

Abstract
It has been proposed that inhibitors of an oncogene's effects on multipotent hematopoietic progenitor cell differentiation may change the properties of the leukemic stem cells and complement the clinical use of cytotoxic drugs. Using zebrafish, we developed a robust in vivo hematopoietic differentiation assay that reflects the activity of the oncogene AML1-ETO. Screening for modifiers of AML1-ETO-mediated hematopoietic dysregulation uncovered unexpected roles of COX-2 and β-catenin-dependent pathways in AML1-ETO function. This approach may open doors for developing therapeutics targeting oncogene function within leukemic stem cells.
INTRODUCTION
The oncogenes that cause many types of leukemia (including acute myelogenous leukemia, AML) function by dysregulating both the proliferation and differentiation of hematopoietic cells. Current treatments for leukemia focus primarily on proliferation, utilizing cytotoxic agents to kill the bulk of leukemic cells, which are highly proliferative. Even after aggressive cytotoxic treatment, 75% of AML patients experience a recurrence within 2 years of remission1. This may be due to the inability of cytotoxic agents to effectively eradicate the leukemic stem cells (LSC), which are less proliferative2,3. Therefore, targeting cell proliferation may be insufficient for eradicating leukemia. Therapies that can reverse the effects of oncogenes on LSC differentiation could be promising alternatives or complements to cytotoxic agents4.
We sought to identify small molecules that target oncogenic function in multipotent hematopoietic progenitor cells (HPCs), especially compounds that can reverse the abnormal cell differentiation that occurs in these cells. Many oncogenes found in leukemia are transcription factors that regulate hematopoietic differentiation5. However, in primary hematopoietic stem cells and in mice, measuring the effects of the oncogenes on hematopoietic differentiation is laborious and requires long incubation times6-9. Thus, these systems are not well suited for high-throughput experimentation. Interestingly, the embryonic zebrafish may be a powerful model that can both recapitulate the effects of oncogenes in multipotent HPCs and enable high-throughput chemical screens10. During development, zebrafish embryos possess blood islands made up of multipotent HPCs11,12. These pools of multipotent HPCs commit to hematopoietic differentiation in synchrony, thus offering unique opportunities to investigate the mechanisms by which oncogenes disrupt hematopoietic differentiation in vivo.
We recently showed that a leukemic oncogene AML1-ETO (AE) robustly converts erythropoiesis to granulopoiesis and blocks the maturation of the granulocytes in the posterior blood island of the embryonic zebrafish13. The cell fate redirection and the differentiation defects evident in the zebrafish also occur in humans expressing AE. The majority of AML patients expressing AE exhibit overproduction of granulocytic blast cells at the expense of other blood cell types14,15. Thus, the zebrafish model of AE may be useful for identifying chemical modifiers of AE's effects on HPCs in vivo.
We utilized the transgenic zebrafish line, Tg(hsp:AML1-ETO), which expresses AE under the control of the zebrafish hsp70 heat shock promoter13. Only 90 minutes after heat-induced AE expression, changes in hematopoietic cell fate are evident by the downregulation of gata1 and scl in the posterior blood island. Within 24 hours, accumulation of mpo+ granulocytic cells is observed13. In this report, using an in vivo chemical suppressor screen, we identify compounds that reverse gata1 downregulation in transgenic AE embryos. The compounds identified from this screen can also suppress AE-induced mpo upregulation, a phenotype that resembles the clinical manifestation of AE-associated leukemias14,15. By studying the mechanisms by which nimesulide (1), one of the compounds identified from the screen, antagonizes AE's effects, we demonstrate the previously unknown roles of COX-2 and β-catenin in AE-mediated hematopoietic differentiation. Our findings suggest the hypothesis that therapeutics that can specifically affect PGE2 signaling or inhibit β-catenin-dependent pathways may provide therapeutic benefit in AML by blocking AE's effects on hematopoietic differentiation. In addition, given the challenge of developing therapeutics directly against oncogenic transcription factors, the method presented herein provides a route to uncover novel therapeutic targets involved in oncogene-regulated hematopoietic differentiation.
RESULTS
Identifying chemical suppressors of the AE phenotype
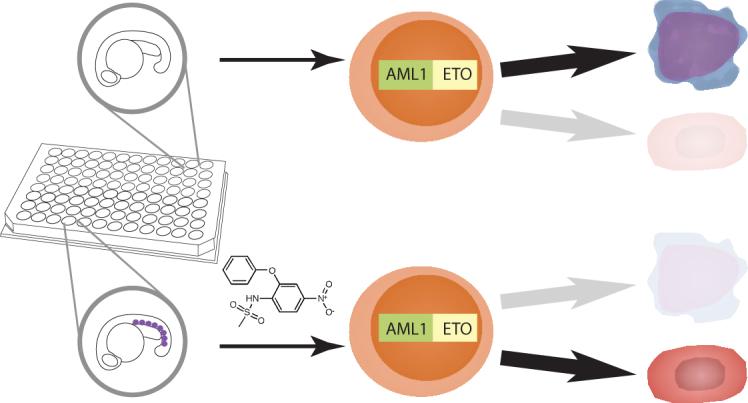
We conducted a chemical screen of 2,000 bioactive compounds to identify small molecules that restore gata1 expression in heat-treated Tg(hsp:AML1-ETO) embryos. A schema of the chemical suppressor screen is shown (Fig. 1). In brief, matings were set up between homozygous Tg(hsp:AML1-ETO) and wild-type fish to obtain heterozygous Tg(hsp:AML1-ETO) embryos. About 30 breeding pairs yielded enough embryos for four to eight 96-well screening plates per week. Five embryos were arrayed into each well, and the chemical library was added at 12−16 hours post-fertilization (hpf) for one hour. To induce AE expression, the plates were incubated at 40 °C for 1 hour. We have previously shown that incubating Tg(hsp:AML1-ETO) embryos at 39−40 °C for 1 hour (as compared to normal fish water temperature at 28.5 °C) leads to the AE phenotype, including downregulation of gata1 expression, in complete penetrance. At 90 minutes after the heat treatment, the embryos were fixed with paraformaldehyde solution for subsequent gata1 staining.
Figure 1.

Screening for chemical suppressors of AE. Homozygous Tg(hsp:AML1-ETO) fish were crossed with wild-type fish to generate thousands of heterozygous Tg(hsp:AML1-ETO) embryos. These embryos were raised for 12−16 hours post fertilization at which point five embryos were arrayed into each well of the 96-well plates. The compounds from the library were added to the plates. An hour later, the plates were heat-shocked at 40 °C for 1 hour to induce AE expression. At 90 minutes after the heat shock, the embryos were processed for in situ hybridization of gata1. Induced expression of AE resulted in lost of gata1+ hematopoietic cells (indicated as purple dots) in the posterior blood islands of the zebrafish embryos. However, the chemical suppressors of AE, such as nimesulide (shown in the figure), antagonized AE's effect, restoring gata1 expression in Tg(hsp:AML1-ETO) embryos.
