

Abstract
The therapeutic effects of inhaled nitric oxide (NO) therapy are thought to be restricted to the pulmonary vasculature because of rapid inactivation of NO by hemoglobin in the bloodstream. However, recent data suggest that inhaled NO may not only be scavenged by the heme iron of hemoglobin but also may react with protein thiols in the bloodstream, including cysteine-93 of the hemoglobin B subunit. Reaction of NO with protein or peptide thiols is termed S-nitrosylation and results in the formation of relatively stable protein S-nitrosothiols that carry NO bioactivity to distal organs. Thus, inhaled NO-induced protein S-nitrosylation may allow inhaled NO to have multiple as yet undiscovered physiologic and pathophysiologic effects outside of the lung. Here we review the immunoregulatory and antimicrobial functions of NO and the potential effects of inhaled NO therapy on host defense.
Keywords: apoptosis, bacteria, inhaled nitric oxide, viruses
Inhaled nitric oxide (NO) is used to treat pulmonary hypertension in both adults and infants. Unlike most vasodilators, inhaled NO selectively vasodilates the pulmonary vasculature without inducing systemic hypotension. The selectivity of inhaled NO has been attributed to the rapid scavenging and inactivation of NO by hemoglobin when NO diffuses from the lung into the bloodstream. However, data from the Stamler laboratory have challenged the idea that hemoglobin merely scavenges NO. Instead, the Stamler group has demonstrated that NO reacts not only with the heme iron but also with cysteine (Cys)-93 on the B subunit of hemoglobin (1, 2). Whereas reactions with the heme iron can inactivate NO, S-nitrosylation of Cys-93 converts hemoglobin into a carrier of NO bioactivity (3, 4). Not only hemoglobin, but also other intravascular proteins such as albumin, may be S-nitrosylated by inhaled NO and transport NO bioactivity to distal organs (Figure 1).
Figure 1.

Transport of NO bioactivity to distal organs after inhaled NO treatment. Inhaled NO diffuses from alveoli into smooth muscle, where it nitrosylates and activates soluble guanylate cyclase (sGC), leading to smooth muscle relaxation and vasodilation. In addition, NO diffuses into pulmonary capillaries, where it undergoes a variety of reactions including reaction with the heme iron of hemoglobin (Hb), leading to formation of nitrate (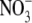 ); reaction with superoxide to form peroxynitrite (ONOO−); reactions with oxygen to form higher oxides of nitrogen, such as N2O3; and reactions with thiols on proteins, such as hemoglobin and albumin to form relatively stable S-nitrosothiols such as S-nitrosohemoglobin (SNO-Hb) and S-nitrosoalbumin (SNO-Alb). Protein S-nitrosothiols transport NO to distal organs.
); reaction with superoxide to form peroxynitrite (ONOO−); reactions with oxygen to form higher oxides of nitrogen, such as N2O3; and reactions with thiols on proteins, such as hemoglobin and albumin to form relatively stable S-nitrosothiols such as S-nitrosohemoglobin (SNO-Hb) and S-nitrosoalbumin (SNO-Alb). Protein S-nitrosothiols transport NO to distal organs.
TRANSPORT OF INHALED NO TO THE INTESTINE DURING ISCHEMIA–REPERFUSION
Animal models of ischemia and reperfusion support the concept that NO bioactivity is transported to distant organs after inhaled NO therapy (5, 6). Exposure of feline intestine to 1 h of ischemia and 1 h of reperfusion results in a decrease in intestinal blood flow, an increase in leukocyte adhesion, and an increase in mucosal barrier leakiness. If animals are ventilated with 80 ppm NO either during or after reperfusion, the abnormalities in intestinal blood flow and leukocyte adhesion are corrected. These data indicate that inhaled NO bioactivity reaches distal organs. However, unlike systemic NO therapy (7), inhaled NO has no effect on mucosal dysfunction induced by ischemia–reperfusion. Thus, inhaled NO bioactivity may not reach the extravascular compartment of distal organs. In support of this hypothesis, S-nitrosothiol levels in the lymph draining the intestinal extravascular space are not increased by inhaled NO therapy. Inhaled NO may be unable to reach extravascular tissue because it is carried to distal organs on proteins such as S-nitrosohemoglobin or S-nitrosoalbumin, which are too large to traverse even an injured endothelial barrier (6). However, the documented effects of inhaled NO on the microvasculature of the gut raise the possibility that inhaled NO may have a variety of as yet undiscovered effects on intravascular physiology and pathophysiology outside of the lung.
