Li J, Thorne LN, Punjabi NM, Sun CK, Schwartz AR, Smith PL, Marino RL, Rodriguez A, Hubbard WC, O'Donnell CP, Polotsky VY. Intermittent hypoxia induces hyperlipidemia in lean mice. Circ Res. 2005;97:698–706. doi: 10.1161/01.RES.0000183879.60089.a9. [DOI] [PubMed] [Google Scholar]
- 19.Sampath H, Ntambi JM. Stearoyl-coenzyme A desaturase 1, sterol regulatory element binding protein-1c and peroxisome proliferator-activated receptor-alpha: independent and interactive roles in the regulation of lipid metabolism. Curr Opin Clin Nutr Metab Care. 2006;9:84–88. doi: 10.1097/01.mco.0000214564.59815.af. [DOI] [PubMed] [Google Scholar]
- 20.Miyazaki M, Kim YC, Gray-Keller MP, Attie AD, Ntambi JM. The biosynthesis of hepatic cholesterol esters and triglycerides is impaired in mice with a disruption of the gene for stearoyl-CoA desaturase 1. J Biol Chem. 2000;275:30132–30138. doi: 10.1074/jbc.M005488200. [DOI] [PubMed] [Google Scholar]
- 21.Ntambi JM, Miyazaki M. Regulation of stearoyl-CoA desaturases and role in metabolism. Prog Lipid Res. 2004;43:91–104. doi: 10.1016/s0163-7827(03)00039-0. [DOI] [PubMed] [Google Scholar]
- 22.Tabor DE, Kim JB, Spiegelman BM, Edwards PA. Identification of conserved cis-elements and transcription factors required for sterol-regulated transcription of stearoyl-CoA desaturase 1 and 2. J Biol Chem. 1999;274:20603–20610. doi: 10.1074/jbc.274.29.20603. [DOI] [PubMed] [Google Scholar]
- 23.Cohen P, Miyazaki M, Socci ND, Hagge-Greenberg A, Liedtke W, Soukas AA, Sharma R, Hudgins LC, Ntambi JM, Friedman JM. Role for stearoyl-CoA desaturase-1 in leptin-mediated weight loss. Science. 2002;297:240–243. doi: 10.1126/science.1071527. [DOI] [PubMed] [Google Scholar]
- 24.Jiang G, Li Z, Liu F, Ellsworth K, Dallas-Yang Q, Wu M, Ronan J, Esau C, Murphy C, Szalkowski D, Bergeron R, Doebber T, Zhang BB. Prevention of obesity in mice by antisense oligonucleotide inhibitors of stearoyl-CoA desaturase-1. J Clin Invest. 2005;115:1030–1038. doi: 10.1172/JCI23962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gutierrez-Juarez R, Pocai A, Mulas C, Ono H, Bhanot S, Monia BP, Rossetti L. Critical role of stearoyl-CoA desaturase-1 (SCD1) in the onset of diet-induced hepatic insulin resistance. J Clin Invest. 2006;116:1686–1695. doi: 10.1172/JCI26991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.MacDonald ML, Singaraja RR, Bissada N, Ruddle P, Watts R, Karasinska JM, Gibson WT, Fievet C, Vance JE, Staels B, Hayden MR. Absence of stearoyl-CoA desaturase-1 ameliorates features of the metabolic syndrome in LDLR-deficient mice. J Lipid Res. 2008;49:217–229. doi: 10.1194/jlr.M700478-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li J, Grigoryev DN, Ye SQ, Thorne L, Schwartz AR, Smith PL, O'Donnell CP, Polotsky VY. Chronic intermittent hypoxia upregulates genes of lipid biosynthesis in obese mice. J Appl Physiol. 2005;99:1643–1648. doi: 10.1152/japplphysiol.00522.2005. [DOI] [PubMed] [Google Scholar]
- 28.Bostrom P, Magnusson B, Svensson PA, Wiklund O, Boren J, Carlsson LM, Stahlman M, Olofsson SO, Hulten LM. Hypoxia converts human macrophages into triglyceride-loaded foam cells. Arterioscler Thromb Vasc Biol. 2006;26:1871–1876. doi: 10.1161/01.ATV.0000229665.78997.0b. [DOI] [PubMed] [Google Scholar]
- 29.Li J, Nanayakkara A, Jun J, Savransky V, Polotsky VY. The Effect of deficiency in SREBP cleavage-activating protein (SCAP) on lipid metabolism during intermittent hypoxia. Physiol Genomics. 2007;31:273–280. doi: 10.1152/physiolgenomics.00082.2007. [DOI] [PubMed] [Google Scholar]
- 30.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsuda M, Korn BS, Hammer RE, Moon YA, Komuro R, Horton JD, Goldstein JL, Brown MS, Shimomura I. SREBP cleavage-activating protein (SCAP) is required for increased lipid synthesis in liver induced by cholesterol deprivation and insulin elevation. Genes Dev. 2001;15:1206–1216. doi: 10.1101/gad.891301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hughes AL, Todd BL, Espenshade PJ. SREBP pathway responds to sterols and functions as an oxygen sensor in fission yeast. Cell. 2005;120:831–842. doi: 10.1016/j.cell.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 33.Semenza GL, Prabhakar NR. HIF-1-dependent respiratory, cardiovascular, and redox responses to chronic intermittent hypoxia. Antioxid Redox Signal. 2007;9:1391–1396. doi: 10.1089/ars.2007.1691. [DOI] [PubMed] [Google Scholar]
- 34.Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 35.Semenza GL. Regulation of physiological responses to continuous and intermittent hypoxia by hypoxia-inducible factor 1. Exp Physiol. 2006;91:803–806. doi: 10.1113/expphysiol.2006.033498. [DOI] [PubMed] [Google Scholar]
