

Summary
Chaperone-mediated autophagy (CMA) is the only type of autophagy in mammalian cells able to selectively degrade cytosolic proteins in lysosomes. CMA is maximally activated in response to stressors such as prolonged starvation, exposure to toxic compounds, or oxidative stress. We have found that CMA activity decreases in aging and in some age-related disorders such as Parkinson’s disease. Impaired CMA under these conditions may be responsible for the accumulation of damaged proteins inside cells and for their higher vulnerability to stressors. In contrast to other forms of autophagy, where substrates are engulfed or sequestered along with other cytosolic components, CMA substrates are translocated one-by-one across the lysosomal membrane. Changes in the levels/activity of the lysosomal components required for substrate translocation can be used to stimulate CMA activity. However, the most unequivocal method to measure CMA is by directly tracking the translocation of substrate proteins into isolated lysosomes.
Keywords: Lysosomes, metabolic labeling, protease protection assay, chaperones, lysosomal membrane proteins
1. Introduction
Two properties differentiate chaperone-mediated autophagy (CMA) from the other types of autophagy in mammalian cells: its selectivity towards a particular pool of cytosolic proteins and the mechanism of delivery of the substrate proteins to lysosomes (1). Only proteins bearing a particular targeting motif in their amino acid sequence, biochemically related to the pentapeptide KFERQ (2), are selectively recognized by the heat shock cognate protein of 70 kDa (hsc70), the chaperone that mediates their delivery to lysosomes for their degradation via CMA (3). It is estimated that about 30% of soluble cytosolic proteins contain this CMA-targeting motif. The already identified substrate proteins include, among others, some glycolytic enzymes (glyceraldehyde-3-phosphate dehydrogenase, aldolase, phosphoglyceromutase), subunits of the 20S proteasome, transcription factors and inhibitors of transcription factors (c-fos, the inhibitor of NFκB [IκB]), cytosolic forms of secretory proteins (α2-microglobulin), calcium-binding proteins (Annexins I, II, IV, and VI), and proteins associated to vesicular trafficking (α-synuclein) (reviewed in refs. 1 and 4).
CMA substrates access the lysosomal lumen directly across the lysosomal membrane. After the CMA targeting motif is recognized by hsc70, the substrate/chaperone complex is targeted to the surface of the lysosomes where it binds to the lysosome-associated membrane protein type 2A (LAMP-2A), a receptor for CMA (5). Once bound, the substrate unfolds (6) and crosses the lysosomal membrane assisted by a lysosomal form of hsc70 (lys-hsc70) present in the lumen (7). Transport is saturable, requires a source of energy (ATP), and is temperature-dependent (binding occurs at temperatures as low as 10°C, but transport is only detected at temperatures above 25°C) (8,9).
The selectivity associated to degradation via CMA seems beneficial under particular conditions in which discrimination between different types of proteins for degradation is required. For example, activation of CMA during prolonged starvation provides the amino acids required for protein synthesis under those conditions but favors degradation of unnecessary proteins against that of proteins essential for cell survival (10,11). Similarly, activation of CMA during mild-oxidative stress (12) or after exposure to protein-modifying toxic compounds (13) allows the selective removal of the proteins damaged or altered under these conditions.
Malfunctioning of CMA has been described in familial forms of Parkinson’s disease where α-synuclein, the protein mutated in this disorder, is delivered to lysosomes for degradation via CMA, but after binding to LAMP-2A it cannot be translocated to the lysosomal lumen resulting in blockage of this pathway (14). CMA activity also decreases with age due to a decrease in the levels of LAMP-2A at the lysosomal membrane (15), contributing thus to the accumulation of abnormal proteins and the higher susceptibility to stressors characteristic of old organisms.
Measurement of changes in the lysosomal levels of the main CMA components (LAMP-2A and hsc70) and of the intracellular abundance and location of the lysosomes active for this pathway (those enriched in the CMA components) provide a general idea about CMA activity in tissues and cells in culture. However, accurate measurement of CMA is only attained by tracking the translocation of CMA substrates into isolated lysosomes using protease-protection assays (8,16-18).
2. Materials
2.1. Isolation of Rat Liver Lysosomes
Wistar rats (200-250 g). To enrich in CMA-active lysosomes, rats should be starved (complete food removal but with water ad libitum) for 24-48 h before lysosomal isolation (minimum 10 h).
Tools: dissection instruments (forceps, scissors, clamps), double cloth gauze, funnels, teflon/glass homogenizer (for motorized homogenizer).
Centrifugation supplies: polycarbonate tubes (30 mL), ultraclear tubes for SW28 (Beckman, Fullerton, CA), SW28 rotor (Beckman).
0.25 M sucrose (American Bioanalytical, Natick, MA) adjusted at pH 7 (with NaOH). All sucrose-containing solutions can be stored at 4°C but for no more than 3 d (as slow hydrolysis of sucrose occurs when stored for longer periods of time).
Metrizamide (Amresco, Solon, OH) prepared as a 85.6% (w/v) stock in water (stock solution adjusted at pH 7.2 with NaOH). Store in aliquots at -20°C in dark (see Note 1).
2.2. Purification of Recombinant GST-hsc70
Glutathione transferase (GST)- hsc70 was purified from XLBlue E. coli bacteria transformed with a plasmid (pGX-2T, Pharmacia, Uppsala, Sweden) containing the cDNA for the fusion protein of human hsc70 (19).
Isopropyl-β-d-thiogalactopyranoside (IPTG, American Bioanalytical) is prepared freshly by dissolving it at 0.1 M in sterile water.