EFFECTS OF NO ON THE FUNCTION AND SIGNALING OF IMMUNE CELLS
Immune cells in the vasculature of distal organs may be one of the targets of inhaled NO therapy. NO has multiple effects on immune cells. For instance, NO alters the T helper (Th)1–Th2 balance (8–10). The Th1 subset of helper T cells synthesizes the inflammatory cytokines IFN-γ and interleukin (IL)-2 whereas the Th2 subset synthesizes cytokines such as IL-4, IL-5, and IL-10. NO decreases Th1 proliferation and IL-2 synthesis while increasing IL-4 synthesis by Th2 cells (8–10). One mechanism by which NO inhibits IL-2 production may be disruption of zinc finger transcription factor Sp1, leading to release of zinc and decreased IL-2 transcription (11). NO-induced enhancement of the Th2 response and inhibition of the Th1 response may promote inflammation in allergic diseases such as asthma but inhibit the inflammatory response to viral and bacterial pathogens (12).
NO also modulates the immune response by regulating leukocyte adhesion and recruitment to sites of infection. As discussed above, inhaled NO has been shown to decrease leukocyte adhesion during ischemia and reperfusion of the gut (5, 6). Decreased leukocyte adhesion and recruitment may have deleterious effects on host defense during infections. However, in models of acute lung injury and Pseudomonas pneumonia, inhaled NO increased leukocyte recruitment when administered with high FiO2 (13, 14). Thus the effects of inhaled NO on leukocyte recruitment may depend on the coadministered FiO2 concentration as well as on the local redox environment of tissues.
One of the best-characterized mechanisms by which NO stimulates the immune response is via S-nitrosylation of the monomeric G protein Ras. Ras is a guanine nucleotide–binding protein that cycles between inactive guanosine diphosphate–bound and active guanosine diphosphate–bound states to stimulate a wide array of cellular processes including lymphocyte proliferation and cytokine production. S-nitrosylation of the redox-active Cys-118 on Ras leads to increased levels of active guanosine triphosphate–bound Ras. Of interest, guanine nucleotide exchange is stimulated not by S-nitrosylation of Cys-118 but by a radical intermediate formed during the process of S-nitrosylation (15–18).
In summary, NO regulates the function of immune cells by a variety of mechanisms, including alteration of the Th1–Th2 balance, regulation of leukocyte recruitment and adhesion, and activation of Ras. As is discussed in more detail below, the net effect of NO on the function of immune cells is likely to be dependent on the specific NO-related species generated within a cell, which in turn is dependent both on the intracellular concentration of NO and the intracellular redox environment.
EFFECTS OF NO ON APOPTOSIS
NO modulates the immune response not only by regulating the function of immune cells but also by regulating apoptosis. Both endogenous NO and inhaled NO have well-documented, generally antiapoptotic, effects in the lung. For instance, endogenous NO production inhibits apoptosis of pulmonary epithelial cells in culture (19) and inhibits pulmonary apoptosis induced by either bleomycin or LPS treatment in vivo (20, 21). In addition, inhaled NO has been shown to inhibit pulmonary apoptosis after hyperoxia (22) or ischemia–reperfusion injury (23).
Inhaled NO may also regulate apoptosis of immune cells in the intravascular compartment of distal organs. NO has both pro- and antiapoptotic effects on immune cells. The mechanisms underlying the proapoptotic effects of NO include inhibition of nuclear factor (NF)-κB through a variety of mechanisms, including S-nitrosylation of NF-κB and inhibitory κB kinase leading to decreased NF-κB–mediated transcription and decreased Bcl-2 expression (24–27), increased p53 expression secondary to NO-mediated inhibition of the proteasome or to direct DNA damage (28) and subsequent increased Bax expression (29, 30), S-nitrosylation–stimulated nuclear translocation of glyderaldehyde-3-phosphate dehydrogenase (GAPDH) (31), increased heme nitrosylation and release of cytochrome c from mitochondria (32, 33), and inhibition of inhibitor of apoptosis protein expression (Figure 2) (34). The mechanisms underlying the antiapoptotic effects of NO include S-nitrosylation and inhibition of caspases (35–37), stimulation of the antiapoptotic activity of thioredoxin via S-nitrosylation (38), and increased expression of heat shock proteins (39) and Bcl-2 (Figure 3) (40).