We scored the hits based on the requirement that at least four out of the five embryos in the well exhibit strong gata1 staining. Based on this stringent criterion, we identified 22 hits during the initial screen, and confirmed 15 hits after re-test (Supplementary Table 1 online). Among these 15 hits, 5 compounds (rotenone (2) and its analogues) are structurally related, whereas the remaining compounds do not have obvious structural similarities. In addition, the known biological effects and uses of these compounds vary widely.
Interestingly, we found that sodium valproate (3) is able to reverse AE's effect in our screen (Supplementary Table 1 online). Valproate is administered clinically as an anticonvulsant and mood-stabilizing drug due to its effects on the function of the neurotransmitter GABA. In addition, valproate is also a histone deacetylase (HDAC) inhibitor16. Since the recruitment of HDAC by the ETO domain of AE is believed to play an important role in AE-mediated pathogenesis, the clinical utility of the HDAC inhibitors against AE-associated AML is currently being investigated. We had previously shown that another HDAC inhibitor, trichostatin A (4), can reverse the effects of AML1-ETO in this zebrafish model13. Valproic acid itself has been shown to induce differentiation and apoptosis of transformed cells and human AML samples expressing AE17-19. The identification of an HDAC inhibitor in our screen supports the validity of the screen and suggests that other compounds identified from the screen may also possess therapeutic potential.
We recognized that some of the compounds identified in this screen may reverse AE's effect by interfering with the inducible expression of AE after heat treatment. For example, cycloheximide (5), also identified from the screen, is likely to block the translation of AE protein, suppressing the AE phenotype. We therefore re-ordered six compounds for follow-up experiments based on the availability of the compounds and our interests (Supplementary Table 1 online). We first conducted Western blot analysis with anti-AML1 antibody and found that rotenone, but not nimesulide, abrogated AE expression in heat-treated Tg(hsp:AML1-ETO) embryos (Fig. 2). Rotenone not only blocks AE expression, but also eliminates the expression of Hsp70 (Fig. 2). Subsequently, we found that rotenone analogue mundoserone (6) and two other compounds including bithionol (7) and dichlorophene (8) also affected AE expression in Tg(hsp:AML1-ETO) embryos. However, AE expression in dicumarol (9)-treated embryos was not affected (Supplementary Table 1 online). Thus, rotenone, rotenone analogues, and at least two of the other compounds identified from the screen retain gata1 expression in Tg(hsp:AML1-ETO) embryos by blocking the heat shock response of the embryonic zebrafish. Two compounds, nimesulide and dicumarol restore gata1 expression without affecting AE expression and therefore received priority for follow up experiments.
Figure 2.

Nimesulide does not affect AE expression in Tg(hsp:AML1-ETO) embryos. Western blot analysis shows that while rotenone (0.15 μM) inhibits heat-induced expression of AE and Hsp70, neither nimesulide (40 μM) nor trichostatin A (TSA, 0.5 μM) affects heat-induced expression of AE and Hsp70. The expression of Akt is used as a reference for protein loading. At the concentrations used, all three compounds suppress the zebrafish AE phenotype. DMSO is used as the vehicle control. WT+, wild-type embryos with heat treatment. AE-, Tg(hsp:AML1-ETO) embryos without heat treatment. AE+, Tg(hsp:AML1-ETO) embryos with heat treatment. The vertical black line indicates the juxtaposition of lanes that were not contiguous in the original gel.
PGE2 modulates AE's effects on hematopoiesis
We sought to understand the mechanism by which nimesulide reverses AE function in Tg(hsp:AML1-ETO) embryos without affecting AE expression. Nimesulide is known to inhibit cyclooxygenase-2. Cyclooxygenase-1 (COX-1) and -2 (COX-2) are two very closely related enzymes that play important roles in the biosynthesis of lipid mediators including prostaglandins. We tested whether other inhibitors of COX-2 are able to block the cell fate change caused by AE. In addition to nimesulide, which effectively restored gata1 expression in Tg(hsp:AML1-ETO) embryos (13/14 embryos), both a selective COX-2 inhibitor NS-398 (10) and a non-selective COX inhibitor indomethacin (INDM, 11) were able to restore gata1 expression in 8/13 and 9/17 embryos, respectively (Fig. 3a-b). Treatment with nimesulide, NS-398 or INDM did not affect gata1 expression in the absence of AE expression (Fig. 3b). These results indicate that the ability to suppress the AE phenotype is not unique to nimesulide but is shared by other COX inhibitors. Interestingly, there are more than 10 compounds in the chemical library we screened (including INDM) that have reported activities against the cyclooxygenases but were not detected in the screen. It is possible that some of these failed to register as hits because they were tested at too low a dose or because the stringency of the assay left out compounds that may exhibit partial rescue of the AE phenotype.
Figure 3.

Cyclooxygenase (COX) inhibitors reverse AE's effects on hematopoietic differentiation. (a) The chemical structures of the selective COX-2 inhibitors nimesulide, NS-398, and the non-selective COX inhibitor indomethacin (INDM). (b) In situ hybridization of gata1. The COX inhibitors restore gata1 expression in Tg(hsp:AML1-ETO) embryos. To induce AE expression, Tg(hsp:AML1-ETO) embryos were incubated at 39 °C for 1 hour at the 16-somite stage (w/ heat). Induced expression of AE results in the loss of gata1 expression as compared to Tg(hsp:AML1-ETO) embryos without heat incubation. Nevertheless, addition of nimesulide (40 μM), NS-398 (25 μM) or INDM (20 μM) reverses AE's effects. All compounds were added to the embryos 1 hour before the heat shock. DMSO was used as the vehicle control. Scale bar, 0.3 mm. (c) In situ hybridization of mpo. NS-398 (25 μM) and INDM (20 μM) antagonize AE's effect, suppressing the accumulation of mpo+ granulocytic cells. The antagonism no longer exists in the presence of prostaglandin E2 (20 μM). All compounds were added at the end of heat shock. Scale bar, 0.3 mm.