Glutathione-agarose (Sigma, St. Louis, MO) is reconstituted as a 50% v/v stock in TBS following manufacture’s instructions.
Ampicillin (American Bioanalytical) is dissolved at 100 mg/mL in sterile water and stored at 4°C.
STE buffer: 10 mM Tris-HCl, pH 8, 150 mM NaCl, 1 mM ethylenediaminete-traacetic acid (EDTA).
Tris buffer saline (TBS): 50 mM Tris-HCl, pH 7.4, 150 mM NaCl.
Lysozyme (Sigma) is prepared fresh by dissolving it at 100 mg/mL in sterile water.
Dithiothreitol (DTT, Sigma) is prepared as 5 M stock in water and stored in aliquots at -20°C.
Triton X-100 (Bio-Rad, Hercules, CA) is prepared as 20% (v/v) stock solution in sterile water and stored at room temperature.
N-Lauryl sarcosine (Sarkosyl, Sigma) is prepared as 15% (w/v) stock solution in sterile water and stored at room temperature.
Elution buffer: 10 mM reduced GSH (Sigma) in 50 mM Tris-HCl, pH 8 is prepared fresh.
Wash buffer: 50 mM Tris-HCl, pH 7.5, 150 mM NaCl.
Thrombin (Sigma) diluted as 10x stock in thrombin cleavage buffer (amount should be adjusted according to the activity of the particular batch used) (optional).
Thrombin cleavage buffer: 2.5 mM CaCl2 in wash buffer (optional).
2.3. Radiolabeling of Substrate Proteins by Reductive Methylation
Purified protein of interest is prepared at 10 mg/mL in the reaction buffer (see Note 2).
Reaction buffer: 10 mM MES (Sigma) pH 7. Store at 4°C.
Bovine serum albumin (BSA) (American Bioanalytical) dissolved at 20 mg/mL in the reaction buffer.
[14C]Formaldehyde (Amersham, Piscataway, NJ) 1-3% aqueous solution in sealed ampoule (1.85-2.29 GBq/mmol, 50-62 mCi/mmol). Store at 4°C.
Sodium cyanoborohydrate (Sigma) (9 mg/mL) in reaction buffer. Prepare fresh.
Sephadex G-25 to G-100 fine (Sigma) (depending on the size of the labeled protein), equilibrated in sterile water following manufacture’s recommendation.
2.4. Protein Degradation Assay
Proteolysis buffer: 10 mM 3-(N-Morpholino)propanesulfonic acid (MOPS, Sigma) pH 7.3, 250 mM sucrose, 1 mM DTT, 5.4 mM cysteine. Prepare fresh.
Energy regenerating system (ERS): prepare a 6x cocktail at these final concentrations 60 mM ATP, 12 mM phosphocreatine (Sigma), 0.3 mg/mL creatine phosphokinase (Sigma), 60 mM MgCl2 in 0.25 M sucrose adjusted at pH 7.3.
Trichloroacetic acid (American Bioanalytical) dissolved at 20% (w/v) in water and stored at room temperature.
BSA (American Bioanalytical) disolved at 20 mg/mL in water.
Millipore multiscreen assay system (Millipore, Bedford, MA), 0.22-μm durapore filter 96-well plates, and polystyrene flat-bottom 96-well plates.
2.5. Protease-Protection Transport Assay
Incubation buffer: 10 mM MOPS pH 7.3, 250 mM sucrose. Prepared fresh and stored at 4°C.
Chymostatin (Sigma) prepared at 10 mM stock dissolved in incubation buffer and stored at -20°C.
ERS (see Subheading 2.4.).
Proteinase K (Sigma) dissolved at 5 mg/mL stock in 10 mM Tris-HCl pH 7.5, 1 mM CaCl2.
4-(2-Aminoethyl)-benzenesulfonyl fluoride hydrochloride (AEBSF, American Analytical) dissolved in water as 1mM stock and stored at -20°C.
Reagents for standard SDS-PAGE and immunoblot.
2.6. Indirect Immunofluorescence
Dulbecco’s modified Eagle’s medium (DMEM) (Sigma) supplemented with 10% newborn calf serum (or any other media that cells require for growth).
Microscope cover slips (22 × 22 mm) (Fisher Scientific, Pittsburgh, PA).
Phosphate-buffered saline (PBS): 1.37 M NaCl, 0.03 M KCl, 0.07 M Na2HPO4, 0.11 M K2HPO4 pH 7.4. Store at room temperature.
Methanol fixing solution: 20% methanol (v/v) (in PBS). Store at -20°C.
Blocking solution: 0.2% (w/v) powdered nonfat milk, 2% newborn calf serum, 0.1 M glycine, 1% BSA, and 0.01% Triton X-100 in PBS. Prepare fresh and maintain at 4°C until use.
Secondary antibodies: Fluorescein-labeled donkey anti-mouse IgM, μ-chain specific, and cyanine5-labeled goat anti-rabbit IgG (H+L chain specific) (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA).
Mounting media: SlowFade Light Antifade Kit with DAPI 1 (Molecular Probes, Invitrogen).