Figure 2.

Proapoptotic effects of NO. NO-related species stimulate apoptosis through a variety of mechanisms, including inhibition of nuclear factor-κB (NF-κB) and stimulation of p53 activity, leading to increased expression of the proapoptotic protein Bax and decreased expression of the antiapoptotic protein Bcl-2, S-nitrosylation–stimulated nuclear translocation of GAPDH, stimulation of cytochrome c (Cyc) release from mitochondria, enhancement of the proapoptotic activity of cytochrome c via heme nitrosylation, and inhibition of inhibitor of apoptosis (IAP) expression. C3 = caspase-3.
Figure 3.

Antiapoptotic effects of NO. NO-related species inhibit apoptosis through a variety of mechanisms, including a cGMP-induced increase in Bcl-2 expression; increased expression of heat shock protein-70 (HSP70), leading to decreased cytochrome c release and decreased apoptosome formation, inhibition of caspase (Casp-3) activity via S-nitrosylation of the caspase catalytic site cysteine, and stimulation of the antiapoptotic activity of thioredoxin via S-nitrosylation of Cys-69.
The of NO concentration plays a key role in determining whether NO stimulates or inhibits apoptosis. In general, higher concentrations of NO induce whereas lower concentrations of NO inhibit apoptosis. For instance, increased generation of NO after apoptotic stimulation elicits S-nitrosylation of GAPDH, which triggers its binding to Siah1 (an E3-ubiquitin ligase), nuclear translocation, and cell death (31). In contrast, low constitutive levels of NO production in nonapoptotic cells lead to S-nitrosylation of the catalytic site cysteine of caspase members (37). Caspases are a family of cysteine proteases that play an essential role in the initiation and execution of apoptotic death pathways. S-nitrosylation of the catalytic site cysteine of caspase members inhibits caspase activity and thereby prevents apoptosis in resting cells.
Thus, NO has multiple targets that can either stimulate or inhibit both the function and apoptotic death of immune cells. The precise effect of inhaled NO on the immune response is likely to depend not only on the concentration of inhaled NO that reaches immune cells but also on the specific redox environment of the cells. The redox environment determines the specific NO-related species that are generated from inhaled NO. Different NO-related species have different biochemical targets and therefore elicit different biological responses. For instance, the presence of electron acceptors, such as transition metals, allows the generation of NO+ equivalents from inhaled NO that S-nitrosylate reduced thiols on caspases and thereby inhibit apoptosis (41). In contrast, superoxide generated intracellularly will react with NO to form the potent oxidant peroxynitrite, which damages DNA, leading to increased p53 levels and stimulation of apoptosis (28). To complicate matters further, inhaled NO can alter NO synthase (NOS) activity in tissues (42–44). Consequently, the intracellular concentration of NO-related species generated after inhaled NO therapy will depend on whether inhaled NO has stimulated or inhibited endogenous NOS activity. Further in vivo studies are needed to help predict the net effect of inhaled NO on the immune response in specific disease states.
ANTIMICROBIAL EFFECTS OF NO
NO not only has immunoregulatory effects but also direct effects on microbes. In general, NO either kills pathogens or inhibits their replication. NO has been reported to inhibit the growth of a wide variety of organisms including viruses, bacteria, parasites, and fungi (45). NO seems to play a particularly important role in the innate immune response to intracellular organisms such as Listeria monocytogenes, Salmonella, and Mycobacterium tuberculosis (46). There are multiple mechanisms involved in the antibacterial properties of nitrogen oxides including inhibition of proteins in the bacterial respiratory chain (47, 48) and disruption of iron–sulfur clusters in bacterial proteins leading to the release of free iron that catalyzes toxic oxidative reactions (49, 50). Moreover, NO and related species inhibit bacterial DNA replication by disrupting zinc metalloproteins involved in DNA replication and by inhibiting ribonucleotide reductase via reactions with a tyrosyl radical in the enzyme (51, 52).
