

Abstract
Objective
To design a new class of selective neuronal nitric oxide synthase (nNOS) inhibitors and demonstrate that administration in a rabbit model for cerebral palsy (CP) prevents hypoxia-ischemia induced deaths and reduces the number of newborn kits exhibiting signs of CP.
Methods
We used a novel computer-based drug design method called fragment hopping to identify new chemical entities, synthesized them, carried out in vitro enzyme inhibition studies with the three isozymes of NOS and in vivo experiments to monitor cardiovascular effects on pregnant rabbit dams, NOS activity and NOx concentration in fetal brain, and assess neurobehavioral effects on kits born to saline- and compound treated dams.
Results
The computer-based design led to the development of powerful and highly selective compounds for inhibition of nNOS over the other isozymes. Following maternal administration in a rabbit model of CP, these compounds were found to distribute to fetal brain, to be non-toxic, without cardiovascular effects, inhibit fetal brain NOS activity in vivo, reduce NO concentration in fetal brain, and dramatically ameliorate deaths and number of newborn kits exhibiting signs of CP.
Interpretation
This approach may lead to new preventive strategies for cerebral palsy.
Cerebral palsy is one of the most severe consequences of hypoxia-ischemia (HI) before birth and is common in premature infants, with 750,000 persons affected in the USA1. It has one of the highest indices of disease burden with direct effects on individual, family, and social institutions (annual cost $8.2 billion2) that last the entire lifetime. There is no known treatment to protect the fetus from hypoxic brain injury leading to cerebral palsy3, despite a reduction in the mortality of high-risk infants 4. Prenatal or fetal HI brain injury has been strongly implicated in the subsequent development of cerebral palsy in premature5 and full-term infants6,7.
Nitric oxide synthase (NOS) comprises a family of enzymes that produces nitric oxide (NO), including neuronal (nNOS), macrophage or inducible (iNOS), and endothelial (eNOS) isozymes. Neuronal NOS knockout neonatal animals are protected from focal HI-induced histopathological brain damage8; elimination of nNOS neurons prior to HI also confers resistance to focal HI-induced histopathological brain damage9. Focal HI-induced histopathological brain damage and locomotor deficits in iNOS knockout animals also are reduced10; however, the expression of nNOS, but not iNOS, is increased dramatically after cerebral HI in the newborn rat11. NO generated by eNOS plays an important role in maintaining blood flow and blood pressure. Animals lacking the eNOS gene have enlarged cerebral infarcts after HI12. Potent nNOS inhibitors that also inhibit eNOS (such as l-Nω-nitroarginine methyl ester) cause sharp increases in blood pressure13. Inhibition of iNOS may negatively affect bacteriocidal, immunoregulatory14 and ischemic preconditioning responses15. These harmful consequences of inhibition of eNOS and iNOS make the present nNOS inhibitors unsatisfactory due to their lack of selectivity and specificity for nNOS.
Using a clinically relevant model of acute placental insufficiency and global fetal HI that results in motor deficits resembling cerebral palsy in humans16,17, we sought a comprehensive approach to identify small molecules that could decrease nNOS-generated NO in HI brain and decrease the motor deficits of cerebral palsy. This therapy would have profound implications for babies at risk for cerebral palsy. Our goal was to develop isoform selective-compounds to control the NO overproduction by nNOS while leaving the microbicidal and immunoregulatory function of iNOS and the vasoprotective function of eNOS undisturbed18,19. We also targeted molecules that were reversible inhibitors of nNOS to prevent potential side effects of irreversible inhibitors, which usually are compounds that react with multiple biological targets. These strategies should allow safe administration to mothers for the treatment of fetal brain HI and prevention of cerebral palsy.
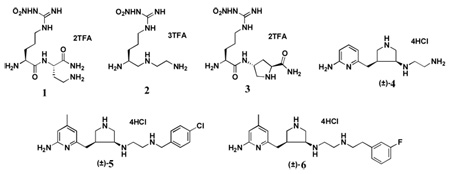
l-Nω-Nitroarginine is well known to be a potent competitive inhibitor of nNOS that binds to the active site of the enzyme and blocks substrate binding, but it shows little selectivity over eNOS20. The methyl ester of l-Nω-nitroarginine, which is a prodrug for L-nitroarginine and is activated by esterase-catalyzed hydrolysis to l-Nω-nitroarginine,21 causes severe hypertension.13 We synthesized dipeptide amides and peptidomimetics built on an l-Nω-nitroarginine scaffold, which has selectivity for nNOS over iNOS, in search of analogues that might have extended binding interactions to differentiate nNOS from eNOS22,23. As a result of these studies, 1–3 (Table 1) were found to be the first highly dual-selective inhibitors of nNOS over both eNOS and iNOS, which should minimize side effects associated with inhibition of those isozymes. Our previous research showed that a single-residue difference in the active site, rat nNOS D597 versus bovine eNOS N368, accounts for a majority of the selectivity of nNOS over eNOS by these compounds24,25. Recently, we developed a new fragment-based de novo design approach termed fragment hopping26. The core of this approach is the determination of the minimal pharmacophoric elements; from these elements, new fragments can be generated and then linked to produce potent molecules. Fragment hopping can explore a much wider chemical diversity space compared with standard fragment-based screening27 and can identify and utilize not only binding sites used to enhance potency but also to identify the specific regions for isozyme selectivity25. Starting from the above studies, a class of potent and highly selective inhibitors of nNOS with new chemical structures and increased lipophilicity was successfully identified. These compounds dramatically diminished the incidence of perinatal deaths and number of newborn rabbit kits exhibiting the signs of cerebral palsy and showed no cardiovascular effects or toxicities, suggesting that a preventive strategy for cerebral palsy may be feasible for humans.