- 36.Li J, Bosch-Marce M, Nanayakkara A, Savransky V, Fried SK, Semenza GL, Polotsky VY. Altered metabolic responses to intermittent hypoxia in mice with partial deficiency of hypoxia-inducible factor-1alpha. Physiol Genomics. 2006;25:450–457. doi: 10.1152/physiolgenomics.00293.2005. [DOI] [PubMed] [Google Scholar]
- 37.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang GL, Semenza GL. Purification and characterization of hypoxiainducible factor 1. J Biol Chem. 1995;270:1230–1237. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 39.Rankin EB, Biju MP, Liu Q, Unger TL, Rha J, Johnson RS, Simon MC, Keith B, Haase VH. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J Clin Invest. 2007;117:1068–1077. doi: 10.1172/JCI30117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walmsley SR, McGovern NN, Whyte MK, Chilvers ER. The HIF/VHL pathway: from oxygen sensing to innate immunity. Am J Respir Cell Mol Biol. 2008;38:251–255. doi: 10.1165/rcmb.2007-0331TR. [DOI] [PubMed] [Google Scholar]
- 41.Yuan G, Nanduri J, Bhasker CR, Semenza GL, Prabhakar NR. Ca2+/calmodulin kinase-dependent activation of hypoxia inducible factor 1 transcriptional activity in cells subjected to intermittent hypoxia. J Biol Chem. 2005;280:4321–4328. doi: 10.1074/jbc.M407706200. [DOI] [PubMed] [Google Scholar]
- 42.Peng YJ, Yuan G, Ramakrishnan D, Sharma SD, Bosch-Marce M, Kumar GK, Semenza GL, Prabhakar NR. Heterozygous HIF-1{alpha} deficiency impairs carotid body-mediated systemic responses and reactive oxygen species generation in mice exposed to intermittent hypoxia. J Physiol. 2006;577:705–716. doi: 10.1113/jphysiol.2006.114033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vink A, Schoneveld AH, Lamers D, Houben AJ, van der GP, van Diest PJ, Pasterkamp G. HIF-1 alpha expression is associated with an atheromatous inflammatory plaque phenotype and upregulated in activated macrophages. Atherosclerosis. 2007;195:e69–e75. doi: 10.1016/j.atherosclerosis.2007.05.026. [DOI] [PubMed] [Google Scholar]
- 45.Chu K, Miyazaki M, Man WC, Ntambi JM. Stearoyl-coenzyme A desaturase 1 deficiency protects against hypertriglyceridemia and increases plasma high-density lipoprotein cholesterol induced by liver X receptor activation. Mol Cell Biol. 2006;26:6786–6798. doi: 10.1128/MCB.00077-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ntambi JM, Miyazaki M. Recent insights into stearoyl-CoA desaturase-1. Curr Opin Lipidol. 2003;14:255–261. doi: 10.1097/00041433-200306000-00005. [DOI] [PubMed] [Google Scholar]
- 47.Attie AD, Krauss RM, Gray-Keller MP, Brownlie A, Miyazaki M, Kastelein JJ, Lusis AJ, Stalenhoef AF, Stoehr JP, Hayden MR, Ntambi JM. Relationship between stearoyl-CoA desaturase activity and plasma triglycerides in human and mouse hypertriglyceridemia. J Lipid Res. 2002;43:1899–1907. doi: 10.1194/jlr.m200189-jlr200. [DOI] [PubMed] [Google Scholar]
- 48.Flowers MT, Groen AK, Oler AT, Keller MP, Choi Y, Schueler KL, Richards OC, Lan H, Miyazaki M, Kuipers F, Kendziorski CM, Ntambi JM, Attie AD. Cholestasis and hypercholesterolemia in SCD1-deficient mice fed a low-fat, high-carbohydrate diet. J Lipid Res. 2006;47:2668–2680. doi: 10.1194/jlr.M600203-JLR200. [DOI] [PubMed] [Google Scholar]
- 49.Wang Y, Kurdi-Haidar B, Oram JF. LXR-mediated activation of macrophage stearoyl-CoA desaturase generates unsaturated fatty acids that destabilize ABCA1. J Lipid Res. 2004;45:972–980. doi: 10.1194/jlr.M400011-JLR200. [DOI] [PubMed] [Google Scholar]
- 50.Ntambi JM, Miyazaki M, Stoehr JP, Lan H, Kendziorski CM, Yandell BS, Song Y, Cohen P, Friedman JM, Attie AD. Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proc Natl Acad Sci USA. 2002;99:11482–11486. doi: 10.1073/pnas.132384699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dobrzyn P, Dobrzyn A, Miyazaki M, Cohen P, Asilmaz E, Hardie DG, Friedman JM, Ntambi JM. Stearoyl-CoA desaturase 1 deficiency increases fatty acid oxidation by activating AMP-activated protein kinase in liver. Proc Natl Acad Sci U S A. 2004;101:6409–6414. doi: 10.1073/pnas.0401627101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de Winther MP, Kanters E, Kraal G, Hofker MH. Nuclear factor kappaB signaling in atherogenesis. Arterioscler Thromb Vasc Biol. 2005;25:904–914. doi: 10.1161/01.ATV.0000160340.72641.87. [DOI] [PubMed] [Google Scholar]
- 53.Stocker R, Keaney JF., Jr. Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84:1381–1478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.