NO also inhibits viral replication via a diverse array of mechanisms (Figure 4). As discussed above, reactions of NO with superoxide lead to the production of the potent oxidant peroxynitrite (ONOO−). Peroxynitrite reacts with capsid proteins on coxsackievirus, leading to the inhibition of viral entry into cells (53). NO also S-nitrosylates and inhibits a variety of viral proteinases that are required for viral replication (54, 55). Finally, NO inhibits transcription factors involved in viral replication (56, 57).
Figure 4.

Antiviral mechanisms of NO. NO reacts with superoxide to form the potent oxidant peroxynitrite. Peroxynitrite nitrates core proteins expressed on the surface of coxsackievirus, leading to the inhibition of viral RNA entry into cells. NO also S-nitrosylates and inhibits viral proteinases that are required for viral replication. In addition, NO inhibits transcription factors involved in viral replication.
In addition to static and cidal effects on pathogens, NO may play a critical role in maintaining pathogen latency. Many organisms, including M. tuberculosis, Leishmania major, Toxoplasma gondii, and Epstein-Barr virus, persist in host cells in a nonreplicative dormant state. Maintenance of latency requires an active host response because latent infections reactivate in immunosuppressed individuals. NO production by host cells may be critical for maintaining latency because inhibition of NO synthesis either by NOS inhibitors or by targeted disruption of the inducible NOS (iNOS) gene leads to reactivation of M. tuberculosis, L. major, T. gondii, and Epstein-Barr virus (56, 58, 59).
Although in general NO exerts antimicrobial effects, not all pathogens are inhibited by NO. Moreover, in some infectious processes NO contributes to disease pathogenesis. For instance, NO may contribute to the pathology of influenza pneumonia because mice with a targeted deletion of iNOS or treated with NOS inhibitors have less severe disease than do control mice (60). Similarly, NOS inhibition improves the course of Mycobacterium avium pneumonia and herpes simplex virus encephalitis in mouse models (61, 62). In these diseases, cytotoxic effects of NO on host cells may contribute to disease pathogenesis.
The effect of inhaled NO on infections in humans is just beginning to be evaluated. In one study, patients with acute lung injury were randomized to receive either placebo or 5 ppm inhaled NO (63). Patients receiving inhaled NO had higher rates of infection than did control subjects. Thus, in some clinical scenarios, inhaled NO may have deleterious effects on host defense. On the other hand, inhaled NO has been reported to decrease bacterial loads in animal models of Pseudomonas pneumonia (64).
Pulmonary tuberculosis is an infectious disease in which inhaled NO is likely to be of therapeutic benefit for several reasons. First, inhaled NO reaches M. tuberculosis in the lung. Second, 90 ppm NO gas kills M. tuberculosis in culture (65). Finally, NOS inhibition or targeted deletion of inducible NOS exacerbates M. tuberculosis infection in animal models, indicating that NO plays an important role in controlling the disease (59, 66). In another study, patients with tuberculosis were treated with 80 ppm inhaled NO for 3 days. Although the inhaled NO therapy was well tolerated, no decrease in bacterial load was noted (67). However, patients with tuberculosis often do not have a therapeutic response to conventional treatment at this early time point. Therefore additional studies using longer courses of inhaled NO therapy are needed to assess its therapeutic efficacy in tuberculosis.
CONCLUSIONS
In summary, NO has multiple immunoregulatory and antimicrobial functions that are likely to be of relevance to inhaled NO therapy. The precise effects of inhaled NO on host defense are likely to be concentration, redox environment, and pathogen dependent. The infections in which inhaled NO is most likely to have therapeutic benefit are diseases such as pulmonary tuberculosis, in which (1) inhaled NO is known to reach the target organ (i.e., the lung or the intravascular compartment of distal organs), (2) NO kills or inhibits the growth of the pathogen, and (3) NO does not contribute to disease pathogenesis. Future studies analyzing inhaled NO treatment in a variety of other infectious diseases are warranted to determine whether inhaled NO is an efficacious antimicrobial agent as well as a pulmonary vasodilator.
Supported by the National Institutes of Health.