Table 1.
Chemical structures of nNOS inhibitors, in vitro NOS inhibition, and the corresponding physicochemical data related to inhibition absorption and biomembrane permeability. All of the NOS isoforms used were recombinant enzymes overexpressed in E.coli and purified as described previously.22,23 Nitric oxide formation from NOS was monitored by the hemoglobin capture assay as described.22,23 NOS assays were recorded on a Perkin–Elmer Lamda 10 UV–visible spectrophotometer. The apparent Ki values were obtained by measuring the percent enzyme inhibition in the presence of 10 µM l-arginine with at least five concentrations of inhibitor. The parameters of the following inhibition equation22 were fitted to the initial velocity data: % inhibition = 100[I]/{[I] + Ki(1+[S]/Km)}. Km values for l-arginine were 1.3 µM (nNOS), 8.2 µM (iNOS), and 1.7 µM (eNOS). The selectivity of an inhibitor was defined as the ratio of the respective Ki values.
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
Ki (µM) |
Selectivity |
ClogP | LogD7.4 | TPSAa | HBAb | HBDc | RBd | ||||
| nNOS | eNOS | iNOS | n/e | n/i | |||||||
| l-NNA | 0.57 | 0.75 | 4.55 | 1.3 | 8 | ||||||
| 1 | 0.13 | 200 | 25 | 1538 | 192 | −7.00 | −5.42 | 224.0 | 10 | 10 | 11 |
| 2 | 0.12 | 314 | 39 | 2577 | 320 | −5.79 | −5.88 | 163.8 | 7 | 8 | 10 |
| 3 | 0.10 | 128 | 29 | 1280 | 290 | −6.27 | −6.57 | 210.0 | 10 | 9 | 8 |
| (±)−4 | 0.388 | 434.5 | 58.4 | 1114 | 150 | −1.33 | −5.16 | 101.0 | 5 | 6 | 5 |
| (±)−5 | 0.085 | 85.2 | 9 | 1002 | 106 | 2.58 | −1.81 | 74.5 | 5 | 5 | 8 |
| (±)−6 | 0.014 | 28 | 4.1 | 2000 | 293 | 2.19 | −2.36 | 74.5 | 5 | 5 | 9 |
topological polar surface area (Å2)
Number of hydrogen bond acceptors
Number of hydrogen bond donors
rotatable bonds
Materials and Methods
Structure-based inhibitor design and synthesis
A novel computer modeling methodology called fragment hopping26 was employed in the structure-based inhibitor design. ClogP values for the inhibitors were calculated by ClogP program version 4.2 (BioByte Corp.http://www.biobyte.com/). Log D values for the inhibitors were calculated by ACD/logD at pH 7.4 (ACD/log D suite, Advanced Chemistry Development Inc. http://www.acdlabs.com). The topological molecular polar surface area (TPSA) was calculated by the atom-based method28. The synthetic route and full experimental procedures are given in the Supplementary Information.
X-ray diffraction data collection and crystal structure determination
The X-ray diffraction data were collected under a liquid nitrogen stream (100 K) with CCD detectors either at Stanford Synchrotron Radiation Laboratory (SSRL, Menlo Park, CA) or at Advanced Light Source (ALS, Berkeley, CA).24,29,30 Raw data were processed with HKL2000. The binding of inhibitor was detected by difference Fourier synthesis. The inhibitor then was modeled into the density using O31 and refined with CNS32. Water molecules were added automatically with CNS and inspected visually in O.
Animal Model
Global HI of fetuses was induced in vivo by uterine ischemia using an aortic balloon catheter in timed pregnant New Zealand white rabbits as published previously17. Blood pressure measurements were made in the right leg. Saline, 5, or 6 were administered to E22 rabbit dams 30 min prior to and 30 min after 40 min of uterine ischemia via the intra-aortic balloon catheter. E22 fetus corresponds to 22–27 weeks in humans from oligodendroglial studies and MRI estimations of white matter maturity. The live newborn kits (postnatal day one; P1) were grouped into severe (postural deficits and/or hypertonia), mild (absence of hypertonia but with other abnormalities), or normal categories based on a standardized neurobehavioral examination16,17.
Inhibition of nNOS
The calcium-dependent NOS activity in brain tissue was measured by following the conversion of [14C]-L-arginine-to [14C]-L-citrulline. NOx were measured using a Sievers 280 Nitric Oxide Chemiluminescent Analyzer (Sievers Instruments, Boulder, CO, USA).
Statistical Analysis
Groups were compared with the Student t-test or Fisher’s exact test and Kendall’s tau-b using SAS version 9.1 for Windows.
Results
Structure-based design of nNOS inhibitors
A pharmacophore-driven strategy for fragment-based de novo design, fragment hopping, which is able to utilize minute structural differences among the NOS isoforms26, was used to design non-peptide small molecule inhibitors. Compounds 5 and 6 were generated from the computer modeling studies and synthesized according to the synthetic route shown in Supplementary Scheme 1. All of the chemical structures of the intermediates and final products were confirmed by 1H NMR, 13CNMR, high-resolution mass spectra, and elemental analysis. All of the products are racemic mixtures.