3. Methods
CMA activity can be measured in confluent cells in culture using pulse-chase experiments. After 2-d labeling with a radiolabeled amino acid (to preferentially label long-lived proteins), CMA can be activated in cultured fibroblasts at about 10 h after serum removal. CMA-dependent degradation is defined under those conditions as that activated by serum removal, which is sensitive to ammonium chloride (the same as any other form of autophagy), but insensitive to phosphatydil-inositol-3-kinase inhibitors (which block macroautophagy) (Fig. 1) (14,20-23). Although this method is useful to discriminate from degradation via macroautophagy, two limitations make it inconvenient to rely only on this method: (1) it only measures activation of CMA in response to serum removal—it does not measure changes in basal CMA activity as it is not possible to differentiate from microautophagy by this approach; (2) it requires the use of inhibitors of macroautophagy, and it is well supported now that maintained blockage of a proteolytic pathway leads to the compensatory upregulation of other proteolytic pathways.
Fig. 1.

Measurement of CMA-dependent protein degradation in culture cells. (A) Cultured cells are labeled by incubation with a radiolabeled amino acid for more than 24 h. At the end of the incubation, cells are extensively washed, and degradation of protein is followed by calculating the percentage of acid precipitable radioactivity at time 0 (protein), converted to acid soluble radioactivity (amino acids and small peptides) at each time. (B) Protein degradation increases after serum removal. The percentage of ammonium chloride-sensitive degradation inhibited by PI3K inhibitors corresponds to macroautophagy, while that insensitive to the inhibitor corresponds to CMA and microautophagy.
The most unequivocal method to measure CMA is by directly tracking the translocation of substrate proteins into lysosomes. CMA can be reproduced in vitro using freshly isolated intact lysosomes by incubating the substrate protein and lysosomes at 37°C in an isotonic media supplemented with hsc70 and an ATP-regenerating system (8,16-18). Using standard protease protection assays, as those commonly used for the study of translocation of proteins across other organelles membrane, the three main steps in CMA (binding, uptake, and degradation) can be separately analyzed. Binding of substrate to the lysosomal membrane can be determined as the amount of substrate associated to the lysosomes recovered by centrifugation, because any protein that has been translocated will be rapidly degraded by the lysosomal proteases (the half-life of substrate proteins in the lysosomal lumen is ∼5 min). Uptake of substrates can be measured in lysosomes pretreated with protease inhibitors. In this case, the substrate remains in the lysosomal lumen after crossing the lysosomal membrane. The amount of substrate translocated into the lysosomal lumen can be determined by comparing the level of total substrate (bound plus translocated) to the substrate bound to the membrane (in lysosomes untreated with protease inhibitors), or by measuring the amount of substrate resistant to cleavage by an exogenously added protease (in lysosomes treated with protease inhibitors) (Fig. 2).
Fig. 2.
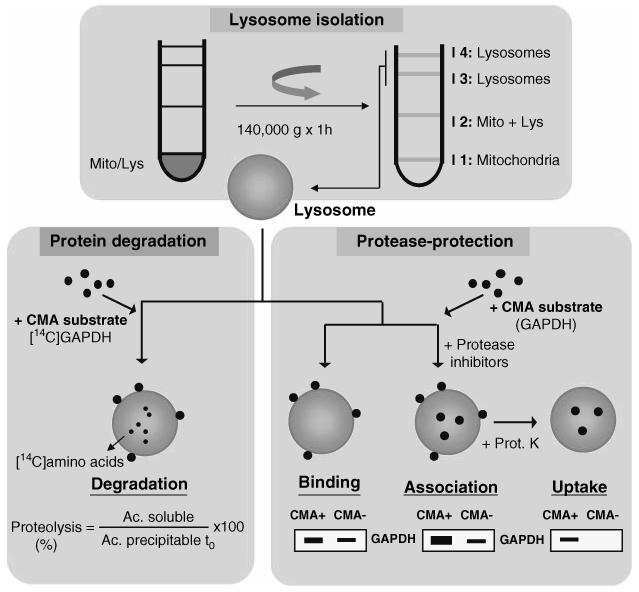
In vitro assay for the measurement of substrate protein translocation into lysosomes via CMA. Lyosome isolation: Lysosomes are isolated from rat liver by centrifugation in a discontinuous gradient of metrizamide of a mitochondria/lysosome enriched fraction obtained by differential centrifugation. Protein degradation: A radiolabeled substrate protein (glyceraldehyde-3-phosphate dehydrogenase [GAPDH] in this example) is incubated with freshly isolated intact lysosomes, and their proteolytic activity is calculated as the percentage of acid precipitable radioactivity at time 0 (protein) transformed to acid soluble radioactivity (amino acids and small peptides) at the end of the incubation. Protease-protection assay: Freshly isolated intact lysosomes, treated or not with protease inhibitors, are incubated with the substrate protein (GAPDH) and at the end of the incubation lysosomes are collected by centrifugation and subjected to SDS-PAGE and immunoblot to detect the amount of substrate protein associated. Binding: substrate associated to lysosomes untreated with protease inhibitors; Association: substrate associated to lysosomes treated with protease inhibitors (bound + internalized); Uptake: substrate associated to lysosomes treated with protease inhibitors after removal of the membrane bound substrate with an exogenous protease (proteinase K [Prot. K] in this example).
Because lysosomal isolation requires a large number of cells and some level of practice in the handling of isolated organelles, we have recently optimized an indirect method to evaluate CMA activity in those tissues or cells in which lysosomal isolation is not feasible. This method is also discussed in this chapter and it is based on the analysis of changes in the population of lysosomes responsible for CMA, which is defined as those containing lys-hsc70 (the lysosomal chaperone required for substrate translocation) in their lumen (12,24). This population can be detected in tissue sections or fixed cultured cells by immunoelectron microscopy with an antibody directed against hsc70 (CMA+ lysosomes are the ones containing the chaperone in their lumen) or by immunofluorescence in cultured cells, as the number of vesicles containing hsc70 and LAMP-2A.