Next, we examined whether the COX inhibitors were able to suppress the accumulation of mpo+ granulocytic cells in Tg(hsp:AML1-ETO) embryos. We used NS-398 instead of nimesulide because the latter caused a significant developmental delay after prolonged treatment. Upon heat treatment, induced expression of AE resulted in accumulation of mpo+ granulocytic cells (Fig. 3c). Compared to the DMSO-treated embryos, NS-398 or INDM-treated embryos showed reduced mpo expression (NS-398, 19/29; INDM, 21/25), indicating that both compounds antagonize AE's effect on hematopoietic differentiation (Fig. 3c). We found that treatment of NS-398 or INDM did not affect the differentiation of mpo+ granulocytic cells in the absence of AE expression (Fig. 3c). Moreover, addition of the major metabolite of the cyclooxygenases in zebrafish, prostaglandin E2 (PGE2, 12)20, reversed the effects of NS-398 and INDM. PGE2 restored mpo upregulation in 14/16 and 11/14 embryos treated with NS-398 and INDM, respectively (Fig. 3c). These results strongly suggest that the biosynthesis of PGE2 is required for AE to exert its effects on zebrafish HPCs.
Interestingly, it was reported recently that PGE2 increases the number of hematopoietic stem cells in zebrafish embryos and enhances the output of multipotent HPCs in in vitro and murine transplantation assays21. These data raise the possibility that PGE2 may not be directly involved in cell fate determination. Instead, PEG2 may promote the expansion of HPCs in Tg(hsp:AML1-ETO) embryos, leading to the accumulation of mpo+ granulocytic cells. We added PGE2 to wild-type embryos at 14 hpf and examined the expression of hematopoietic genes, including gata1, scl, mpo and l-plastin, at various stages. We found that PGE2 does not affect the levels of expression of the genes tested (Supplementary Figure 1 online). On the other hand, we show that PGE2 synergizes with AE in inducing the accumulation of mpo+ cells (Supplementary Figure 2 online). When a mild heat treatment (37 °C for 1 hour instead of 39−40 °C for 1 hour) was applied to Tg(hsp:AML1-ETO) embryos, the typical AE phenotype was not observed. However, combination of PGE2 and the mild heat treatment resulted in the full AE phenotype, including downregulation of gata1 (18/30 embryos) and upregulation of mpo (25/33 embryos). Thus, PGE2 by itself does not induce hematopoietic cell fate changes in wild-type zebrafish embryos, nor does it increase the numbers of specified hematopoietic cells in 1−2 day-old zebrafish embryos. These data suggest that some other downstream component(s) of AE signaling may collaborate with PGE2 to mediate the observed hematopoietic effects of AE.
COXs are important downstream mediators of AE's effects
To test whether AE regulates the expression of the genes encoding COXs, we used real-time PCR to examine the expression profiles of the hematopoietic cells isolated from either wild-type or Tg(hsp:AML1-ETO) embryos that have been subjected to the same heat treatment. The hematopoietic cells were extracted at two different time point, 22 and 40 hpf. One ortholog of the gene encoding COX-1 (ptgs1) and two orthologs of the gene encoding COX-2 (ptgs2a and ptgs2b) have been identified in the zebrafish20,22. We found that the expression of ptgs2b was induced 3−10 fold in Tg(hsp:AML1-ETO) embryos at 22 hpf, only 2 hours after the induction of AE expression, and its level returned to close to the wild-type level at 40 hpf (Fig. 4a-b). In contrast, the expression of ptgs2a was not affected at 22 hpf, but it became upregulated 4−8 fold in the Tg(hsp:AML1-ETO) embryos at 40 hpf. Moreover, the expression of ptgs1 in Tg(hsp:AML1-ETO) embryos was similar to that in the wild-type embryos at 22 hpf but reduced to about 30% compared to the wild-type embryos at 40 hpf. These data indicate that expression of AE upregulates the expression of both COX-2 genes but not the COX-1 gene in hematopoietic cells. This finding agrees with the known characteristics of the COX genes. In mammals, COX-1 is expressed constitutively to serve housekeeping functions, whereas COX-2 is inducible by various stimuli under both physiological and pathological conditions23.
Figure 4.

The hematopoietic phenotype of AE involves induction of the genes that encode both zebrafish COX-2 isoforms. (a) Real-time PCR indicates that ptgs2b is upregulated in the hematopoietic cells of Tg(hsp:AML1-ETO) embryos at 2 hours after heat shock. Total RNA was extracted from the hematopoietic cells isolated from heat-treated wild-type and Tg(hsp:AML1-ETO) embryos at 22 hpf. The expression of ptgs1, ptgs2a, and ptgs2b was evaluated by real-time PCR analysis using two independent primer sets for each gene, and was normalized to gapdh expression. The fold change indicates the ratio of mRNA expression between Tg(hsp:AML1-ETO) and wild-type embryos. ptgs1, 2.09±0.0.91, 1.22±0.08; ptgs2a, 0.62±0.21; 1.04±0.03; ptgs2b, 10.31±1.39, 3.60±1.25 (mean±SEM). (b) Real-time PCR indicates that ptgs2a is upregulated in the hematopoietic cells of Tg(hsp:AML1-ETO) embryos at 40 hpf. ptgs1, 0.26±0.12, 0.28±0.11; ptgs2a, 3.86±0.36; 7.6±0.98; ptgs2b, 1.28±0.39, 1.45±0.81 (mean±SEM). (c) In situ hybridization of gata1. Knockdown of ptgs1, ptgs2a, or ptgs2b partially restored gata1 expression in heat-treated Tg(hsp:AML1-ETO) embryos. Scale bar, 0.3 mm. (d) In situ hybridization of mpo. Knockdown of ptgs2a or ptgs2b suppresses AE-induced mpo expression. Control, non-injected embryos. Scale bar, 0.3 mm.
Next, we examined whether the AE-induced hematopoietic differentiation effects were altered by knockdown of the COX genes using antisense morpholino oligonucleotides (MO). We found that knockdown of any of the COX genes partially restored gata1 expression (ptgs1, 14/35; ptgs2a, 14/30; ptgs2b, 10/41) in heat-treated Tg(hsp:AML1-ETO) embryos (Fig. 4c). However, knockdown of ptgs1 was able to suppress the upregulation of mpo in only 17 % of Tg(hsp:AML1-ETO) embryos (7/41), whereas knockdown of ptgs2a and ptgs2b suppressed the upregulation of mpo in 72 and 57 % of the embryos (ptgs2a, 34/47; ptgs2b, 20/35) expressing AE (Fig. 4d). It has been shown that various degrees of ptgs1 knockdown can cause phenotypes ranging from gastrulation arrest to specific vascular defects in the trunk region in zebrafish embryos, whereas specific phenotypes from knockdown of ptgs2a or ptgs2b have not been reported20,24,25. At the concentrations of MOs that we injected (100−400 μM), we observed normal levels of gata1 and mpo expression in Tg(hsp:AML1-ETO) embryos that were injected with MOs against any of the three COX genes but were not subjected to the heat treatment (Fig. 4c-d). These results indicate that knockdown of the COX genes suppresses AE-mediated hematopoietic effects, but that knockdown of COX-2 is much more effective than knockdown of COX-1. Knockdown of ptgs1 can partially rescue gata1 expression, suggesting that the level of PGE2 provided by the constitutive ptgs1 expression aids in at least one of AE's early effects. However, knockdown of either of the two genes encoding COX-2 proteins, ptgs2a and ptgs2b, is more effective in suppressing AE-induced mpo expression at the later time, indicating that the expression of the COX-2 genes induced by AE is critical for AE's hematopoietic effects in the zebrafish.