NOS inhibition and X-ray crystallographic analysis
The evolution of the design of specific inhibitors is found in the kinetic data and the corresponding physicochemical properties of 1–6 shown in Table 1. When compared to peptidomimetic compounds 1–3, heterocyclic compounds 5 and 6 show higher inhibitory potency for nNOS. The nNOS selectivities of 5 and 6 over eNOS and iNOS are similar to those of 1–3. Compared to 4, compounds 5 and 6 show much higher nNOS inhibitory activity and possess similar or better nNOS/eNOS selectivity.
The racemic mixture of 5 was used in co-crystallization with rat nNOS (Figure 1; see Supplemental Material Table 1 for the X-ray data collection and structure refinement statistics).
Figure 1.



Comparison of the binding modes of 3 and 5 in nNOS. The atomic coordinates for Figure 1 have been deposited in the Protein Data Bank, accession number 3B3O for Figure 1A. The data collection and refinement statistics are summarized in Supporting Information Table 1. AutoDock 3.0.5 was employed to perform the docking calculations.38 For the protein structure (PDB code 1P6J24), polar hydrogen atoms were added, and Kollman united atom charges were assigned45. Hydrogens were also added to the heme and H4B, and charges were calculated by the Gasteiger–Marsili method46. The charge of the Fe atom bound to heme was assigned +3. The nonpolar hydrogen atoms of heme and H4B were removed manually and their charges were united with the bonded carbon atoms. The 3D structures of the ligands were built and partial atomic charges were also calculated using the Gasteiger–Marsili method. The rotatable bonds in the ligands were defined using another AutoDock 3.0 auxiliary program, AutoTors, which also unites the nonpolar hydrogens and partial atomic charges to the bonded carbon atoms. The grid maps were calculated using AutoGrid. The dimensions of the grid box encompass all of the active site residues shown in Figure 1, and the grid spacing was set to 0.375 Å. Docking was performed using the Lamarckian genetic algorithm (LGA), and the pseudo-Solis and Wets method were applied for the local search. The detailed procedure used has been described previously25. The structural waters that are involved in inhibitor binding are included. (A) Binding conformation of 5 in nNOS (PDB id: 3B3O). The Fo – Fc omit electron density for the inhibitor is shown contoured at 3.0σ; (B) Superimposition of the predicted conformation of 5 (gold) and the actual binding conformation (crystal structure) of 5 (blue); (C) Binding conformation of 3 in the crystal structure of nNOS (PDB id: 1P6J24).
Blood pressure homeostasis
Because of the importance of eNOS to the regulation of cardiovascular activity, the effect of maternal administration of 5 and 6 on blood pressure and heart rate in 22 day-gestation (E22) New Zealand white rabbit dams was determined. Figure 2 shows there was no effect on heart rate or on systolic or diastolic blood pressure by treatment with 6 (data for 5 are similar and are not shown).
Figure 2.

Effect of 6 on heart rate and blood pressure in New Zealand white rabbits. Blood pressure and heart rate were measured intermittently every 5 min with a blood pressure instrument (Sensor Devices, Waukesha, WI) on the right leg. Each drug group contained 8 dams. The heart rate (HR), systolic (sBp) and diastolic (dBp) blood pressure were measured in the right leg every 10 min. After a period of baseline measurements, 6 (0.204 mg in 30 ml saline) or saline (30 ml) was administered over a period of 30 min into the lower aorta below the renal arteries and above the uterine arteries. A balloon catheter was inserted and then 40 min of uterine ischemia was induced by inflation of the balloon catheter in the lower aorta. The balloon catheter was removed and then 6 in 30 ml of saline (or 30 ml saline) was administered over a period of 30 min into the lower aorta as before.
In vivo NOS inhibition and NOx production
Since NO is always present in brain and is a substrate for reactive nitrogen species produced by HI, we investigated the mechanism of injury from pre-existing NO by lowering NO levels at the start of HI using nNOS-selective inhibitors prior to HI. The NOS activity and NOx levels in fetal brain were determined following maternal administration of 6 to E22 rabbit dams 30 min before and 30 min after HI to (1) show 6 permeates the fetal brain, and (2) that 6 inhibits NOS activity in the fetal brain. The NOS activity assay in fetal brain homogenates assessed total constitutive NOS activity (i.e., both nNOS and eNOS). However, the in vitro studies indicated that eNOS is very poorly inhibited by compound 6. This also was demonstrated by the lack of in vivo effects on blood pressure. Therefore, changes in NOS activity correspond to changes in nNOS activity. As shown in Figure 3 (A, B), NOS inhibition was observed in fetal brain after acute treatment with 6 was completed. The corresponding decrease in NOx concentration (Figure 3C, D) also indicated strong NOS inhibition had occurred.
Figure 3.