It is also possible to indirectly track changes in CMA activity by measuring changes in the major components of this pathway (hsc70 and LAMP-2A) in lysosomes. It is important to point out that total cellular levels of these components do not necessarily correlate with changes in CMA, because both proteins are also present in other cellular compartments and play other roles in the cell. It is thus necessary to determine that the increase or decrease in these components takes place in the lysosome-associated fraction. Protein levels usually result more accurate measurement than changes in mRNA levels. Hsc70 is constitutively expressed and we have not found changes at the transcriptional level in any condition associated to changes in CMA activity. In the case of LAMP-2A, conditions such as oxidative stress associate to transcriptional upregulation of LAMP-2A (12), but in other conditions, such as prolonged starvation, the increase in levels of LAMP-2A in the lysosomal compartment does not require de novo synthesis of the protein (25).
3.1. Isolation of Rat Liver Lysosomes
Livers from two starved rats are washed extensively with cold 0.25 M sucrose (see Note 3), weighed, and then minced.
The minced livers are homogenized in three volumes of 0.25 M sucrose/g of liver with 8-10 strokes at maximum speed. The homogenates are filtered through double gauze, and four volumes of cold 0.25 M sucrose are added.
After centrifugation of the homogenate at 6800g for 5 min at 4°C, the supernatant is collected into a clean tube (avoid collecting the white floating material close to the pellet, which mainly consists of heavy mitochondria). The pellet is gently resuspended with a dry glass test tube filled with ice (“cold finger”) in starting volume of 0.25 M sucrose and spun under the same conditions. Both supernatants are then pooled together.
The pooled supernatants are centrifuged at 17,000g for 10 min and the pellet resuspended with the “cold finger” in dry to guarantee complete resuspension. After adding 3.5 volumes of 0.25 M sucrose/g liver to the resuspended pellet, it is centrifuged under the same conditions (this step assures that any cytosolic component trapped in the initial pellet is released into the supernatant).
The pellet from the previous centrifugation, highly enriched in light mitochondria and lysosomes (mitochondrial-lysosomal fraction), is then resuspended with the “cold finger,” and 1 mL 0.25 M sucrose/3 g liver and two volumes of 85.6% Metrizamide are added and gently mixed with the sample to a final concentration of 57% metrizamide (see Note 4).
Each 10 mL of the 57% metrizamide light mitochondrial-lysosomal fraction are loaded at the bottom of an ultraclear tube and a discontinuous metrizamide gradient is generated on top by overlying: 6 mL of 32.8% Metrizamide, 10 mL 26.3% Metrizamide, and 11 mL 19.8% Metrizamide (all diluted in water pH 7.3). The tube is filled up to 3 mm of the border with 0.25 M sucrose, and it is subjected to centrifugation in a SW 28 rotor at 141,000g for 1 h at 4°C (see Note 5).
After centrifugation, white to brownish material is clearly visible in each of the interphases. Mitochondria are preferentially located in the first interphase of the gradient (starting from the bottom); the following interphase is enriched in a mixture of mitochondria and lysosomes. Lysosomes are present in the third (I3) and fourth (I4) interphases, which are collected separately (approx 3-4 mL) with a Pasteur pipet in 30-mL polycarbonate tubes (Fig. 2).
After dilution in five volumes of 0.25 M sucrose, the fractions are washed by centrifugation at 37,000g for 10 min at 4°C.
The pellet of I3 is carefully resuspended with a blunt Pasteur pipet in 1 mL 0.25 M sucrose and spun at 10,000g for 5 min at 4°C. The pellet of this centrifugation is preferentially enriched in secondary lysosomes with low CMA activity (lacking hsc70 in their lumen). The supernatant of the I3 centrifugation is used to resuspend the pellet of the I4 fraction to produce the secondary lysosomes with high CMA activity (enriched in hsc70) (if resuspended in 0.4 mL, the final protein concentration is 10 mg/mL approx.) (see Notes 6 and 7).
3.2. Purification of Recombinant GST-hsc70
Bacteria strains carrying the pGx-GST-hsc70 plasmid are grown overnight at 37°C with agitation in Luria Broth (LB) with 100 μg/mL ampicillin. Cultures are refreshed next morning by 1/10 dilution in LB ampicillin and incubated under the same conditions.
When the cultures are visibly turbid (OD600 = 0.3), IPTG is added to a final concentration of 0.1 mM and they are incubated for an additional 4-8 h.
Bacteria are collected by centrifugation (7000g for 5 min) and the pellet is washed with 1/10 of the starting volume of STE with 100 μg/mL lysozyme.
After incubation for 15 min on ice, the samples are adjusted to 5 mM DTT and are lysed by the addition of 1.5% sarkosyl (N-lauryl sarcosine) and sonication on ice (1 min in a bath sonicator).
Lysates are subjected to centrifugation (10,000g 5 min at 4°C) and the supernatant is adjusted to 2% Triton X-100 (to minimize association of bacterial proteins with hsc70) and vortexed gently.
A 1/50 starting volume of GSH-agarose beads (50% v/v in TBS) is added to the supernatant and after incubation for 15 min at 4°C on a shaker, the beads are collected by centrifugation and washed 6-8 times with ice-cold TBS (see Note 8).
The final beads slurry is resuspended in the same volume of wash buffer supplemented with reduced GSH and incubated for 30 min at room temperature to elute the protein from the beads. After spinning, the eluted protein is recovered in the supernatant. If desired the GSH can be removed by dialysis against washing buffer without GSH.
