

Abstract
The sensitivity of glycan analysis using nano-liquid chromatography interfaced with electrospray ionization mass spectrometry (ESI-MS) increases with the decrease of the mobile phase flow rate, accompanied by reduced ion suppression. In this study, we describe the preparation and performance of high efficiency 10 μm i.d. amine-bonded poly(vinylbenzyl chloride-divinylbenzene) hydrophilic interaction (HILIC) porous layer open tubular (PLOT) columns operated at 20 nL/min for the separation and analysis of glycan mixtures. HILIC-PLOT columns with a uniform porous polymer layer were reproducibly prepared (~4% RSD in retention time from column to column) via in-situ polymerization, followed by one step modification with ethylenediamine. When coupled on-line with negative ESI-MS, low detection limits (0.3 fmol) for a 3-sialyl-tetrasaccharide were achieved using a 2.5 m × 10 μm i.d. HILIC-PLOT column. A dextran ladder standard was used to evaluate the performance of the column, and high efficiency separation was achieved with detection of the dextrans up to G22 from ~50 fmol amounts injected. As an example of the high sensitivity of the column, MS6 characterization of glycan structures was possible from the injection of 10 fmol of a neutral and sialylated glycan. As another example of high sensitivity LC-MS analysis of 3 ng of a PNGase F digest of ovalbumin allowed 28 N-linked glycans to be confidently identified from a single analysis. High quality MS/MS spectra for each ovalbumin glycan were acquired and manually interpreted for structure analysis. The HILIC PLOT column is a very promising approach for LC-MS analysis of glycans at the ultratrace level.
1. INTRODUCTION
Glycosylation is one of the most important post-translational modifications (PTMs) of proteins. It is estimated that more than 50% of proteins are glycosylated, and glycosylation is known to play a crucial role in numerous biological processes, including antibody recognition, intracellular transport, cell recognition, cell-cell interactions [1–3]. Comprehensive characterization of glycoproteins, including glycosylation site identification and glycan structure determination, has long been a challenge [4–8]. Usually glycoprotein characterization requires purification of a protein, chemical or enzymatic release of glycans, followed by analysis of the pooled glycans using a variety of techniques, such as high performance liquid chromatography (LC), capillary electrophoresis (CE), mass spectrometry (MS), and nuclear magnetic resonance (NMR) [2,5]. Matrix-assisted laser desorption/ionization (MALDI)-MS and electrospray ionization (ESI)-MS have become the key tools for glycan analysis, mainly due to their high sensitivity and capability for structure determination [2,4,9–11]. Due to the potential loss of the sialic acid moiety during MALDI analysis, derivatization is usually required for MALDI-MS of sialylated glycans [12–14], making analysis of limited amounts of sample problematic. Thus, ESI-MS coupled with LC separation is becoming a method of choice for analysis of native glycans at the trace level [15,16].
Separation of native glycans is challenging due the complexity of their structures and their hydrophilicity. CE [5,17–18], graphitized carbon chromatography (GCC) [6,15,19,20], and hydrophilic interaction chromatography (HILIC) [16,21–26] have been widely used in the separation of glycans. On-line coupling of CE with MS allows fast and sensitive glycan analysis and the ability to separate isomeric species. GCC columns provide high resolving power; however, extensive sample cleanup prior to the analysis is often required to remove sample contaminants such as the deglycosylated proteins to prevent irreversible adsorption of protein on the column. HILIC separation, based on polar analytes partitioning between the bulk eluent and a water-enriched layer [26], has emerged as an appealing alternative to GCC for glycan separation. In the HILIC mode, glycans are retained in high organic buffer and eluted by increasing water content, while the deglycosylated proteins and most peptides are not retained on the column. The mobile phases used for HILIC separation are compatible with ESI-MS, and thus particularly suitable for glycan analysis at trace level. Among the various materials for HILIC, amine- and amide-bonded stationary phases are among the most popular for glycan separation [27–30].
For positive electrospray ionization, the ionization efficiency for glycans was reported to be ~2 orders of magnitude lower than that for peptides at conventional flow rates (μL/min level) [31]. In addition, ESI of hydrophilic compounds, such as glycans, can be easily suppressed by surface active analytes, such as peptides. Lower flow rates result in smaller electrospray droplet sizes [32,33] that have a higher surface-to-volume ratio, allowing more efficient desolvation. Importantly, the increased charge per analyte at low flow rate enables more effective ionization, especially for neutral analytes, in part due to limited number of ions per small droplet [31,32] As a result, enhanced sensitivity can be achieved at lower flow rates, along with reduced ion suppression [31–36].