Conflict of Interest Statement: J.B.M. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Luchsinger BP, Rich EN, Gow AJ, Williams EM, Stamler JS, Singel DJ. Routes to S-nitroso-hemoglobin formation with heme redox and preferential reactivity in the β subunits. Proc Natl Acad Sci USA 2003;100:461–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gow AJ, Luchsinger BP, Pawloski JR, Singel DJ, Stamler JS. The oxyhemoglobin reaction of nitric oxide. Proc Natl Acad Sci USA 1999;96:9027–9032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robinson JM, Lancaster JR Jr. Hemoglobin-mediated, hypoxia-induced vasodilation via nitric oxide: mechanism(s) and physiologic versus pathophysiologic relevance. Am J Respir Cell Mol Biol 2005;32:257–261. [DOI] [PubMed] [Google Scholar]
- 4.Gow AJ. Nitric oxide, hemoglobin, and hypoxic vasodilation. Am J Respir Cell Mol Biol 2005;32:479–482. [DOI] [PubMed] [Google Scholar]
- 5.Fox-Robichaud A, Payne D, Hasan SU, Ostrovsky L, Fairhead T, Reinhardt P, Kubes P. Inhaled NO as a viable antiadhesive therapy for ischemia/reperfusion injury of distal microvascular beds. J Clin Invest 1998;101:2497–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kubes P, Payne D, Grisham MB, Jourd-Heuil D, Fox-Robichaud A. Inhaled NO impacts vascular but not extravascular compartments in postischemic peripheral organs. Am J Physiol 1999;277:H676–H682. [DOI] [PubMed] [Google Scholar]
- 7.Payne D, Kubes P. Nitric oxide donors reduce the rise in reperfusion-induced intestinal mucosal permeability. Am J Physiol 1993;265:G189–G195. [DOI] [PubMed] [Google Scholar]
- 8.Taylor-Robinson AW, Liew FY, Severn A, Xu D, McSorley SJ, Garside P, Padron J, Phillips RS. Regulation of the immune response by nitric oxide differentially produced by T helper type 1 and T helper type 2 cells. Eur J Immunol 1994;24:980–984. [DOI] [PubMed] [Google Scholar]
- 9.Ianaro A, O'Donnell CA, Di Rosa M, Liew FY. A nitric oxide synthase inhibitor reduces inflammation, down-regulates inflammatory cytokines and enhances interleukin-10 production in carrageenin-induced oedema in mice. Immunology 1994;82:370–375. [PMC free article] [PubMed] [Google Scholar]
- 10.Chang RH, Feng MH, Liu WH, Lai MZ. Nitric oxide increased interleukin-4 expression in T lymphocytes. Immunology 1997;90:364–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berendji D, Kolb-Bachofen V, Meyer KL, Grapenthin O, Weber H, Wahn V, Kroncke KD. Nitric oxide mediates intracytoplasmic and intranuclear zinc release. FEBS Lett 1997;405:37–41. [DOI] [PubMed] [Google Scholar]
- 12.Romagnani S. Lymphokine production by human T cells in disease states. Annu Rev Immunol 1994;12:227–257. [DOI] [PubMed] [Google Scholar]
- 13.Kermarrec N, Chollet-Martin S, Beloucif S, Faivre V, Gougerot-Pocidalo MA, Payen DM. Alveolar neutrophil oxidative burst and β2 integrin expression in experimental acute pulmonary inflammation are not modified by inhaled nitric oxide. Shock 1998;10:129–134. [PubMed] [Google Scholar]
- 14.Jean D, Maitre B, Tankovic J, Meignan M, Adnot S, Brun-Buisson C, Harf A, Delclaux C. Beneficial effects of nitric oxide inhalation on pulmonary bacterial clearance. Crit Care Med 2002;30:442–447. [DOI] [PubMed] [Google Scholar]
- 15.Lander HM, Ogiste JS, Pearce SF, Levi R, Novogrodsky A. Nitric oxide–stimulated guanine nucleotide exchange on p21ras. J Biol Chem 1995;270:7017–7020. [DOI] [PubMed] [Google Scholar]
- 16.Lander HM, Hajjar DP, Hempstead BL, Mirza UA, Chait BT, Campbell S, Quilliam LA. A molecular redox switch on p21ras: structural basis for the nitric oxide-p21ras interaction. J Biol Chem 1997;272:4323–4326. [DOI] [PubMed] [Google Scholar]