3.3. Radiolabeling by Reductive Methylation
The protein of interest is dissolved in reaction buffer to a final concentration of 3 mg/mL, and [14C]formaldehyde (250 μCi) and sodium cyanoborohydrate (final concentration of 1.8 mg/mL in reaction buffer) are added. Handling of this amount of isotope requires the use of filtered tips, double closure microfuge tubes, and disposal of all materials as solid radioactive residues waste.
After incubation of the reaction in a final volume of 500 μL at 25°C for one hour, the radiolabeled protein is separated from the nonincorporated isotope by gel filtration through the pertinent Sephadex matrix (according to the molecular weight of the protein). We routinely use mini-spin columns packed with the matrix (1 mL approx.) previously blocked with 20 mg/mL BSA (5 vol) (to prevent nonspecific binding) and equilibrated with the reaction buffer (10 vol). Spinning time is adjusted depending on the protein of interest and the characteristics of the mini-spin column.
The eluted radiolabeled protein is collected in separate aliquots (one for each spin with the column first loaded with the reaction mixture, and then with similar volumes of reaction buffer—three times).
The amount of radiolabeled protein and free isotope in each aliquot (total 4 aliquots) is determined by measurement of the radioactivity associated to the protein precipitated with TCA. Briefly, an aliquot of each fraction is diluted (1:100) in reaction buffer, and 50 μL of this dilution are added to a microfuge tube containing 50 μL of 20% TCA and 30 μL of 20 mg/mL BSA. The samples are incubated at 4°C for 30 min and then spun at 15,000g for 15 min at 4°C. The supernatant of each tube is collected in a scintillation vial, and the pellet is washed with other 50 μL of 20% TCA. After centrifugation in the same conditions, the second supernatant is added to the vial, while the pellet is resuspended in 50 μL of 0.1 M NaOH and then transferred to a different scintillation vial. After adding the pertinent amount of scintillation liquid, samples are counted in a scintillation β-counter. The amount of acid precipitable radioactivity (protein) in the sample is calculated by the formula: [(dpm pellet/(dpm pellet + dpm supernatant)) *100], and the amount of radioactivity incorporated by microgram of protein (dpm/μg) by measuring the amount of protein in the collected aliquots by the Lowry procedure (26). Reasonable specific activities are in the order of 1 - 100 × 106 dpm/nmol. Radiolabeled proteins are stored in small aliquots (10 μL) in double sealing microfuge tubes at -80°C for months (avoid repeated freezing and thawing).
3.4. Protein Degradation Assay
Incubations are carried out in a 0.22-μm filtered durapore 96-well plate previously wet with sterile water for 10 min at room temperature. Freshly isolated rat liver lysosomes (25 μg protein) (10 μL of a 1:4 dilution in proteolysis buffer) are incubated with 10 μL of radiolabeled protein (2000 dpm/μL), 1 μL of the (6x) ERS and 10 μg/mL GST-hsc70 in a final volume of 60 μL (adjusted with proteolysis buffer) for 30 min at 37°C. One separate well should contain the same reagents, except for the lysosomes, to determine the amount of protein spontaneously cleaved (blank).
At the end of the incubation 90 μL of 20% TCA and 30 μL of 20 mg/mL BSA are added to each well to stop the reaction. After incubation at 4°C for at least 30 min, the acid-soluble radioactivity is collected in a polystyrene 96-well plate using the Millipore multiscreen vacuum system. The flow throughs (acid-soluble) are collected in separate scintillation vials and counted.
Proteolysis is calculated as the amount of acid precipitable radioactivity (protein) transformed in acid-soluble radioactivity (amino acids and small peptides) at the end of the incubation: [((dpm flow through sample - dpm flow through blank) / dpm pellet time 0)*100] (see Note 9).
3.5. Protease-Protection Transport Assay
Freshly isolated rat liver lysosomes are incubated with 100 μM chymostatin for 10 min on ice. Depending on the substrate, a protease inhibitor cocktail (x100) can be used instead (10 mM leupeptine, 10 mM AEBSF, 1 mM pepstatin and 100 mM EDTA).
Transport assay is carried out in 0.5-mL microfuge tubes by adding freshly isolated rat liver lysosomes treated or not with protease inhibitor (100 μg protein), GAPDH, or another protein of interest (50 μg), 5 μL of (x6) ERS and 10 μg/mL GST-hsc70 in 30 μL final volume of incubation buffer. Samples are incubated for 20 min at 37°C.
At the end of the incubation half of the tubes containing lysosomes treated with chymostatin are cooled down on ice (1 min) and proteinase K is added (5 μL). Samples are maintained on ice for 10 min, AEBSF is added (5 μL) and all samples are centrifuged at 25,000g for 5 min at 4°C.
Supernatants are aspirated and pellets are washed with 100 μL of incubation buffer by centrifugation under the same conditions. The final pellet is resuspended in 30 μL of Laemmli buffer (27) with protease inhibitors, boiled for 5 min at 95°C and subjected to SDS-PAGE and immunoblot for GAPDH. A lane with 1/10 of the amount of GAPDH added should be included in the immunoblot.
Densitometric analysis is performed on the immunoblots directly (if developed by the alkaline phosphatase method) or using the film from an exposure that does not contain saturated bands (if developed by the chemiluminiscence method). For each antibody a standard curve with increasing concentrations of antigen is generated to determine the linear range. These values allow the calculation of (1) binding as the percentage of total added GAPDH associated to lysosomes untreated with protease inhibitors, (2) sssociation as the percentage of GAPDH recovered in lysosomes treated with protease inhibitors, and (3) uptake, calculated as either the difference between association and binding or the amount or the percentage of GAPDH associated to lysosomes treated with proteinase inhibitors after proteinase K treatment (see Note 10).