Effect on nitric oxide synthase and nitric oxide concentration in fetal rabbit brain after maternal administration of 6 to the distal aorta of New Zealand white rabbit dams at 22 days gestation (E22, term is 31.5 days). The calcium-dependent NOS activity in brain tissue was measured by following the conversion of [14C]-L-arginine-to [14C]-L-citrulline. Briefly, ~250 mg of tissue was homogenized in 250 µl of chilled 50 mM Hepes buffer (pH 7.4) containing a cocktail of protease inhibitors (Complete, Roche). After precipitating tissue debris by centrifugation at 12,500 rpm for 10 min, the supernatant was transferred to a clean tube. A 50 µl aliquot was added to a freshly prepared 150 µl solution containing 50 mM HEPES buffer (pH 7.4) containing 0.1 mM L-arginine (313 µCi/µmol), calcium (0.2 mM), calmodulin (20 µg/ml), 0.5 mM NADPH, and 10 µM tetrahydrobiopterin. The reaction mixture was incubated for 0, 30, and 60 min at 37 °C, and the reaction was stopped with 100 µl of a cold solution of 200 mM HEPES, pH 5.5, containing 10 mM EGTA in an ice bath. [14C]-L-citrulline was isolated from the incubation mixtures using a Dowex 50 (Na+ form) column eluted with double distilled water, and the activity was determined after counting in a scintillation counter. Values were normalized per mg protein of supernatant. NOx were measured using a Sievers 280 Nitric Oxide Chemiluminescent Analyzer (Sievers Instruments, Boulder, CO, USA). Acid and reducing agents (vanadium III chloride in 1M hydrochloric acid) at 95 °C were added to brain samples, converting NO3− and NO2− to NO. An inert gas was then used to purge NO from the solution, which was detected by chemiluminescence, with a sensitivity limit of detection of 20 nmol. Standard curves were constructed, and the amount of NO3− and NO2− in brain was determined. Measurements of constitutive NOS activity (A, B) and NOx concentration (C, D) (in nmol/mg protein) were made either immediately after hypoxia (A and C) or after 24 h (B and D). *Student t test: p < 0.050 for the acute NOS activity; p < 0.020 for NOx at the acute time point; and p < 0.007 for NOx at the 24 h time point.
Neurobehavioral assessment
We investigated whether compounds 5 and 6 could ameliorate the postnatal motor deficits suggestive of cerebral palsy (Figure 4). The most striking difference was that the P1 kits from saline-treated dams had a large incidence of discovered fetal/neonatal deaths (16/34; 47%) but no deaths (0/49) were observed from 5- and 6-treated animals. Of the kits from saline-treated dams that came to term (18/34), severe neurobehavioral abnormalities occurred in 12/18 (67%) compared to only 7/49 (14%) in those from dams treated with 5 or 6. Furthermore, the 5- and 6-treated animals exhibited a remarkably larger number (83 and 69%, respectively) of normal postnatal P1 kits (in two litters, all 19 kits were normal); only 9% (3/34) of the kits from saline-treated dams were born normal. Kit gender did not have an affect on the degree of neurobehavioral abnormality. None of the compounds caused any detectable changes in vital signs, blood pressure, or apparent changes in general appearance in the rabbit dams. When 6 was administered after HI, there was no significant improvement in motor deficits or deaths: 2 normal, 2 mild, 7 severe and 20 stillbirths.
Figure 4.

Neurobehavioral assessment of rabbit kits exposed to hypoxia-ischemia (HI) and treated with compounds 5 and 6. Global HI of fetuses was induced in vivo by uterine ischemia in timed pregnant New Zealand white rabbits (Myrtle’s Rabbits, Thompson Station, Tennessee) at 70% gestation (E22) as described previously16,17. Briefly, the dams were anesthetized with intravenous fentanyl (75 µg/kg/h) and droperidol (3.75 mg/kg/h), followed by spinal anesthesia with 0.75% bupivicaine. An arterial balloon catheter was introduced into the left femoral artery and advanced 10 cm into the descending aorta. Drugs or saline were administered via this catheter for 30 min. The balloon was then inflated for 40 min, inducing continuous uterine ischemia, which resulted in fetal HI. At the end of the ischemia, the balloon was deflated, and the drugs or saline were administered for another 30 min. The catheter was then removed. The dams were allowed to deliver in a nest box at term (31.5 days). Each newborn kit’s behavior was videotaped and scored by two blinded observers as published previously16,17. A videotape of the results is provided in the Supplementary Material. Tactile sensation was tested by stroking the side of the kit’s face with a cotton swab. Locomotion on a flat surface was assessed by grading the amount of spontaneous movement of head, trunk and limbs. Tone was assessed by active flexion and extension of the fore and hind limbs and scored (0–4) using a modified Ashworth scale. The righting reflex was assessed when the kits were placed on their backs. Suck and swallow were assessed by introduction of formula (Similac with iron®, Abbott Labs, Abbott Park, IL) into the kit’s mouth with a plastic pipette. Olfaction was tested by aversive response to a cotton swab soaked with peppermint or pure ethanol. Compounds 5 and 6 were administered to E22 dams via the balloon catheter at 75 times the Ki. Neurobehavioral examination was conducted at postnatal day 1 (P1 = E32), and categorization was made principally on the presence of postural deficits, hypertonia, and other neurobehavioral abnormalities16,17. Both 5 and 6 were significantly different from saline (p>0.0001 for all adverse outcome and also for outcome in survivors for both drugs, Fisher’s exact test). Kendall’s tau-b 95% confidence intervals for protection against any adverse outcome, including death, was 0.537–0.806 and 0.537–0.790; and for severity of motor deficits in survivors was 0.352 to 0.836 and 0.333–0.785 for 5 and 6, respectively.