- 17.Williams JG, Pappu K, Campbell SL. Structural and biochemical studies of p21Ras S-nitrosylation and nitric oxide-mediated guanine nucleotide exchange. Proc Natl Acad Sci USA 2003;100:6376–6381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heo J, Campbell SL. Mechanism of p21Ras S-nitrosylation and kinetics of nitric oxide-mediated guanine nucleotide exchange. Biochemistry 2004;43:2314–2322. [DOI] [PubMed] [Google Scholar]
- 19.Janssen YM, Soultanakis R, Steece K, Heerdt E, Singh RJ, Joseph J, Kalyanaraman B. Depletion of nitric oxide causes cell cycle alterations, apoptosis, and oxidative stress in pulmonary cells. Am J Physiol 1998;275:L1100–L1109. [DOI] [PubMed] [Google Scholar]
- 20.Davis DW, Weidner DA, Holian A, McConkey DJ. Nitric oxide- dependent activation of p53 suppresses bleomycin-induced apoptosis in the lung. J Exp Med 2000;192:857–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rudkowski JC, Barreiro E, Harfouche R, Goldberg P, Kishta O, D'Orleans-Juste P, Labonte J, Lesur O, Hussain SN. Roles of iNOS and nNOS in sepsis-induced pulmonary apoptosis. Am J Physiol Lung Cell Mol Physiol 2004;286:L793–L800. [DOI] [PubMed] [Google Scholar]
- 22.Howlett CE, Hutchison JS, Veinot JP, Chiu A, Merchant P, Fliss H. Inhaled nitric oxide protects against hyperoxia-induced apoptosis in rat lungs. Am J Physiol 1999;277:L596–L605. [DOI] [PubMed] [Google Scholar]
- 23.Yamashita H, Akamine S, Sumida Y, Inoue M, Sawada T, Nagayasu T, Oka T. Inhaled nitric oxide attenuates apoptosis in ischemia–reperfusion injury of the rabbit lung. Ann Thorac Surg 2004;78:292–297. [DOI] [PubMed] [Google Scholar]
- 24.Matthews JR, Botting CH, Panico M, Morris HR, Hay RT. Inhibition of NF-κB DNA binding by nitric oxide. Nucleic Acids Res 1996;24:2236–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marshall HE, Stamler JS. Nitrosative stress–induced apoptosis through inhibition of NF-κB. J Biol Chem 2002;277:34223–34228. [DOI] [PubMed] [Google Scholar]
- 26.Marshall HE, Stamler JS. Inhibition of NF-κB by S-nitrosylation. Biochemistry 2001;40:1688–1693. [DOI] [PubMed] [Google Scholar]
- 27.Reynaert NL, Ckless K, Korn SH, Vos N, Guala AS, Wouters EF, van der Vliet A, Janssen-Heininger YM. Nitric oxide represses inhibitory κB kinase through S-nitrosylation. Proc Natl Acad Sci USA 2004;101:8945–8950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Messmer UK, Brune B. Nitric oxide–induced apoptosis: p53-dependent and p53-independent signalling pathways. Biochem J 1996;319:299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Glockzin S, von Knethen A, Scheffner M, Brune B. Activation of the cell death program by nitric oxide involves inhibition of the proteasome. J Biol Chem 1999;274:19581–19586. [DOI] [PubMed] [Google Scholar]
- 30.Tamatani M, Ogawa S, Niitsu Y, Tohyama M. Involvement of Bcl-2 family and caspase-3–like protease in NO-mediated neuronal apoptosis. J Neurochem 1998;71:1588–1596. [DOI] [PubMed] [Google Scholar]
- 31.Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol 2005;7:665–674. [DOI] [PubMed] [Google Scholar]
- 32.Schonhoff CM, Gaston B, Mannick JB. Nitrosylation of cytochrome c during apoptosis. J Biol Chem 2003;278:18265–18270. [DOI] [PubMed] [Google Scholar]
- 33.Brookes PS, Salinas EP, Darley-Usmar K, Eiserich JP, Freeman BA, Darley-Usmar VM, Anderson PG. Concentration-dependent effects of nitric oxide on mitochondrial permeability transition and cytochrome c release. J Biol Chem 2000;275:20474–20479. [DOI] [PubMed] [Google Scholar]
- 34.Manderscheid M, Messmer UK, Franzen R, Pfeilschifter J. Regulation of inhibitor of apoptosis expression by nitric oxide and cytokines: relation to apoptosis induction in rat mesangial cells and RAW 264.7 macrophages. J Am Soc Nephrol 2001;12:1151–1163. [DOI] [PubMed] [Google Scholar]