Preparation of a small i.d. LC columns is key to successful operation of LC at low nL/min of flow rate. Although efforts have been made in recent years to prepare ultranarrow bore LC columns, success has been limited due part to difficulty in manufacture and limited reproducibility [37–39]. Following gas chromatographic columns [40], porous layer open tubular (PLOT) LC columns [41–45], especially polymer based PLOT columns [46–48], are attractive because of their high separation efficiency, stability, and ease of preparation. Recently, our laboratory reproducibly prepared 10 μm i.d. high efficiency poly(styrene-divinylbenzene) (PS-DVB) reversed-phase (RP) PLOT columns for ultratrace proteomic analysis [49–51].
In this work, we have prepared 10 μm i.d. amine-bonded HILIC-PLOT columns in a simple and robust fashion. When coupled on-line with ESI/MS/MS, the columns demonstrated high resolving power for native glycans with the detection limit in the low fmol to sub-fmol level. Examples are presented to demonstrate the potential of the HILIC-PLOT/ESI/MSn approach for the structural analysis of acidic as well as neutral glycans from low fmol amounts injected.
2. EXPERIMENTAL
2.1 Materials
Fused silica capillary tubing was purchased from Polymicro Technologies (Phoenix, AZ, USA). Vinylbenzyl chloride (VBC), divinylbenzene (DVB), ethylenediamine, methanol, ethanol, formic acid (HPLC grade), 3-(trimethoxysilyl)propyl methacrylate, 2, 2′-diphenyl-1-picrylhydrazyl (DPPH), 2, 2′-azobisoisobutyronitrile (AIBN), lacto-N-tetraose, dextran ladder standard, and 3-sialyl-tetrasaccharide were obtained from Sigma-Aldrich (St. Louis, MO, USA). Trisialylated galactosylated triantennary glycan was purchased from ProZyme (San Leandro, CA, USA). Ammonium acetate, acetonitrile (HPLC grade), and deionized water (HPLC grade) were from Thermo Fisher Scientific (Fair Lawn, NJ, USA). Standard glycoprotein ovalbumin and enzyme PNGase F were from Sigma-Aldrich (St. Louis, MO, USA).
2.2 Direct infusion studies of ESI-MS response
Coated ESI emitters (360 μm o.d., 20 μm i.d. fused silica with 2, 5, and 10 μm i.d. spray tip, New Objective, Woburn, MA, USA) were used for the wide range of flow rates. ESI-MS experiments were performed on an LTQ ion trap mass spectrometer (Thermo Fisher, San Jose, CA, USA). The temperature of the MS heated capillary inlet was fixed at 245 °C. Both the electrospray voltage and the ESI emitter position were adjusted to provide maximum MS response at each flow rate, and a stereo-microscope was used to monitor the electrospray stability. The MS response of a mixture of angiotensin (10−6 mol/L), lacto-N-tetraose (10−5 mol/L) and 3-sialyl-tetrasaccharide (10−5 mol/L) was measured by infusing solution at flow rates from 20 nL/min to 1 μL/min generated using Dionex Ultimate 3000 nano HPLC system (Sunnyvale, CA, USA). The samples were dissolved in 0.1% (v/v) formic acid, 50% (v/v) acetonitrile in water. MS scans with an m/z range of 150–1500 were acquired for 1 min, starting 20 min after each flow rate change to assure that the flow rate had stabilized. The MS intensities reported for the peptide and glycan represent the average values during 1 minute acquisition.
2.3 Preparation of a 10 μm i.d. HILIC-PLOT Column
Fused-silica capillary tubing with 10 μm i.d. (~ 3 meters) was first washed overnight with 1.0 mol/L NaOH, then with water, next with 1.0 mol/L hydrochloric acid, and then washed again with water and acetonitrile using a home made pneumatic pressure bomb operating at ~1000 psi. The capillary was dried with nitrogen at ~1000 psi to remove residual water and acetonitrile. 30% (v/v) 3-(trimethoxysilyl)propyl methacrylate and 0.5% (w/v) 2, 2-diphenyl-1-picrylhydrazyl (DPPH) in anhydrous N,N-dimethylformamide (DMF) was freshly prepared and filled into the 10 μm i.d. pretreated capillary with a pressure bomb at ~300 psi.. Both ends of the capillary were sealed with a septum, and the capillary was placed in an oven at 110 °C for 6 – 10 h. Then, the capillary was washed with acetonitrile and blown dry with nitrogen at 1000 psi [49].