Discussion
The aim of the structure-based design described here was to identify highly potent and nNOS-selective inhibitors, which would allow for much smaller doses to be administered, leading to fewer side effects and decreased cardiovascular toxicity. The crystal structures of the oxygenase domains for all three isoforms of NOS are now known33, which opened the way for structure-based inhibitor design. There are three key challenges to nNOS inhibitor design: (1) Selectivity: the active sites of NOS isozymes are highly conserved. Sixteen out of 18 residues within 6 Å of the substrate in the active site are identical (Supplemental Material Figure 1), and the side chain of one of the dissimilar amino acids of nNOS, namely S585 (the numbering of residues is based on rat nNOS if not specified), points out of the substrate-binding site, implying that, within that space, only one residue is different between nNOS and eNOS. We previously found that this single-residue difference in the active site, rat nNOS D597 versus bovine eNOS N368, accounts for a majority of the selectivity of nNOS over eNOS24,25,29 (2) Inhibitor potency: the very large active site of nNOS, 1000 Å3~1100 Å3, makes it difficult to design an inhibitor that can contact the protein surface well enough to produce good binding; (3) Permeability: 18 of 30 residues/cofactor side chains that point into the active site of NOS are polar or charged, and clusters of acidic residues/cofactor side chains, especially residues E592 and D597, the heme propionate groups, and two low pKa polar side chains (residues Y562 and Y588), form a strongly acidic local environment (Supplemental Material Figure 2). This environment requires the inhibitor to contain positively charged electrostatic or hydrogen bond donor groups, such as amines, which creates a problem in designing a cell-permeable exogenous inhibitor. Multiple numbers of positively charged hydrogen bond donors are always unfavorable for diffusion through biomembranes, like the blood brain barrier (BBB)34.
To enhance the potency, isozyme selectivity, and potential bioavailability of these compounds, we developed fragment hopping,26 a novel fragment-based de novo design approach, which generated a class of potent and highly selective inhibitors of nNOS with new chemical structures. Residues in the active site of nNOS critical for ligand binding were analyzed by Multiple Copy Simultaneous Search (MCSS)35 and GRID36. The structural differences in the active site among the three NOS isoforms were analyzed by the GRID/CPCA method25. The minimal pharmacophoric elements were then extracted according to the results of the GRID and MCSS analyses. The fragments that match the minimal pharmacophoric elements were generated, and the molecules were constructed using LUDI library design and LUDI fragment connection37. The designed molecules were docked into the active site using the AutoDock 3.0.5 program38, and the binding scores were evaluated with the commercially-available Cscore program (SYBYL Molecular Modeling Package, available at http://www.tripos.com). Finally, a property-based drug design strategy was used to evaluate the ADME/Tox effect of the molecules39. If the binding score and/or property score of the molecules did not meet the requirements, the molecule was re-constructed, re-docked, and re-scored, until the new molecules gave satisfactory results. Compounds 5 and 6 were generated from the above computer modeling studies. The pKa value of the 2-amino-4,6-dimethylpyridine moiety is 7.140; therefore, this fragment could act as a charge switch. In the small intestine the fragment could be in its neutral form, which is favorable for absorption; in the NOS active site the local acidic environment (rat nNOS E592) would convert it into the protonated (positively charged) form, which is favorable for binding. The pyrrolidine fragment was chosen as the substitute for the α-amino group of 1–3 for two reasons. First, inhibitors 1–3 adopt distinctly different conformations in eNOS and nNOS24. By replacing the inhibitor α-amino group with a rigid pyrrolidine secondary amine, the pyrrolidine amino group is locked in a conformation for enhanced interactions with D597, favoring nNOS interaction. Second, a secondary amino group is more lipophilic and has less polar surface area, compared to a primary amino group, which is better for in vivo inhibitor delivery28. The ethylenediamine fragment was chosen to form an electrostatic interaction and hydrogen bond with the heme propionate groups. Another reason to choose this fragment is because the nitrogen atom in the pyrrolidine and the terminal nitrogen atom of the ethylenediamine fragment are expected to be higher pKa groups and, therefore, are protonated and positively charged; this causes the nitrogen atom in the middle to have a low pKa and to be neutral, which is favorable for the inhibitor to permeate biomembranes. The introduction of halogen-substituted phenyl groups in 5 and 6 increases the lipophilicity and decreases the polar surface area, making them much more favorable for biomembrane permeability, compared to the primary amino group in 428. On the other hand, the introduction of halogen atoms at the para or meta positions of the phenyl group blocks/decreases metabolic degradation of the phenyl group41.
The racemic mixture of 5 was used in cocrystallization with rat nNOS. As expected, only one of the cis enantiomers, the (3’S, 4’S)-isomer, was bound to the active site. The binding conformation of 5 in the crystal structure is very similar to that predicted in the fragment-based de novo design (Figure 1B) and is very similar to the binding modes of 1–3 (Figure 1C). The 2-amino-4-methylpyridine group of 5 forms two H-bonds and electrostatic interactions with the carboxylic acid of E592, just as the nitroguanidino groups of 1–3 do. The nitrogen atom of the pyrrolidine ring of 5 forms a direct electrostatic interaction with E592 and is involved in a H-bond network with the carboxylic acid of D597 via two structural water molecules (Figure 1B), just as the α-amino groups of 1–3 do (Figure 1C). The NH group of the ethylenediamine fragment of 5 that is attached to the pyrrolidine ring forms a hydrogen bond to the heme propionate group, just as the amido NH groups of 1 and 3 and the secondary amino group of 2 do. The other nitrogen atom of the ethylenediamine fragment of 5 is involved in electrostatic interactions with heme propionate groups. The predicted physicochemical properties of 5 and 6, which are related to biomembrane permeability and absorption, are superior to those of the dipeptide amide compounds 1–3 and the prototype molecule 4 (Table 1) and meet the basic virtual requirements for oral bioavailability and transport ability through the BBB. This suggests that 5 and 6 evolved well from test molecule 4 and might have better activities in vivo35.