- 35.Dimmeler S, Haendeler J, Nehls M, Zeiher AM. Suppression of apoptosis by nitric oxide via inhibition of interleukin-1β-converting enzyme (ICE)-like and cysteine protease protein (CPP)-32–like proteases. J Exp Med 1997;185:601–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim YM, Talanian RV, Billiar TR. Nitric oxide inhibits apoptosis by preventing increases in caspase-3–like activity via two distinct mechanisms. J Biol Chem 1997;272:31138–31148. [DOI] [PubMed] [Google Scholar]
- 37.Mannick JB, Hausladen A, Liu L, Hess DT, Zeng M, Miao QX, Kane LS, Gow AJ, Stamler JS. Fas-induced caspase denitrosylation. Science 1999;284:651–654. [DOI] [PubMed] [Google Scholar]
- 38.Haendeler J, Hoffmann J, Tischler V, Berk BC, Zeiher AM, Dimmeler S. Redox regulatory and anti-apoptotic functions of thioredoxin depend on S-nitrosylation at cysteine 69. Nat Cell Biol 2002;4:743–749. [DOI] [PubMed] [Google Scholar]
- 39.Kim YM, de Vera ME, Watkins SC, Billiar TR. Nitric oxide protects cultured rat hepatocytes from tumor necrosis factor-α-induced apoptosis by inducing heat shock protein 70 expression. J Biol Chem 1997;272:1402–1411. [DOI] [PubMed] [Google Scholar]
- 40.Genaro AM, Hortelano S, Alvarez A, Martinez C, Bosca L. Splenic B lymphocyte programmed cell death is prevented by nitric oxide release through mechanisms involving sustained Bcl-2 levels. J Clin Invest 1995;95:1884–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stamler JS, Lamas S, Fang FC. Nitrosylation: the prototypic redox-based signaling mechanism. Cell 2001;106:675–683. [DOI] [PubMed] [Google Scholar]
- 42.Black SM, Heidersbach RS, McMullan DM, Bekker JM, Johengen MJ, Fineman JR. Inhaled nitric oxide inhibits NOS activity in lambs: potential mechanism for rebound pulmonary hypertension. Am J Physiol 1999;277:H1849–H1856. [DOI] [PubMed] [Google Scholar]
- 43.Ross GA, Oishi P, Azakie A, Fratz S, Fitzgerald RK, Johengen MJ, Harmon C, Hendricks-Munoz K, Xu J, Black SM, et al. Endothelial alterations during inhaled NO in lambs with pulmonary hypertension: implications for rebound hypertension. Am J Physiol Lung Cell Mol Physiol 2005;288:L27–L35. [DOI] [PubMed] [Google Scholar]
- 44.Weinberger B, Fakhrzadeh L, Heck DE, Laskin JD, Gardner CR, Laskin DL. Inhaled nitric oxide primes lung macrophages to produce reactive oxygen and nitrogen intermediates. Am J Respir Crit Care Med 1998;158:931–938. [DOI] [PubMed] [Google Scholar]
- 45.Fang FC. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat Rev Microbiol 2004;2:820–832. [DOI] [PubMed] [Google Scholar]
- 46.Fang FC. Host/pathogen interactions: mechanisms of nitric oxide–related antimicrobial activity [perspectives series]. J Clin Invest 1997;99:2818–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pacelli R, Wink DA, Cook JA, Krishna MC, DeGraff W, Friedman N, Tsokos M, Samuni A, Mitchell JB. Nitric oxide potentiates hydrogen peroxide–induced killing of Escherichia coli. J Exp Med 1995;182:1469–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stevanin TM, Ioannidis N, Mills CE, Kim SO, Hughes MN, Poole RK. Flavohemoglobin Hmp affords inducible protection for Escherichia coli respiration, catalyzed by cytochromes bo′ or bd, from nitric oxide. J Biol Chem 2000;275:35868–35875. [DOI] [PubMed] [Google Scholar]
- 49.Flint DH, Tuminello JF, Emptage MH. The inactivation of Fe–S cluster containing hydro-lyases by superoxide. J Biol Chem 1993;268:22369–22376. [PubMed] [Google Scholar]