NS-398 reverses AE effects in human cells
To investigate whether COX-2 also plays a role in AE-mediated hematopoietic dysregulation in mammalian cells, we obtained two previously established clones of human myelogenous leukemia K562 cells expressing either GFP or AE-GFP6. It has been shown that K562 cells can spontaneously differentiate into either erythroid or myeloid cells in cell culture, and that the expression of AE-GFP decreases the erythroid differentiation efficiency of these cells. Using real-time RT-PCR, we found that PTGS2 expression in the AE-GFP+ cell clone was 4.798±0.373 fold higher than in the cell clone expressing GFP (Fig. 5a). However, the expression levels of PTGS1 were similar (0.962±0.169 fold) in both cell clones. Moreover, while AE-GFP expression reduced erythroid differentiation efficiency as measured by benzidine staining for the presence of hemoglobin, NS-398 treatment restored erythroid differentiation of the AE-GFP+ cell clone (Fig. 5b). These data indicate that, consistent with the findings in the zebrafish, AE upregulates the gene encoding COX-2 and reduces erythroid differentiation, while inhibition of COX-2 blocks the AE-dependent differentiation defect in human multipotent hematopoietic cells.
Figure 5.

Erythroid differentiation of human myelogenous leukemia K562 cells is attenuated by AE via a COX-2-dependent mechanism. (a) Real-time PCR indicates that expression of AE causes upregulation of PTGS2 but not PTGS1 in K562 cells. K562 cells containing stable integration of the vector pLRT-GFP (clone B9) or the AE expression vector pLRT-AE (clone D8) were harvested for RNA extraction. The expression of PTGS1 and PTGS2 was evaluated by real-time PCR analysis and normalized to 18S RNA expression. The fold change indicates the ratio of mRNA expression between D8 and B9 cells. PTGS1, 0.962±0.169; PTGS2, 4.798±0.373 (mean±SEM). (b) Erythroid differentiation efficiency as scored by benzidine staining. While AE suppresses erythroid differentiation of K562 cells, inhibition of COX-2 by NS-398 (75 μM) reverses AE's effect. K562 clone B9 (Control) and clone D8 (AE) were treated with either DMSO or NS-398 for 4 days before staining. Control-DMSO, 24.5±0.65; Control-NS-398, 25±0.58; AE-DMSO, 16.25±1.03; AE-NS-398, 25.75±1.89 (mean±SEM). *p<0.01 (two-tailed t-test).
AE activates β-catenin-TCF-dependent transcription
So far, very little is known about the potential role of PGE2 in leukemogenesis. However, it has been clearly demonstrated that PGE2 plays an important role in the pathogenesis of colon cancers26,27. COX-2 upregulation has been observed in many epithelial tumors and tumor cell lines including the intestinal epithelia of colon cancer patients harboring adenomatous polyposis coli (APC) mutations (see review28). Meanwhile, PGE2 has been shown to activate β-catenin-dependent signaling, promoting the proliferation of the cells containing APC mutations29-31. Thus, we investigated whether a similar signaling pathway is also being utilized by AE in hematopoietic cells. Using a β-catenin-TCF reporter (TOPflash) assay, we show that overexpression of AE induces reporter activity about 12 fold (12.29±1.09, p=0.0005) in K562 cells (Fig. 6a). In addition, AE-induced reporter activity can be reversed by co-expression of a dominant negative form of TCF, dnTCF (2.93±0.3, p=0.0012), and by the COX-2 inhibitor NS-398 (5.29±0.57, p=0.0047) (Fig. 6a). Furthermore, addition of either PGE2 or 16,16-dimethyl prostaglandin E2 (dmPGE2, 13, a PGE2 analog with a long half-life) also induced β-catenin-TCF-dependent transcription in K562 cells (PGE2, 2.44±0.15, p=0.0008; dmPGE2, 9.06±1.02, p=0.0014) (Fig. 6b). Co-expression of dnTCF suppressed the reporter activities upregulated by PGE2 (0.83±0.1, p=0.0008) and by dmPGE2 (1.89±0.38, p=0.0028). These data suggest that AE activates β-catenin signaling through COX-mediated prostaglandin synthesis.
Figure 6.

AE activates β-catenin-dependent transcription through COX-2. (a) Expression of AE induces TOPflash luciferase activity, which can be suppressed by overexpression of a dominant-negative form of TCF (dnTCF) or by NS-398 (75 μM). The empty vector (pCS2) or the expression vector for AE (pCS2-AE) was transfected into K562 cells along with TOPflash and pRL-tk which encodes Renilla luciferase for normalizing the transfection efficiency. The results are shown in relative TOPflash luciferase activities after normalization. pCS2/control, 1±0.09; pCS2+dnTCF, 0.73±0.03; pCS2+NS-398, 0.58±0.01; pCS2-AE/control, 12.29±1.09, pCS2-AE+dnTCF,2.93±0.3; pCS2-AE+NS-398, 5.29±0.57 (mean±SEM). *p<0.01 (two-tailed t-test). (b) 16,16-dimethylprostaglandin E2 (dmPGE2) activates β-catenin-TCF-dependent transcription. K562 cells were transfected with TOPflash and pRL-tk. At 4 hours after transfection, PGE2 (20 μM), dmPGE2 (20 μg ml−1) or DMSO was added to the cells. The luciferase activities were measured two days after the transfection. In addition, the dmPGE2-induced TOPflash activity can be suppressed by overexpression of dnTCF. control, 1±0.05; control+dn-TCF, 0.56±0.03; PGE2, 2.44±0.15; PGE2+dnTCF, 0.83±0.1; dmPGE2, 9.06±1.02; dmPGE2+dnTCF, 1.89±0.38 (mean±SEM). *p<0.01 (two-tailed t-test).