Since identification of fetal HI brain injury is still a huge problem, a preventive strategy for treating mothers may be immediately feasible. A preventive drug has to have a high therapeutic index, and have minimal side effects. Because of the high selectivity of 5 and 6 for nNOS relative to eNOS (1000- and 2000-fold, respectively), there are no detrimental cardiovascular effects (Figure 2). Neither the heart rate nor the blood pressures of the dams was affected by these compounds, unlike the cardiovascular effects observed with poorly selective compounds, such as l-Nω-nitroarginine methyl ester (L-NAME)13 and 7-nitroindazole42. This large therapeutic index of 5 and 6 offers an exciting possibility of a preventive strategy for pregnant mothers showing signs of some fetal distress.
NO is being produced all of the time, and the NO present at the beginning of HI could form reactive nitrogen species, a pathway facilitated by superoxide produced by nNOS43 . Oxidative damage during HI and immediately post-HI may be the trigger that causes all of the damage that determines the motor deficits since MRI changes during HI can predict which fetuses are destined to be hypertonic.44 Maternal administration of 5 and 6 leads to inhibition of fetal brain NOS activity (Figure 3 A,B) and a decrease in the production of fetal brain NO (Figure 3C,D). This demonstrates the ability of these compounds to distribute to fetal brain and show in vivo NOS inhibition.
A truly striking difference was observed in the neurobehavioral assessment of kits born to 5- and 6-treated dams relative to those from the saline control dams. The P1 kits from dams treated with 5 or 6 had a remarkable decrease in adverse outcome, including deaths (reduced to none) and severe abnormalities including hypertonia/postural deficits when compared to saline-treated dams. A large majority of kits from 5- and 6-treated dams were normal. None of the compounds caused any detectable systemic toxicity in the rabbit dams. No other compounds reported exhibit these remarkable protective properties in the cerebral palsy model. Our results indicate that 5 and 6 are safe, powerful neuroprotective agents against HI-fetal brain injury, and the compounds are stable during the ischemic event. We did not observe protection from post-HI treatment of dams with 6, but this is not surprising considering that the upregulation of NOS and increased NO produced occurs prior to the inhibitors reaching the fetal brain, so they can only block NO produced from that point on (and not the critical NO produced before). The relatively poor potencies, selectivities, and pharmacokinetic (ADME) profiles of available inhibitors have precluded a convincing demonstration of their efficacy clinically.
We have followed a cohort of rabbit kits treated with compound 6 from P1 to P5. The kits that were not hypertonic remained non-hypertonic (10/10). The severely affected remained severely affected (3/6) or died (3/6). Swimming tests confirm the persistence of the protective neurobehavioral effect (data not shown). Future studies need to be done on the regional protection of the nNOS inhibitors,
In conclusion, using the binding modes of our earlier dipeptide inhibitors with nNOS and the application of our novel fragment hopping approach, we effectively utilized a one-residue difference in the nNOS active site relative to eNOS and iNOS to evolve new bioavailable inhibitors. These inhibitors exhibit low nanomolar potency for nNOS and 1000–2000-fold selectivity for nNOS over eNOS. The newly designed compounds distribute readily to the fetal brain, inhibit NOS activity and decrease NO concentration in vivo, appear to be nontoxic, without detrimental cardiovascular effects, and show a remarkable protection of fetal rabbit kits from the HI induced phenotype of cerebral palsy. These potent inhibitors may enable a preventive strategy for high-risk mothers to ameliorate the pathophysiological cascades resulting in postnatal cerebral palsy.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for grants from the National Institutes of Health to R.B.S. (GM49725), S.T. (NS43285 and NS051402), T.L.P. (GM57353), J.V.V. (HL067244 and NS54017), and Dr. Bettie Sue Masters (GM52419, in whose laboratory P.M. and L.J.R. work), as well as the Robert A. Welch Foundation to Dr. Bettie Sue Masters (AQ1192).