3.6. Indirect Immunofluorescence
Cells grown on sterile cover slips at the bottom of 6-well plates until semi-confluent (50-70% confluence) are washed (twice) with serum-free culture medium (to remove all remaining IgGs in the serum), and fixed in -20°C methanol (20% in PBS) for 1 min at room temperature (7). Cells can be stored in PBS containing 0.02% NaN3 at 4°C in dark for weeks.
To start the immunofluorescence procedure, PBS is aspirated and 2 mL of blocking solution are added per well. Blockage is completed by incubating the slides for 30 mins at room temperature and washing the slides (three times) with PBS.
The primary antibody (rabbit IgG anti-LAMP-2A (Zymed Laboratories, Invitrogen, Carlsbad, CA) is diluted 1:100 in filtered 0.1% BSA in PBS. Slides are placed facing up in a Petri plate layered with Parafilm and 25 μL of diluted primary antibody are spotted on top of each slide. Incubation with the antibody is carried out in a wet chamber for 1 h at room temperature.
Slides are then extensively washed with PBS by picking up the slides with flat forceps and dipping them sequentially in four different reservoirs with 200 mL PBS each.
After aspirating the excess of PBS in the slides, they are incubated with 25 μL of the secondary antibody (diluted in filtered 0.1% BSA in PBS) for 30 min at room temperature in the wet chamber. Better results are obtained if the dilutions of the secondary antibodies are spun down before adding to the slides to remove any possible aggregated fluorochrome.
After extensive washes (as described in step 4), slides are incubated with 25 μL of the other primary antibody (mouse IgM anti-hsc70 (Abcam Inc., Cambridge, MA)) diluted 1:150 in filtered 0.1% BSA in PBS, under the same conditions (one h at room temperature in wet chamber).
After extensive washes (as described in step 4), slides are incubated with 25 μL of the secondary antibody (diluted in filtered 0.1% BSA in PBS) for 30 min at room temperature in the wet chamber as described in step 5, and washed (as described in step 4).
Slides are mounted by placing them face down on top of 15 μL of DAPI-containing mounting media spotted on a glass slide, and sealed with nail polish to prevent drying.
Slides are visualized in a fluorescence microscope (Axoivert 200, Carl Zeiss Ltd. Thornwood, NY), and captured images are subjected to deconvolution with the manufacture’s software. Colocalization of the two antibodies is quantified in thresholded images with the JACoP plug-in of the Image J software (NIH, MD). The mean distance of the vesicles positive for each antibody to the nucleus is calculated with the “straight lane tool” and the “analyze particles” function of the Image J software, by drawing lanes from the most distant vesicle positive for each antibody to the nucleus and computing the particle distribution (distance and density). An average of six different radial lines per cell and 20 cells per field is usually calculated to determine changes in the intracellular distribution of CMA + lysosomes (see Note 11).
4. Notes
Metrizamide has to be dissolved in the dark (beaker wrapped with foil paper) slowly to avoid the formation of solid clumps of undissolved material. We usually start with half of the final volume of water and keep adding metrizamide in little amounts while stirring. Adjusting the pH of the solution should be done with 0.01 M NaOH once the metrizamide is dissolved (wait until it stabilizes before adding the next drop). Our laboratory has tested different replacements for metrizamide (sucrose, premade solutions of Percoll or Nycodenz), but in all cases the level of enrichment of the lysosomes more active for CMA was considerably low and the stability of the lysosomal membrane (critical for the uptake experiments) was compromised.
We have used more than 20 different proteins for transport assays. The method of radiolabeling by reductive methylation is similar for all of them, but the size of the molecular exclusion matrix (Sephadex) for the separation of the labeled protein from the non-incorporated isotope should be chosen based on the molecular weight of the protein. As an example we have used Sephadex G-50 for GAPDH (144,000 kDa) and Sephadex G-25 for Ribonuclease A (16,000 kDa).
Liver perfusion is not strictly necessary. Extensive washing of the livers with sucrose removes most of the contaminant blood, and the remaining blood cells are eliminated in the pellet of the first centrifugation. Two critical factors to obtain a fraction of lysosomes with preserved lysosomal membrane stability are the temperature of isolation (all solutions should be kept on ice to prevent proteases released during the homogenization for damaging the cytosolic face of the lysosomal membrane) and the time required for the whole procedure. Eliminating liver perfusion shortens the time without compromising the purity of the lysosomal fraction.
Pipetting the lysosomal fraction through regular plastic 1000-μL micropipet tips reduces the lysosomal membrane stability considerably. We prefer instead to use 10-mL plastic pipets (wide mouth) when possible. Pipetting up and down the sucrose/metrizamide mixture four to five times should be enough to homogeneously mix the two solutions.
As pointed out before, it is critical to keep the temperature as low as possible (without freezing) to prevent protease activation. In order to do that when preparing the discontinuous metrizamide gradient, the tube should be placed inside an ice bucket (or a bigger tube filled with ice), leaving visible only the region where the metrizamide gradient is being formed. A gradient maker can be used if preferred, but with a little practice it is possible to overlay the different density metrizamide solutions with a Pasteur pipet and a rubber bulb without mixing of the phases.