β-catenins are required for AE's hematopoietic effects
To confirm the roles of β-catenin in AE-regulated hematopoietic differentiation, we performed genetic knockdown of the genes encoding β-catenin-1 and β-catenin-2 (ctnnb1 and ctnnb2) in Tg(hsp:AML1-ETO) embryos. It has been shown that β-catenin-1 and β-catenin-2 possess overlapping and non-overlapping functions in regulating dorsoventral patterning and that knockdown of either ctnnb1 or ctnnb2 can cause developmental defects in zebrafish embryos32. We injected antisense morpholino oligonucleotides against either ctnnb1 or ctnnb2 at 0.5 mM and found that the injected embryos looked grossly normal except for a subset of animals that exhibit a different tail morphology compared to the non-injected embryos (Fig. 7). Most importantly, knockdown of either gene did not affect the expression of gata1 at 20 hpf, or the expression of mpo at 36 hpf in the absence of AE (Fig. 7a-b). However, knockdown of either ctnnb1 or ctnnb2 compromised AE's effects, restoring gata1 expression (10/12 and 11/12 embryos, respectively) and suppressing mpo upregulation (19/49 and 17/37, respectively) in heat-treated Tg(hsp:AML1-ETO) embryos (Fig. 7a-b).
Figure 7.

The hematopoietic differentiation effects caused by AE are dependent on β-catenin function. (a-b) In situ hybridization of gata1 (a) and mpo (b). Knockdown of either β-catenin1 ctnnb1 MO) or β-catenin2 (ctnnb2 MO) restores gata1 expression and suppresses mpo upregulation in the presence of AE. Control, non-injected embryos. Scale bar, 0.3 mm.
In addition, we tested whether pharmacological activation of β-catenin signaling by a GSK-3β inhibitor BIO (14) could enhance AE's effects on gata1 and mpo expression33. Activation of β-catenin signaling may cause toxicity during early development34,35. Thus, BIO was added to Tg(hsp:AML1-ETO) embryos at 14 hpf after gastrulation was complete. We found that BIO treatment did not affect the gross morphology of the embryos or the expression of gata1 or mpo in the absence of AE expression (Supplementary Figure 3 online). Nevertheless, BIO treatment caused the downregulation of gata1 (20/30 embryos) and upregulation of mpo (21/32 embryos) in Tg(hsp:AML1-ETO) embryos incubated in a mild heat shock condition (BIO, 37 °C 1h), even though the control Tg(hsp:AML1-ETO) embryos (DMSO, 37 °C 1h) did not exhibit the AE phenotype. These data indicate that, along with COX-2, β-catenin plays an important role in AE-regulated hematopoietic differentiation.
DISCUSSION
Previously, we demonstrated that early zebrafish embryos can be used as a simple and efficient surrogate model to detect the hematopoietic differentiation defects induced by the expression of the leukemic oncogene AE. During normal zebrafish development, HPCs in the posterior blood island differentiate synchronously, providing a population of highly-visible, manipulable HPCs that are useful for testing the effects of oncogenes on hematopoietic differentiation. Inducing AE expression in early zebrafish embryos causes rapid, highly-penetrant cell fate switching, converting erythropoiesis to myelopoiesis in multipotent HPCs13. Exploiting this fact and the amenability of zebrafish to small molecule screening, we were able to screen a small molecule library and discover compounds that antagonize the activity of AE in the HPCs. It is likely that a similar approach might be used to efficiently detect the hematopoietic differentiation effects mediated by other leukemic oncogenes and to identify compounds that antagonize those oncogenes.
We chose the heat shock promoter to drive the expression of AE in the zebrafish. The heat shock promoter is able to confer a rapid and strong induction of AE, while keeping the basal expression low enough to sustain the viability of the transgenic animals. However, in our follow-up experiments, we found 4 compounds (rotenone, mundoserone, bithionol, and dichlorophene) that affect the heat shock response in the zebrafish embryos. These 4 compounds and 3 other rotenone analogs (β-dihydrorotenone (15), deguelin (16), and α-toxicarol (17)) also identified in our screen are therefore unlikely to be true antagonists of AE function. The large number of hits that affect the heat shock response may be unique to the chemical library that we used. Nevertheless, our data suggest that future studies may benefit from choosing other promoters that are less susceptible to chemical inhibition.
From our screen, we identified a selective COX-2 inhibitor nimesulide that does not affect the inducible expression of AE, but antagonizes the effects of AE on hematopoietic differentiation. Further experimentation pointed to a critical role for COX expression and PGE2 in AE's hematopoietic effects. We demonstrate that expression of AE activates the transcription of a β-catenin-TCF reporter construct in K562 cells through COX2-dependent signaling. Although we have not investigated the mechanisms by which PGE2 activates β-catenin signaling in our systems, several potential mechanisms have been shown in vitro as well as in vivo29-31,36,37. Most importantly, our results indicate that, in addition to inhibition of COX-2-dependent signaling, inhibition of β-catenin signaling may be sufficient to antagonize AE's effects on multipotent HPC differentiation.
The exact functions of PGE2 and β-catenin activation are still unclear. It has been shown that PGE2 causes the expansion of definitive hematopoietic stem cell markers in zebrafish embryos at 36 hpf21. In addition, inhibitors of GSK-3β have been shown to increase the repopulating efficiency of hematopoietic stem cells38. In principle, PGE2 and β-catenin activation may therefore augment AE's effect by expanding the progenitors exhibiting an altered cell fate. However, addition of PGE2 or BIO by themselves did not cause the erythroid-myeloid cell fate change in wild-type zebrafish embryos, nor did they lead to accumulation of mpo+ granulocytes. Thus, the activities of AE are not due exclusively to their effects on the size of the hematopoietic progenitor pool, nor is increasing PGE2 levels sufficient to emulate all of AE's functions. It is likely that another downstream target(s) of AE is also required for the observed hematopoietic differentiation defects. Future mode-of-action studies on other chemical suppressors of AE identified in this screen (such as dicumarol) as well as conducting a larger scale chemical screen may help identify additional contributors to AE-mediated hematopoietic dysregulation.
Our results identify essential roles of PGE2 and β-catenin in AE-dysregulated hematopoietic differentiation. Inhibitors of these pathways may therefore exert therapeutic benefits in treating human AML. As a cautionary note, activation of PGE2 and β-catenin pathways have been shown to cause expansion of the HSC pool and to enhance the repopulating efficiency of the HSC, respectively21,39. Thus, inhibition of these two pathways might be hypothesized to compromise the function of normal HSCs. On the other hand, several lines of evidence contradict this idea. For example, it has been shown that conditional knockout of β-catenin in bone marrow does not affect hematopoiesis or HSC function40. Similarly, despite the presence of renal, cardiac, and gastric abnormalities, both COX-1 and COX-2 null mice can grow to adulthood. There are no reported hematopoietic defects in these mice despite altered inflammatory responses to some inflammatory stimuli41-43. It remains to be determined whether or not blocking the hematopoietic differentiation effects of AE in multipotent HPCs would offer therapeutic benefit to patients with AE-associated AML. The findings presented here provide impetus to test that hypothesis in future studies using mammalian models.