The preparation of the stationary phase is schematically summarized in Figure 1A. A polymerization solution was prepared containing of 5 mg of AIBN, 200 μL vinylbenzyl chloride, 200 μL divinylbenzene, and 600 μL methanol. The solution was degassed by sonication for 5 min and then filled into the silanized capillary with the pressure bomb at ~300 psi. Both ends of the capillary were sealed with septa, and the capillary was heated at 74 °C for ~16 h in a water bath. The column was then fully washed with acetonitrile to remove the solvent as well as the unreacted monomer. For in-situ modification, ethylenediamine was forced through the poly(vinylbenzyl chloride-divinylbenzene) PLOT column with the pressure bomb at ~150 psi, and the column was merged into a water bath at 70 °C. The reaction was allowed to proceed for 24 h under continuous flow of ethylenediamine. After the reaction was complete, the column was washed with acetonitrile and stored by immerging both ends of the capillary into water for later use.
Figure 1.

Synthesis steps for the preparation of 10 μm i.d. amine-bonded HILIC-PLOT columns (A), and scanning electron micrographs (SEM) of the cross-section of a HILIC PLOT column prepared by using 40% methanol as solvent (B).
2.4 NanoLC-ESI-MS using 10 μm i.d. HILIC-PLOT Column
HPLC separations were performed using an Agilent 1100 Series pump (Agilent Technologies, Santa Clara, CA, USA). Mobile phase A (0.1% (v/v) formic acid, 10% (v/v) water in acetonitrile) and mobile phase B (0.1% (v/v) formic acid, 40% (v/v) water in acetonitrile) were used for the gradient separation. To evaluate the performance of the HILIC-PLOT column, the column was carefully butt-to-butt connected to a coated spray tip (360 μm o.d., 20 μm i.d. fused silica with a 5 μm i.d. spray tip, 2–3 cm long) with a PicoClear connector (New Objective), with the electrospray voltage being applied directly on the spray tip. A PEEK tee (Upchurch Scientific, Oak Harbor, WA, USA) was used as a flow splitter. 2 μL of sample solution was first loaded into the sample loop. After splitting, ~4 nL of sample was directly loaded onto the PLOT column at a split ratio of 1:500. Flow rates of the HILIC-PLOT column were measured by connecting 50 μm i.d. fused-silica capillary tubing to the exit of the PLOT column and determining the volume of mobile phase A that flowed for a given period of time through the tubing. An LTQ ion trap mass spectrometer (Thermo Fisher) with a heated capillary temperature of 245 °C was used in all experiments with a maximum ion injection time set at 50 ms. In all data-dependent MS/MS analysis, a full MS scan between 400–2000 m/z was followed by five MS/MS scans for the 5 most abundant ions. A collision energy setting of 35% was applied for ion fragmentation. In MSn analysis, a full MS scan between 400–2000 m/z was followed by further fragmentation of the highest intensity fragment ion in each scan with AGC target settings of 3 × 104 for MS and 5000 for MSn scans.
2.5 Sample preparation
The N-glycans from ovalbumin were enzymatically released in Eppendorf tubes. Typically, 0.1 mg of glycoprotein to be deglycosylated was dissolved in 45 μL of 10 mM sodium phosphate (pH 7.5), followed by addition of 1 μL of PNGase F. The reaction mixture was incubated for 1 day at 37 °C, and the digestion mixture was then injected into the PLOT column for HILIC-nanoLC/MS/MS analysis without further cleanup.
3. RESULTS AND DISCUSSION
In this study, we have introduced an amine-bonded HILIC-PLOT column coupled to a linear ion trap MS for high sensitivity separation and analysis of native glycans. First, a neutral and a sialylated standard glycan, mixed with angiotensin (10:10:1) were used to examine the influence of flow rate on glycan ionization efficiencies and ion suppression by infusing the sample into the LTQ MS. Significantly increased ionization efficiency of glycans was observed at flow rates below 50 nL/min, along with reduced ion suppression. Use of these substances for column characterization revealed good column to column reproducibility of 4% in retention time. Then, we separated and analyzed a dextran ladder standard as well as glycans released from ovalbumin on a HILIC-PLOT column coupled with ESI-MS at the flow rate of 20 nL/min. High resolution with low femtomole detection limits were reached.