As in any isolation procedure, it is essential to test the purity, enrichment, and stability of the isolated organelles. We analyze systematically for the activity of two different lysosomal enzymes (hexosaminidase and β-N-acetylglycosaminidase) in the homogenate and in the lysosomal fraction (which allows us to calculate the recovery and enrichment in lysosomal enzymes of the fraction). As possible contamination markers we measure lactic dehydrogenase activity (cytosolic marker), succinyl dehydrogenase activity (mitochondrial marker), and catalase activity (peroxisomal marker) using standard fluorimetric and colorimetric procedures as described before (28). Preparations with more than 1% of contamination are discarded. To test the stability of the lysosomal membrane, we centrifuge an aliquot of 5 μL of the lysosomal preparation at 25,000g for 5 min at 4°C, and measure the hexosaminidase activity in the supernatant (outside lysosomes) and in the pellet (inside lysosomes). Preparations with more than 10% of hexosaminidase activity outside lysosomes are discarded and they cannot be used for uptake experiments.
Secondary lysosomes for CMA uptake assays can also be isolated from cultured cells as described in detail in ref. 28. Cell lysis is a critical step in that procedure in order to guarantee proper lysosomal membrane stability. Nitrogen cavitation is the most effective method to disrupt the collected cells without altering the organelle’s membranes. Although sonication or mechanical homogenization is appropriate for isolation of other intracellular organelles, they consistently result in higher than 10% lysosomal membrane breakage after isolation.
Although we have not found differences between GST-hsc70 and hsc70 in their ability to recognize CMA substrates and target them for degradation to lysosomes, if desired, the GST tag can be cleaved by incubation with thrombin in cleavage buffer. The amount of GST-hsc70 can be estimated running a small aliquot of the bead-bound fraction on SDS-PAGE and staining with Coomassie blue. After incubation of the beads with 0.2 to 1% w/w thrombin/fusion protein for one hour at 25°C, they are washed three to four times with wash buffer where the cleaved protein is collected.
It is essential to determine that degradation is taking place inside the lysosomal lumen and not as result of the lysosomal enzymes released into the media. We systematically incubated lysosomes in parallel filtered plates at 37°C in the absence of radiolabeled substrate and measure (1) the hexosaminidase activity present at different times in the flow through (our values right after isolation are around 1-2% of the total lysosomal activity and reach 5-7% after a 30 min incubation) and (2) the proteolytic activity of the flow through when incubated with the same amount of radiolabeled protein as the experimental samples in the same conditions (to determine the percentage of degradation due to release of lysosomal enzymes). Experiments in which hexosaminidase activity or proteolysis by extralysosomal content is more than 10% of the one obtained with the total lysosomal fraction are discarded.
Proper controls should be added to verify that the treatment with proteinase K is effective but it is not disrupting the lysosomal membrane. We usually include an extra tube in which Triton X-100 (0.5% final concentration) is added to the samples in the presence of proteinase K. Under these conditions, the substrate is accessible to the protease and should be completely degraded. Persistence of undegraded GAPDH indicates that the amount of proteinase K is not enough to completely degrade GAPDH. To determine that the degradation by proteinase K is efficient we routinely monitor cleavage of the cytosolic tail of LAMP-2A (accessible to proteinase K) by immunoblot with a specific antibody. Persistence of the LAMP-2A tail indicates that the amount of proteinase K is not enough to remove all the GAPDH bound to the lysosomal membrane.
Methanol fixation is required to eliminate the soluble cytosolic hsc70 and to preserve the membrane-bound form. Routine negative controls in these studies include slides incubated with (1) only one of the secondary antibodies, (2) both secondary antibodies together, and (3) the primary LAMP-2A antibody with the secondary antibody for hsc70 and vice versa. Activation of CMA associates to the mobilization of the hsc70/LAMP-2A-enriched lysosomes from the periphery of the cell to the perinuclear area (Fig. 3). If the treatment of the cells results in changes in their shape/volume, values should be normalized to the total cellular area (calculated with the “freehand selection” and the “measure” tool of ImageJ).
Fig. 3.

Changes in the components and intracellular distribution of lysosomes active for CMA with the activation of this autophagic pathway. (A) Confluent cells with low CMA activity (i.e., growth in the presence of serum) or with high CMA activity (i.e., after prolonged (>10 h) serum removal) are subjected to immunofluorescence for LAMP-2A and hsc70 (the two main CMA components). Under conditions when CMA is maximally activated, the lysosomes active for CMA (those enriched in LAMP-2A and hsc70) relocate to the perinuclear region. (B) Increase in CMA activity usually results in an increase in the amount of CMA-active lysosomes, and this can be quantified as an increase in the colocalization of hsc70 and LAMP-2A in vesicular structures. (C) The mobilization of the CMA-active lysosomes from the cell periphery toward the perinuclear region can be quantified by drawing straight lines and comparing the distribution of the population of LAMP-2A/hsc70-positive vesicles with respect to the nucleus (distance to nucleus measured in μm).
Acknowledgments
We would like to gratefully acknowledge the members of our laboratory for their valuable suggestions. Research in our laboratory is supported by National Institutes of Health/National Institute of Aging grants AG021904 and AG19834 and an Ellison Medical Foundation Award.