Many attributes of the zebrafish have made this model organism suitable for in vivo, high-throughput chemical screens10. Furthermore, in vivo chemical screens may simultaneously provide information about the efficacy as well as the toxicity of each compound, increase the possibility of identifying pro-drugs (compounds that need to be metabolized to become active), and identify compounds that work in a non-cell autonomous fashion. The effects of several human oncogenes other than AE have also been studied in the zebrafish (see review44). These studies and ours collectively show that many human oncogenes are able to elicit phenotypes relevant to human cancers in either embryonic or adult zebrafish. The molecular mechanisms of many of these oncogenes remain poorly understood. Nevertheless, phenotype-based chemical screens in the zebrafish represent a simple and efficient option for drug discovery, even when validated downstream therapeutic targets are not yet available.
METHODS
Chemicals
The SPECTRUM library (Microsource Discovery Systems, Inc.) containing 2,000 known bioactive compounds was used for the screening. The compounds for the follow-up experiments including rotenone and nimesulide were purchased from Microsource Discovery Systems. NS-398 and indomethacin were purchased from Calbiochem.
Screening for AE antagonists using the in vivo hematopoietic differentiation assay
All zebrafish experiments were approved by the Massachusetts General Hospital Subcommittee on Research Animal Care. Homozygous Tg(hsp:AML1-ETO) fish were crossed with wild-type fish to generate heterozygous Tg(hsp:AML1-ETO) embryos. At 12−16 hours post fertilization (hpf), the chorions were removed by pronase (0.5 μg ml−1) treatment. Subsequently, the embryos were rinsed thoroughly with E3 buffer (5 mM NaCl, 0.17 mM Kcl, 0.33 mM CaCl2, 0.33 mM MgSO4). Five embryos were manually arrayed into each well of the 96-well plates containing 250 μl of E3. A half microliter of the compound library was then added to reach a final concentration of 20 μM. One hour later, the plates were heat-shocked in a water bath at 40 °C for 1 hour to induce AE expression. The plates were then moved to a 28.5 °C incubator for 90 minutes. The embryos were fixed in 4% paraformaldehyde-1X PBS solution and were then subjected to in situ hybridization of gata1.
Heat treatments, compound treatments and morpholino injections of zebrafish embryos
For Supplementary Fig. 2 & 3, heat treatments were performed at 37 °C for 1 hour at 18 hours post fertilization (hpf). For all the other experiments, heat treatments were performed at 39−40 °C for 1 hour at 16−18 hpf. For gata1 staining, the compounds were added at 1 hour before heat treatments and the embryos were fixed at 90 minutes after heat treatments. For mpo staining, the compounds were added right at the end of heat treatments and the embryos were fixed at 36−40 hpf.
Microinjections were performed as described previously45. Antisense morpholino oligonucleotides (MO) for ctnnb1 (MORPH0756), ptgs1 (MORPH1203) and ptgs2a (MORPH0943) were purchased from Open Biosystems. The MOs for ctnnb2 (5’-CCTTTAGCCTGAGCGACTTCCAAAC) and ptgs2b (5’-AGGCTTACCTCCTGTGCAAACCACG) were purchased from Gene Tools. The MOs for ptgs2b were injected at 250 μM. The MOs for ptgs1 and ptgs2a were injected at 400 μM in Fig. 4c and at 100 μM in Fig. 4d. The MOs for ctnnb1 and ctnnb2 were injected at 500 μM.
In situ hybridization of zebrafish embryos
In situ hybridization was performed as previously described46. For screening plates, in situ hybridization was done manually up to the probe hybridization step. The subsequent steps were done using a liquid handling machine (BioLane™ HTI, Holle & Huttner AG). Zebrafish gata1, mpo, l-plastin and scl probes were synthesized as previously described47,48.
RNA expression measured by real-time PCR
For RNA expression in the hematopoietic cells isolated from zebrafish embryos, the hematopoietic cells were isolated from zebrafish embryos as described previously13. RNA was isolated using RNAqueous®-Micro kit (Ambion) according to the manufacturer's protocol. First-strand cDNA was synthesized using SuperScript™ III Reverse Transcriptase (Invitrogen) and random hexamers. The cDNA was then used for real-time PCR with Power SYBR® Green PCR Master Mix (Applied Biosystems) on ABI Prism 7000 machine. Zebrafish gene expression was normalized to gapdh levels. For RNA expression in the cultured cells, RNA was isolated using Trizol® Reagent (Invitrogen) according to the manufacturer's protocol. The remaining reactions were as described above. Human gene expression was normalized to 18S RNA levels. Primer sequences for zebrafish genes are ptgs1 primer set #1, 5’-TATGGCTTGGAGAAGCTGGT and 5’- CGATTCAACGATGACCCTCT; ptgs1 primer set #2, 5’-CATCCTTCGCAGAATTGACA and 5’-ATTTCCACCATGCTTTCACC; ptgs2a primer set #1, 5’-TGGATCTTTCCTGGTGAAGG and 5’-GAAGCTCAGGGGTAGTGCAG; ptgs2a primer set #2, 5’-CCAGACAGATGCGCTATCAA and 5’-GACCGTACAGCTCCTTCAGC; ptgs2b primer set #1, 5’-CAGGAAACGCTTCAACATGA and 5’-CAGCATAAAGCTCCACAGCA; ptgs2b primer set #2, 5’-CCCTGTCAGAATCGAGGTGT and 5’-TTGGGAGAAGGCTTCAGAGA; gapdh, 5’-AGGCTTCTCACAAACGAGGA and 5’-GATGGCCACAATCTCCACTT. Primer sequences for human genes are PTGS1, 5’-TTGCCTTCTTTGCACAACAC and 5’-CATAAATGTGGCCGAGGTCT; PTGS2, 5’-CTCCTGTGCCTGATGATTGC and 5’-GGGATGAACTTTCTTCTTAG; 18S RNA, 5’-CGGCTACCACATCCAAGGAA and 5’-GCTGGAATTACCGCGGCT.
Cell culture and the luciferase reporter assay
The cell clones B9 and D8 of human myelogenous leukemia K562 cells containing stable integration of pLRT-GFP and pLRT-AE, respectively, have been published previously6. The cells were maintained in RPMI 1690 with 10% FBS and 8 μg ml−1 Blasticidin S (Calbiochem). For transfection, the cells were plated in 24-well plates at 2×105 cells per ml and transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. In all of the luciferase assay experiments, the cells were transfected with β-catenin-TCF reporter TOPflash (Upstate) and pRL-TK Renilla luciferase (Promega) for normalizing the transfection efficiency. For some experiments, pCS2, pCS2-AE49, and/or pCDNA-Myc-DeltaN-TCF450 (dnTCF) were co-transfected with the reporter constructs as designated in the figures. For drug treatments, the compounds were added to the medium at 4 hours after transfection. The luciferase reporter assays were performed two days after transfection using the Dual-Luciferase® Reporter Assay System (Promega).