3.1 ESI-MS Analysis of Glycans at Low nL/min Flow Rates
ESI ionization efficiency and ion suppression are strongly dependent on the physical and chemical properties of the analytes (chargeability and surface activity) as well as on factors, such as liquid flow rate and spray tip. Due to low surface activity and poor chargeability of glycans, especially for the neutral glycans, ESI-MS analysis of glycans at conventional flow rates result in poor response factors [22,24,26]. We investigated the influence of flow rate, especially very low flow rates, on the positive ESI-MS response of oligosaccharides in a mixture with a peptide. A mixture of 10−6 M of angiotensin, 10−5 M of lacto-N-tetraose and 10−5 M of 3-sialyl-tetrasaccharide (1:10:10) in 0.1% (v/v) formic acid, 50% (v/v) acetonitrile in water was used for the ESI-MS study, and the mixture was directly infused at a flow rate of 200 nL/min, and the results are shown in Figure 2A. The m/z and charge state of the dominant peptide and glycan ions in the ESI positive mode were 523.9 [M+2H]2+ for angiotensin, 349.82 [NG+H+Na]2+, 707.96 [NG+H]+ for lacto-N-tetraose, and 820.84 [SG+H]+, 430.21 [SG+K+H]2+ for 3-sialyl-tetrasaccharide, where NG and SG are the neutral and sialylated glycans, respectively. Although the concentration of each glycan is 10 fold higher than the peptide, the MS intensity of angiotensin is still roughly 5 fold greater, mainly due to lower ioinization efficiencies of the glycans and ion suppression effects. More comparable ion intensities were found for angiotensin and the glycans ions in the negative ESI mode where the m/z values and charge state, respectively, were 1044.5 [M−H] for angiotensin−, 706.16 [NG−H]−, 742.30 [NG+Cl]−, and 751.97 [NG+HCO2]− for lacto-N-tetraose, and 819.3 [SG−H]− for the sialylated glycan,(Figure 2B). The neutral glycan was distributed in three different forms, while only a single m/z ion for the sialylated glycan was found because of its ease of deprotonation. In addition, the MS spectrum in the negative mode was found to contain less noise than the positive mode.
Figure 2.

(A) Positive and (B) negative infusion ESI-MS/MS spectra of a mixture of angiotensin, lacto-N-tetraose, and 3-sialyl-tetrasaccharide. Concentration: 10−6 M for angiotensin, 10−5 M for 3-sialyl-tetrasaccharide and lacto-N-tetraose (1:10:10). SG and NG represent sialylated and neutral glycan, respectively. Flow rate: 200 nL/min. See Experimental Section for details.
We then studied by infusion the effect of liquid flow rate on the ionization efficiency of glycans in the negative ESI mode. Figure 3A shows the ionization efficiency, defined as the MS intensity divided by the analyte amount consumed per unit time, of angiotensin and the two glycans as a function of flow rate. The ionization efficiencies for all three species increased dramatically at low flow rates down to 20 nL/min. Note, however, the ionization efficiency of the two glycans was still lower than that of angiotensin even in the negative ESI mode. Figure 3B shows the ionization efficiencies as a function of flow rate of the two glycans relative to that of angiotensin. The ionization efficiency ratio of each glycan to angiotensin improved as much as ~5-fold as the flow rate decreased, indicating reduced ion suppression for the glycans at the lower flow rate. The results of Figure 3A and 3B demonstrate that nanoESI/MS at flow rates of 20 nL/min or lower can significantly improve the response factors for underivatized glycans, in agreement with that shown by others [32,52].
Figure 3.

Comparison of ion intensities for angiotensin (10−6 M), lacto-N-tetraose (10−5 M), and 3-sialyl-tetrasaccharide (10−5 M) (1:10:10) at different liquid flow rates: (A) ionization efficiencies of angiotensin, lacto-N-tetraose, and 3-sialyl-tetrasaccharide as a function of flow rate; (B) ionization efficiency ratio of 3-sialyl-tetrasaccharide and lacto-N-tetraose to angiotensin at different flow rates. I, F, and C represent intensity, flow rate, and concentration, respectively.