REFERENCES
- 1.Lorenz JM. The outcome of extreme prematurity. Semin Perinatol. 2001;25:348–359. doi: 10.1053/sper.2001.27164. [DOI] [PubMed] [Google Scholar]
- 2.Koman LA, Smith BP, Shilt JS. Cerebral palsy. Lancet. 2004;363:1619–1631. doi: 10.1016/S0140-6736(04)16207-7. [DOI] [PubMed] [Google Scholar]
- 3.Winter S, Autry A, Boyle C, Yeargin-Allsopp M. Trends in the prevalence of cerebral palsy in a population-based study. Pediatrics. 2002;110:1220–1225. doi: 10.1542/peds.110.6.1220. [DOI] [PubMed] [Google Scholar]
- 4.Tyson JE, Gilstrap LC. Hope for perinatal prevention of cerebral palsy. JAMA. 2003;290:2730–2732. doi: 10.1001/jama.290.20.2730. [DOI] [PubMed] [Google Scholar]
- 5.O'Shea TM. Cerebral palsy in very preterm infants: new epidemiological insights. Ment Retard Dev Disabil Res Rev. 2002;8:135–145. doi: 10.1002/mrdd.10032. [DOI] [PubMed] [Google Scholar]
- 6.Cowan F, Rutherford M, Groenendaal F, Eken P, Mercuri E, Bydder GM, Meiners LC, Dubowitz LMS, de Vries LS. Origin and timing of brain lesions in term infants with neonatal encephalopathy. Lancet. 2003;361:736–742. doi: 10.1016/S0140-6736(03)12658-X. [DOI] [PubMed] [Google Scholar]
- 7.Robertson C, Finer N. Term infants with hypoxic-ischemic encephalopathy: outcome at 3.5 years. Dev Med Child Neurol. 1985;27:473–484. doi: 10.1111/j.1469-8749.1985.tb04571.x. [DOI] [PubMed] [Google Scholar]
- 8.Ferriero DM, Holtzman DM, Black SM, Sheldon RA. Neonatal mice lacking neuronal nitric oxide synthase are less vulnerable to hypoxic-ischemic injury. Neurobiol Dis. 1996;3:64–71. doi: 10.1006/nbdi.1996.0006. [DOI] [PubMed] [Google Scholar]
- 9.Ferriero DM, Sheldon RA, Black SM, Chuai J. Selective destruction of nitric oxide synthase neurons with quisqualate reduces damage after hypoxia-ischemia in the neonatal rat. Pediatr Res. 1995;38:912–918. doi: 10.1203/00006450-199512000-00014. [DOI] [PubMed] [Google Scholar]
- 10.Iadecola C, Zhang F, Casey R, et al. Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J Neurosci. 1997;17:9157–9164. doi: 10.1523/JNEUROSCI.17-23-09157.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van den Tweel ERW, Nijboer C, Kavelaars A, et al. Expression of nitric oxide synthase isoforms and nitrotyrosine formation after hypoxia-ischemia in the neonatal rat brain. J Neuroimmunol. 2005;167:64–71. doi: 10.1016/j.jneuroim.2005.06.031. [DOI] [PubMed] [Google Scholar]
- 12.Huang Z, Huang PL, Ma J, et al. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J Cereb Blood Flow Metab. 1996;16:981–987. doi: 10.1097/00004647-199609000-00023. [DOI] [PubMed] [Google Scholar]
- 13.Zicha J, Pechánová O, Dobesová Z, Kunes J. Hypertensive response to chronic NG-nitro-L-arginine methyl ester (L-NAME) treatment is similar in immature and adult Wistar rats. Clin Sci. 2003;105:483–489. doi: 10.1042/CS20030078. [DOI] [PubMed] [Google Scholar]
- 14.Wei X-q, Charles IG, Smith A, Ure J, Feng G-J, Huang F-p, Xu D, Mullers W, Moncada S, Liew FY. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature. 1995;375(6530):408–411. doi: 10.1038/375408a0. [DOI] [PubMed] [Google Scholar]
- 15.Cho S, Park EM, Zhou P, et al. Obligatory role of inducible nitric oxide synthase in ischemic preconditioning. J Cereb Blood Flow Metab. 2005;25:493–501. doi: 10.1038/sj.jcbfm.9600058. [DOI] [PubMed] [Google Scholar]
- 16.Derrick M, Luo NL, Bregman JC, et al. Preterm fetal hypoxia-ischemia causes hypertonia and motor deficits in the neonatal rabbit: a model for human cerebral palsy? J Neurosci. 2004;24:24–34. doi: 10.1523/JNEUROSCI.2816-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan S, Drobyshevsky A, Jilling T, et al. Model of cerebral palsy in the perinatal rabbit. J Child Neurol. 2005;20:972–979. doi: 10.1177/08830738050200120801. [DOI] [PubMed] [Google Scholar]
- 18.Erdal EP, Litzinger EA, Jinwon S, et al. Selective neuronal nitric oxide synthase inhibitors. Curr Top Med Chem. 2005;5:603–624. doi: 10.2174/1568026054679317. [DOI] [PubMed] [Google Scholar]
- 19.Tafi A, Angli L, Venturini G, et al. Computational studies of competitive inhibitors of nitric oxide synthase (NOS) enzymes: towards the development of powerful and isoform-selective inhibitors. Curr Med Chem. 2006;13:1929–1946. doi: 10.2174/092986706777585031. [DOI] [PubMed] [Google Scholar]
- 20.Furfine ES, Harmon MF, Paith JE, Garvey EP. Selective Inhibition of Constitutive Nitric Oxide Synthase by L-NG-nitroarginine. Biochemistry. 1993;32:8512–8517. doi: 10.1021/bi00084a017. [DOI] [PubMed] [Google Scholar]
- 21.Pfeiffer S, Leopold E, Schmidt K, et al. Inhibition of Nitric Oxide Synthase by NG-Nitro-L-arginine Methyl Ester (L-NAME): Requirement for Bioactivation to the Free Acid, NG-Nitro-L-arginine. Br J Pharmacol. 1996;118:1433–1440. doi: 10.1111/j.1476-5381.1996.tb15557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hah J-M, Martásek P, Roman LJ, Silverman RB. Aromatic reduced amide and peptidomimetics as selective inhibitors of neuronal nitric oxide synthase. J Med Chem. 2003;46:1661–1669. doi: 10.1021/jm0202932. [DOI] [PubMed] [Google Scholar]