Benzidine staining of cultured cells
K562 cell clones B9 (GFP) and D8 (AE-GFP) were plated in a 96-well plate at 2×105 cells per ml. DMSO or NS-398 (75 μM) was added to the culture medium. On the fourth day after the drug treatment, benzidine staining was performed to detect cell hemoglobinization. Fifty microliters of cells was mixed with 10 μl bezidine reagent (0.6% H2O2, 0.5 M acetic acid, 0.2 % benzidine dihydrochloride). The percentage of benzidine-positive cells was determined under a light microscope. Each experiment was performed in triplicate and 100 cells were counted per sample.
Supplementary Material
ACKNOWLEDGEMENTS
We thank ER Plovie and MN Rivera (Massachusetts General Hospital) and Hans Clevers (Hubrecht Institute) for providing reagents, CL Tsai, C Sachidanandan and the members of the Developmental Biology Laboratory for helpful discussion. J.-R.J.Y. is supported by a Career Development Award (AG031300) from the National Institute of Aging. The authors received financial support from the National Cancer Institute (CA118498 to R.T.P.) and the Claflin Distinguished Scholar Award (to J.-R.J.Y.).
REFERENCES
- 1.Redaelli A, Botteman MF, Stephens JM, Brandt S, Pashos CL. Economic burden of acute myeloid leukemia: a literature review. Cancer Treat Rev. 2004;30:237–247. doi: 10.1016/j.ctrv.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Guan Y, Gerhard B, Hogge DE. Detection, isolation, and stimulation of quiescent primitive leukemic progenitor cells from patients with acute myeloid leukemia (AML). Blood. 2003;101:3142–3149. doi: 10.1182/blood-2002-10-3062. [DOI] [PubMed] [Google Scholar]
- 3.Terpstra W, et al. Fluorouracil selectively spares acute myeloid leukemia cells with long-term growth abilities in immunodeficient mice and in culture. Blood. 1996;88:1944–1950. [PubMed] [Google Scholar]
- 4.Wang JC, Dick JE. Cancer stem cells: lessons from leukemia. Trends Cell Biol. 2005;15:494–501. doi: 10.1016/j.tcb.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 5.Tenen DG. Disruption of differentiation in human cancer: AML shows the way. Nat Rev Cancer. 2003;3:89–101. doi: 10.1038/nrc989. [DOI] [PubMed] [Google Scholar]
- 6.Choi Y, Elagib KE, Delehanty LL, Goldfarb AN. Erythroid inhibition by the leukemic fusion AML1-ETO is associated with impaired acetylation of the major erythroid transcription factor GATA-1. Cancer Res. 2006;66:2990–2996. doi: 10.1158/0008-5472.CAN-05-2944. [DOI] [PubMed] [Google Scholar]
- 7.Schwieger M, et al. AML1-ETO inhibits maturation of multiple lymphohematopoietic lineages and induces myeloblast transformation in synergy with ICSBP deficiency. J Exp Med. 2002;196:1227–1240. doi: 10.1084/jem.20020824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fenske TS, et al. Stem cell expression of the AML1/ETO fusion protein induces a myeloproliferative disorder in mice. Proc Natl Acad Sci U S A. 2004;101:15184–15189. doi: 10.1073/pnas.0400751101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Guzman CG, et al. Hematopoietic stem cell expansion and distinct myeloid developmental abnormalities in a murine model of the AML1-ETO translocation. Mol Cell Biol. 2002;22:5506–5517. doi: 10.1128/MCB.22.15.5506-5517.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zon LI, Peterson RT. In vivo drug discovery in the zebrafish. Nat Rev Drug Discov. 2005;4:35–44. doi: 10.1038/nrd1606. [DOI] [PubMed] [Google Scholar]
- 11.Galloway JL, Wingert RA, Thisse C, Thisse B, Zon LI. Loss of gata1 but not gata2 converts erythropoiesis to myelopoiesis in zebrafish embryos. Dev Cell. 2005;8:109–116. doi: 10.1016/j.devcel.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Rhodes J, et al. Interplay of pu.1 and gata1 determines myelo-erythroid progenitor cell fate in zebrafish. Dev Cell. 2005;8:97–108. doi: 10.1016/j.devcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 13.Yeh JR, et al. AML1-ETO reprograms hematopoietic cell fate by downregulating scl expression. Development. 2008;135:401–410. doi: 10.1242/dev.008904. [DOI] [PubMed] [Google Scholar]
- 14.Yamasaki H, et al. High degree of myeloid differentiation and granulocytosis is associated with t(8;21) smoldering leukemia. Leukemia. 1995;9:1147–1153. [PubMed] [Google Scholar]
- 15.Nakamura H, et al. Morphological subtyping of acute myeloid leukemia with maturation (AML-M2): homogeneous pink-colored cytoplasm of mature neutrophils is most characteristic of AML-M2 with t(8;21). Leukemia. 1997;11:651–655. doi: 10.1038/sj.leu.2400618. [DOI] [PubMed] [Google Scholar]
- 16.Gottlicher M. Valproic acid: an old drug newly discovered as inhibitor of histone deacetylases. Ann Hematol. 2004;83(Suppl 1):S91–S92. doi: 10.1007/s00277-004-0850-2. [DOI] [PubMed] [Google Scholar]
- 17.Gottlicher M, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. Embo J. 2001;20:6969–6978. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu S, et al. Targeting AML1/ETO-histone deacetylase repressor complex: a novel mechanism for valproic acid-mediated gene expression and cellular differentiation in AML1/ETO-positive acute myeloid leukemia cells. J Pharmacol Exp Ther. 2007;321:953–960. doi: 10.1124/jpet.106.118406. [DOI] [PubMed] [Google Scholar]
- 19.Bug G, et al. Effect of histone deacetylase inhibitor valproic acid on progenitor cells of acute myeloid leukemia. Haematologica. 2007;92:542–545. doi: 10.3324/haematol.10758. [DOI] [PubMed] [Google Scholar]
- 20.Grosser T, Yusuff S, Cheskis E, Pack MA, FitzGerald GA. Developmental expression of functional cyclooxygenases in zebrafish. Proc Natl Acad Sci U S A. 2002;99:8418–8423. doi: 10.1073/pnas.112217799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.North TE, et al. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature. 2007;447:1007–1011. doi: 10.1038/nature05883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishikawa TO, Griffin KJ, Banerjee U, Herschman HR. The zebrafish genome contains two inducible, functional cyclooxygenase-2 genes. Biochem Biophys Res Commun. 2007;352:181–187. doi: 10.1016/j.bbrc.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williams CS, Mann M, DuBois RN. The role of cyclooxygenases in inflammation, cancer, and development. Oncogene. 1999;18:7908–7916. doi: 10.1038/sj.onc.1203286. [DOI] [PubMed] [Google Scholar]