References
- 1.Massey A, Zhang C, Cuervo A. Chaperone-mediated autophagy in aging and disease. Curr. Top. Dev. Biol. 2006;73:205–235. doi: 10.1016/S0070-2153(05)73007-6. [DOI] [PubMed] [Google Scholar]
- 2.Dice J. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem. Sci. 1990;15:305–309. doi: 10.1016/0968-0004(90)90019-8. [DOI] [PubMed] [Google Scholar]
- 3.Chiang H, Terlecky S, Plant C, Dice J. A role for a 70 kDa heat shock protein in lysosomal degradation of intracellular protein. Science. 1989;246:382–385. doi: 10.1126/science.2799391. [DOI] [PubMed] [Google Scholar]
- 4.Majeski A, Dice J. Mechanisms of chaperone-mediated autophagy. Int. J. Biochem. Cell Biol. 2004;36:2435–2444. doi: 10.1016/j.biocel.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Cuervo A, Dice J. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273:501–503. doi: 10.1126/science.273.5274.501. [DOI] [PubMed] [Google Scholar]
- 6.Salvador N, Aguado C, Horst M, Knecht E. Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. J. Biol. Chem. 2000;275:27447–27456. doi: 10.1074/jbc.M001394200. [DOI] [PubMed] [Google Scholar]
- 7.Agarraberes F, Terlecky S, Dice J. An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J. Cell. Biol. 1997;137:825–834. doi: 10.1083/jcb.137.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Terlecky S, Dice J. Polypeptide import and degradation by isolated lysosomes. J. Biol. Chem. 1993;268:23490–23495. [PubMed] [Google Scholar]
- 9.Aniento F, Papavassiliou AG, Knecht E, Roche E. Selective uptake and degradation of c-fos and v-fos by rat liver lysosomes. FEBS Lett. 1996;390:47–49. doi: 10.1016/0014-5793(96)00625-4. [DOI] [PubMed] [Google Scholar]
- 10.Wing S, Chiang HL, Goldberg AL, Dice JF. Proteins containing peptide sequences related to KFERQ are selectively depleted in liver and heart, but not skeletal muscle, of fasted rats. Biochem. J. 1991;275:165–169. doi: 10.1042/bj2750165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cuervo A, Knecht E, Terlecky S, Dice J. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am. J. Physiol. 1995;269:C1200–C1208. doi: 10.1152/ajpcell.1995.269.5.C1200. [DOI] [PubMed] [Google Scholar]
- 12.Kiffin R, Christian C, Knecht E, Cuervo A. Activation of chaperone-mediated autophagy during oxidative stress. Mol. Biol. Cell. 2004;15:4829–4840. doi: 10.1091/mbc.E04-06-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cuervo A, Hildebrand H, Bomhard E, Dice J. Direct lysosomal uptake of alpha2-microglobulin contributes to chemically induced nephropathy. Kidney Int. 1999;55:529–545. doi: 10.1046/j.1523-1755.1999.00268.x. [DOI] [PubMed] [Google Scholar]
- 14.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 15.Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. J. Biol. Chem. 2000;275:31505–31513. doi: 10.1074/jbc.M002102200. [DOI] [PubMed] [Google Scholar]
- 16.Cuervo AM, Dice JF, Knecht E. A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J. Biol. Chem. 1997;272:5606–5615. doi: 10.1074/jbc.272.9.5606. [DOI] [PubMed] [Google Scholar]
- 17.Cuervo A, Terlecky S, Dice J, Knecht E. Selective binding and uptake of ribonuclease A and glyceraldehyde-3-phosphate dehydrogenase by isolated rat liver lysosomes. J. Biol. Chem. 1994;269:26374–26380. [PubMed] [Google Scholar]
- 18.Aniento F, Roche E, Cuervo AM, Knecht E. Uptake and degradation of glyceraldehyde-3-phosphate dehydrogenase by rat liver lysosomes. J. Biol. Chem. 1993;268:10463–10470. [PubMed] [Google Scholar]
- 19.Dworniczak B, Mirault M. Structure and expression of a human gene coding for a 71 kd heat shock ‘cognate’ protein. Nucl. Acids Res. 1987;15:5181–5197. doi: 10.1093/nar/15.13.5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Auteri J, Okada A, Bochaki V, Dice J. Regulation of intracellular protein degradation in IMR- 90 human diploid fibroblasts. J. Cell Physiol. 1983;115:159–166. doi: 10.1002/jcp.1041150210. [DOI] [PubMed] [Google Scholar]
- 21.Finn PF, Dice JF. Ketone bodies stimulate chaperone-mediated autophagy. J. Biol. Chem. 2005;280:25864–25870. doi: 10.1074/jbc.M502456200. [DOI] [PubMed] [Google Scholar]
- 22.Fuertes G, Martin De Llano J, Villarroya A, Rivett A, Knecht E. Changes in the proteolytic activities of proteasomes and lysosomes in human fibroblasts produced by serum withdrawal, amino-acid deprivation and confluent conditions. Biochem. J. 2003;375:75–86. doi: 10.1042/BJ20030282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM. Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA. 2006;103:5905–5910. doi: 10.1073/pnas.0507436103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cuervo A, Dice J. Unique properties of lamp2a compared to other lamp2 isoforms. J. Cell Sci. 2000;113:4441–4450. doi: 10.1242/jcs.113.24.4441. [DOI] [PubMed] [Google Scholar]
- 25.Cuervo A, Dice J. Regulation of lamp2a levels in the lysosomal membrane. Traffic. 2000;1:570–583. doi: 10.1034/j.1600-0854.2000.010707.x. [DOI] [PubMed] [Google Scholar]
- 26.Lowry O, Rosebrough N, Farr A, Randall R. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 27.Laemmli U. Cleavage of structural proteins during the assembly of the head of the bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 28.Storrie B, Madden E. Isolation of subcellular organelles. Methods Enzymol. 1990;182:203–225. doi: 10.1016/0076-6879(90)82018-w. [DOI] [PubMed] [Google Scholar]