- 23.Gómez-Vidal JA, Martásek P, Roman LJ, Silverman RB. Potent and selective conformationally restricted neuronal nitric oxide synthase inhibitors. J Med Chem. 2004;47:703–710. doi: 10.1021/jm030297m. [DOI] [PubMed] [Google Scholar]
- 24.Flinspach ML, Li H, Jamal J, et al. Structural basis for dipeptide amide isoform-selective inhibition of neuronal nitric oxide synthase. Nat Struct Mol Biol. 2004;11:54–59. doi: 10.1038/nsmb704. [DOI] [PubMed] [Google Scholar]
- 25.Ji H, Li H, Flinspach M, et al. Computer modeling of selective regions in the active site of nitric oxide synthases: implication for the design of isoform-selective inhibitors. J Med Chem. 2003;46:5700–5711. doi: 10.1021/jm030301u. [DOI] [PubMed] [Google Scholar]
- 26.Ji H, Stanton BZ, Igarashi J, et al. Minimal pharmacophoric elements and fragment hopping, an approach directed at molecular diversity and isozyme selectivity. Design of selective neuronal nitric oxide synthase inhibitors. J Am Chem Soc. 2008;130:3900–3914. doi: 10.1021/ja0772041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hajduk PJ, Greer J. A decade of fragment - based drug design: strategic advances and lessons learned. Nat Rev Drug Disc. 2007;6:211–219. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- 28.Ertl P, Rohde B, Selzer P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem. 2000;43:3714–3717. doi: 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]
- 29.Flinspach M, Li H, Jamal J, et al. Structures of the neuronal and endothelial nitric oxide synthase heme domain with D-nitroarginine-containing dipeptide inhibitors bound. Biochemistry. 2004;43:5181–5187. doi: 10.1021/bi0361867. [DOI] [PubMed] [Google Scholar]
- 30.Li H, Flinspach ML, Igarashi J, et al. Exploring the binding conformations of bulkier dipeptide amide inhibitors in constitutive nitric oxide synthases. Biochemistry. 2005;44:15222–15229. doi: 10.1021/bi0513610. [DOI] [PubMed] [Google Scholar]
- 31.Jones TA, Zou J-Y, Cowan SW, Kjeldgaarrd M. Improved methods for building models in electron density and the location of errors in these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 32.Brunger AT, Adams PD, Clore GM, et al. Crystallography & NMR System: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 33.Li H, Poulos TL. Structure-function studies on nitric oxide synthases. J Inorg Biochem. 2005;99:293–305. doi: 10.1016/j.jinorgbio.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 34.Norinder U, Haeberlein M. Computational approaches to the prediction of the blood-brain distribution. Adv Drug Deliv Rev. 2002;54:291–313. doi: 10.1016/s0169-409x(02)00005-4. [DOI] [PubMed] [Google Scholar]
- 35.Miranker A, Karplus M. Functionality maps of binding sites: a multiple copy simultaneous search method. Proteins. 1991;11:29–34. doi: 10.1002/prot.340110104. [DOI] [PubMed] [Google Scholar]
- 36.Goodford PJ. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J Med Chem. 1985;28:849–857. doi: 10.1021/jm00145a002. [DOI] [PubMed] [Google Scholar]
- 37.Böhm H-J. The computer program LUDI: a new method for the de novo design of enzyme inhibitors. J Comput-Aided Mol Des. 1992;6:61–78. doi: 10.1007/BF00124387. [DOI] [PubMed] [Google Scholar]
- 38.Morris GM, Goodsell DS, Halliday RS, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comp Chem. 1998;19:1639–1662. [Google Scholar]
- 39.van de Waterbeemd H, Smith DA, Beaumont K, Walker DK. Property-based design: optimization of drug absorption and pharmacokinetics. J Med Chem. 2001;44:1313–1333. doi: 10.1021/jm000407e. [DOI] [PubMed] [Google Scholar]
- 40.Paudler WW, Blewitt HL. Ten π electron nitrogen heterocyclic compounds. V. The site of protonation and N-methylation of imidazo[1,2-α]pyridines and the planarity of the ring system. J Org Chem. 1966;31:1295–1298. [Google Scholar]
- 41.Müller K, Faeh C, Diederich F. Fluorine in pharmaceuticals: looking beyond intuition. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 42.Ishida A, Trescher WH, Lange MS, Johnston MV. Prolonged supression of brain nitric oxide synthase activity by 7-nitroindazole protects against cerebral hypoxic-ischemic injury in neonatal rat. Brain Dev. 2001;23:349–354. doi: 10.1016/s0387-7604(01)00237-6. [DOI] [PubMed] [Google Scholar]
- 43.Vásquez-Vivar J, Hogg N, Martasek P, et al. Tetrahydrobiopterin-dependent inhibition of superoxide generation from neuronal nitric oxide synthase. J Biol Chem. 1999;274:26736–26742. doi: 10.1074/jbc.274.38.26736. [DOI] [PubMed] [Google Scholar]
- 44.Drobyshevsky A, Derrick M, Prasad PV, et al. Fetal brain magnetic resonance imaging response acutely to hypoxia-ischemia predicts postnatal outcome. Ann Neurol. 2007;61:307–314. doi: 10.1002/ana.21095. [DOI] [PubMed] [Google Scholar]
- 45.Weiner SJ, Kollman PA, Case DA, et al. A new force field for molecular mechanical simulation of nucleic acid and proteins. J Am Chem Soc. 1984;106:765–784. [Google Scholar]
- 46.Gasteiger J, Marsili M. Iterative paratial equalization of orbital electronegativity – a rapid access to atomic charges. Tetrahedron. 1980;36:3219–3228. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.