- 24.Cha YI, et al. Cyclooxygenase-1-derived PGE2 promotes cell motility via the G-protein-coupled EP4 receptor during vertebrate gastrulation. Genes Dev. 2006;20:77–86. doi: 10.1101/gad.1374506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cha YI, Kim SH, Solnica-Krezel L, Dubois RN. Cyclooxygenase-1 signaling is required for vascular tube formation during development. Dev Biol. 2005;282:274–283. doi: 10.1016/j.ydbio.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 26.Sonoshita M, et al. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in Apc(Delta 716) knockout mice. Nat Med. 2001;7:1048–1051. doi: 10.1038/nm0901-1048. [DOI] [PubMed] [Google Scholar]
- 27.Oshima M, et al. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell. 1996;87:803–809. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- 28.Cha YI, DuBois RN. NSAIDs and cancer prevention: targets downstream of COX-2. Annu Rev Med. 2007;58:239–252. doi: 10.1146/annurev.med.57.121304.131253. [DOI] [PubMed] [Google Scholar]
- 29.Wang D, et al. Prostaglandin E(2) promotes colorectal adenoma growth via transactivation of the nuclear peroxisome proliferator-activated receptor delta. Cancer Cell. 2004;6:285–295. doi: 10.1016/j.ccr.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 30.Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005;310:1504–1510. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]
- 31.Shao J, Jung C, Liu C, Sheng H. Prostaglandin E2 Stimulates the beta-catenin/T cell factor-dependent transcription in colon cancer. J Biol Chem. 2005;280:26565–26572. doi: 10.1074/jbc.M413056200. [DOI] [PubMed] [Google Scholar]
- 32.Lyman Gingerich J, Westfall TA, Slusarski DC, Pelegri F. hecate, a zebrafish maternal effect gene, affects dorsal organizer induction and intracellular calcium transient frequency. Dev Biol. 2005;286:427–439. doi: 10.1016/j.ydbio.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 33.Meijer L, et al. GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem Biol. 2003;10:1255–1266. doi: 10.1016/j.chembiol.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 34.van de Water S, et al. Ectopic Wnt signal determines the eyeless phenotype of zebrafish masterblind mutant. Development. 2001;128:3877–3888. doi: 10.1242/dev.128.20.3877. [DOI] [PubMed] [Google Scholar]
- 35.Stachel SE, Grunwald DJ, Myers PZ. Lithium perturbation and goosecoid expression identify a dorsal specification pathway in the pregastrula zebrafish. Development. 1993;117:1261–1274. doi: 10.1242/dev.117.4.1261. [DOI] [PubMed] [Google Scholar]
- 36.Fujino H, West KA, Regan JW. Phosphorylation of glycogen synthase kinase-3 and stimulation of T-cell factor signaling following activation of EP2 and EP4 prostanoid receptors by prostaglandin E2. J Biol Chem. 2002;277:2614–2619. doi: 10.1074/jbc.M109440200. [DOI] [PubMed] [Google Scholar]
- 37.Hino S, Tanji C, Nakayama KI, Kikuchi A. Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase stabilizes beta-catenin through inhibition of its ubiquitination. Mol Cell Biol. 2005;25:9063–9072. doi: 10.1128/MCB.25.20.9063-9072.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trowbridge JJ, Xenocostas A, Moon RT, Bhatia M. Glycogen synthase kinase-3 is an in vivo regulator of hematopoietic stem cell repopulation. Nat Med. 2006;12:89–98. doi: 10.1038/nm1339. [DOI] [PubMed] [Google Scholar]
- 39.Reya T, et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423:409–414. doi: 10.1038/nature01593. [DOI] [PubMed] [Google Scholar]
- 40.Cobas M, et al. Beta-catenin is dispensable for hematopoiesis and lymphopoiesis. J Exp Med. 2004;199:221–229. doi: 10.1084/jem.20031615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dinchuk JE, et al. Renal abnormalities and an altered inflammatory response in mice lacking cyclooxygenase II. Nature. 1995;378:406–409. doi: 10.1038/378406a0. [DOI] [PubMed] [Google Scholar]
- 42.Langenbach R, et al. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell. 1995;83:483–492. doi: 10.1016/0092-8674(95)90126-4. [DOI] [PubMed] [Google Scholar]
- 43.Morham SG, et al. Prostaglandin synthase 2 gene disruption causes severe renal pathology in the mouse. Cell. 1995;83:473–482. doi: 10.1016/0092-8674(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 44.Patton EE, Zon LI. Taking human cancer genes to the fish: a transgenic model of melanoma in zebrafish. Zebrafish. 2005;1:363–368. doi: 10.1089/zeb.2005.1.363. [DOI] [PubMed] [Google Scholar]
- 45.Nasevicius A, Ekker SC. Effective targeted gene ‘knockdown’ in zebrafish. Nat Genet. 2000;26:216–220. doi: 10.1038/79951. [DOI] [PubMed] [Google Scholar]
- 46.Schulte-Merker S, Ho RK, Herrmann BG, Nusslein-Volhard C. The protein product of the zebrafish homologue of the mouse T gene is expressed in nuclei of the germ ring and the notochord of the early embryo. Development. 1992;116:1021–1032. doi: 10.1242/dev.116.4.1021. [DOI] [PubMed] [Google Scholar]
- 47.Thompson MA, et al. The cloche and spadetail genes differentially affect hematopoiesis and vasculogenesis. Dev Biol. 1998;197:248–269. doi: 10.1006/dbio.1998.8887. [DOI] [PubMed] [Google Scholar]
- 48.Bennett CM, et al. Myelopoiesis in the zebrafish, Danio rerio. Blood. 2001;98:643–651. doi: 10.1182/blood.v98.3.643. [DOI] [PubMed] [Google Scholar]
- 49.Kalev-Zylinska ML, et al. Runx1 is required for zebrafish blood and vessel development and expression of a human RUNX1-CBF2T1 transgene advances a model for studies of leukemogenesis. Development. 2002;129:2015–2030. doi: 10.1242/dev.129.8.2015. [DOI] [PubMed] [Google Scholar]
- 50.van de Wetering M, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241–250. doi: 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.