- 50.Keyer K, Imlay JA. Superoxide accelerates DNA damage by elevating free-iron levels. Proc Natl Acad Sci USA 1996;93:13635–13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schapiro JM, Libby SJ, Fang FC. Inhibition of bacterial DNA replication by zinc mobilization during nitrosative stress. Proc Natl Acad Sci USA 2003;100:8496–8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lepoivre M, Fieschi F, Coves J, Thelander L, Fontecave M. Inactivation of ribonucleotide reductase by nitric oxide. Biochem Biophys Res Commun 1991;179:442–448. [DOI] [PubMed] [Google Scholar]
- 53.Padalko E, Ohnishi T, Matsushita K, Sun H, Fox-Talbot K, Bao C, Baldwin WM III, Lowenstein CJ. Peroxynitrite inhibition of coxsackievirus infection by prevention of viral RNA entry. Proc Natl Acad Sci USA 2004;101:11731–11736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saura M, Zaragoza C, McMillan A, Quick RA, Hohenadl C, Lowenstein JM, Lowenstein CJ. An antiviral mechanism of nitric oxide: inhibition of a viral protease. Immunity 1999;10:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cao W, Baniecki ML, McGrath WJ, Bao C, Deming CB, Rade JJ, Lowenstein CJ, Mangel WF. Nitric oxide inhibits the adenovirus proteinase in vitro and viral infectivity in vivo. FASEB J 2003;17:2345–2346. [DOI] [PubMed] [Google Scholar]
- 56.Mannick JB, Asano K, Izumi K, Kieff E, Stamler JS. Nitric oxide produced by human B lymphocytes inhibits apoptosis and Epstein-Barr virus reactivation. Cell 1994;79:1137–1146. [DOI] [PubMed] [Google Scholar]
- 57.Gao X, Tajima M, Sairenji T. Nitric oxide down-regulates Epstein-Barr virus reactivation in epithelial cell lines. Virology 1999;258:375–381. [DOI] [PubMed] [Google Scholar]
- 58.Stenger S, Donhauser N, Thuring H, Rollinghoff M, Bogdan C. Reactivation of latent leishmaniasis by inhibition of inducible nitric oxide synthase. J Exp Med 1996;183:1501–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, Nathan CF. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc Natl Acad Sci USA 1997;94:5243–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Akaike T, Noguchi Y, Ijiri S, Setoguchi K, Suga M, Zheng YM, Dietzschold B, Maeda H. Pathogenesis of influenza virus-induced pneumonia: involvement of both nitric oxide and oxygen radicals. Proc Natl Acad Sci USA 1996;93:2448–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gomes MS, Florido M, Pais TF, Appelberg R. Improved clearance of Mycobacterium avium upon disruption of the inducible nitric oxide synthase gene. J Immunol 1999;162:6734–6739. [PubMed] [Google Scholar]
- 62.Fujii S, Akaike T, Maeda H. Role of nitric oxide in pathogenesis of herpes simplex virus encephalitis in rats. Virology 1999;256:203–212. [DOI] [PubMed] [Google Scholar]
- 63.Taylor RW, Zimmerman JL, Dellinger RP, Straube RC, Criner GJ, Davis K Jr, Kelly KM, Smith TC, Small RJ. Low-dose inhaled nitric oxide in patients with acute lung injury: a randomized controlled trial. JAMA 2004;291:1603–1609. [DOI] [PubMed] [Google Scholar]
- 64.Webert KE, Vanderzwan J, Duggan M, Scott JA, McCormack DG, Lewis JF, Mehta S. Effects of inhaled nitric oxide in a rat model of Pseudomonas aeruginosa pneumonia. Crit Care Med 2000;28:2397–2405. [DOI] [PubMed] [Google Scholar]
- 65.Long R, Light B, Talbot JA. Mycobacteriocidal action of exogenous nitric oxide. Antimicrob Agents Chemother 1999;43:403–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Flynn JL, Scanga CA, Tanaka KE, Chan J. Effects of aminoguanidine on latent murine tuberculosis. J Immunol 1998;160:1796–1803. [PubMed] [Google Scholar]
- 67.Long R, Jones R, Talbot J, Mayers I, Barrie J, Hoskinson M, Light B. Inhaled nitric oxide treatment of patients with pulmonary tuberculosis evidenced by positive sputum smears. Antimicrob Agents Chemother 2005;49:1209–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]