3.2 Preparation and Characterization of the HILIC-PLOT Column
High performance separations of complex glycan mixtures prior to MS detection are often needed. As noted earlier, hydrophilic interaction chromatography (HILIC), where hydrophilic analytes are retained in high organic solvent and eluted in increasing amounts of water, has emerged as an important method of separation of glycans [16,21,23]. In the present study, we prepared HILIC-PLOT columns based on our success with PS-DVB reversed-phase PLOT columns [49]. For the HILIC-PLOT column, styrene was replaced by vinylbenzyl chloride to generate a PLOT column suitable for surface modification. As discussed in our previous paper [49], selection of a suitable solvent is the key to successful preparation of a PLOT column. Figure 1B shows a scanning electron microscopy (SEM) photograph of a cross section of a vinylbenzyl chloride based PLOT column when using methanol (40% v/v) as polymerization solvent, and a uniform layer was observed. The thickness of the layer was estimated to be roughly 0.5 – 1 μm, which is similar to that of the PS-DVB PLOT column [49]. The chloromethyl groups of the poly(VBC-DVB) layer can be readily modified via a single step reaction with primary amine such as ethylenediamine. The resulting modified column provides a primary amino group and a secondary amine for HILIC separation. The presence of the amino groups allow HILIC based separations and also benefits the separation of silylated glycans from neutral glycans via anion exchange interaction. The presence of amino groups could potentially lead to formation of glycosylamine with the aldehyde at the reducing end of glycans, thus resulting in the loss of glycans or shorter lifetime of the column [2,23]. However, this is not a problem in our study due to the acidified mobile phase (pH 2–3), leading to positively charged amino groups that do not react with aldehyde.
3.3 HILIC NanoLC/ESI/MS of Glycan
A 2.5 m × 10 μm i.d. HILIC-PLOT column was characterized and used throughout the study. Due to the high permeability of the HILIC PLOT column, the 2.5 m long column provided a flow rate of 20 nL/min at a pressure of only 700 psi with solvent A. At such a low flow rate, the HILIC-PLOT column is particularly valuable for the glycan analysis when combined with ESI/MS. Initially, a dextran ladder standard was used to evaluate the separation performance of the HILIC-PLOT column. Figure 4 shows a gradient nanoLC/ESI/MS analysis of the dextran ladder using the 2.5 m × 10 μm i.d. HILIC-PLOT column. The linear dextrans, consisting of various glucose units, were eluted in the order of the oligosaccharide chain length. It can be seen that the glucose trimer (G3) eluted first followed by dextrans with increasing chain length. Due to the high ionization efficiency at 20 nL/min flow rate, dextrans up to G22 could be detected using only 50 fmol injection amount.
Figure 4.

High efficiency nanoLC-ESI-MS/MS analyses of a dextran ladder standard using 2.5 m × 10 μm i.d. HILIC-PLOT column. ~50 fmol amount was split loaded on the PLOT column. Gradient: mobile phase A (0.1% (v/v) formic acid, 10% (v/v) water in acetonitrile) to 100% B (0.1% (v/v) formic acid, 40% (v/v) water in acetonitrile) in 75 min with data collection initiated at the start of the gradient. Flow rate: 20 nL/min at an inlet pressure of 700 psi.
3.4 Reproducibility
The HILIC PLOT column was made in two simple synthetic steps, leading to robust column preparation. The column-to-column reproducibility was evaluated using a 30 min gradient elution (0 to 100% of solvent B) of a mixture of two standard glycans, trisialylated galactosylated triantennary and 3-sialyl-tetrasaccharide. Three columns were prepared separately, and the column to column reproducibility of the retention time was found to be better than 4% (Table 1). After retention time normalization, the relative retention time was less than 2%. In addition, the reproducibility of the chromatographic peak width at half-height, which was measured using extracted ion chromatogram, was found to be 19 ± 1.5 s for the three HILIC-PLOT columns using the same gradient and sample.
Table 1.
Column-to-Column Reproducibilitya
| retention time (min) |
|||||
|---|---|---|---|---|---|
| m/z | Column 1 | Column 2 | Column 3 | RSD, % | NRSD*, %b |
| 959.3 | 24.03 | 24.71 | 25.92 | 3.81 | 1.36 |
| 843.2 | 25.41 | 26.32 | 27.14 | 3.23 | 0.74 |
| 843.1 | 26.41 | 27.24 | 28.31 | 3.49 | 1.03 |
| 673.1 | 28.28 | 29.77 | 30.23 | 3.35 | |
Mixture of tri-sialylated-galactosylated triantennary and 3-sialyl-tetrasaccharide was used to test the column-to-column reproducibility of three 2.5 m × 10 μm i.d. HILIC-PLOT columns. For other conditions, see Figure 5.
NRSD* represents the RSD of relative retention time normalized to the ion of m/z = 673.1.
3.5 Loading Capacity
One of the model sialylated glycans, 3-sialyl-tetrasaccharide, was used to determine the loading capacity of the HILIC PLOT column. The sample solution at various concentrations was split loaded on the HILIC-PLOT column, followed by a 30-min gradient separation. The loading capacity of the HILIC-PLOT column was evaluated by measuring the increase in the peak width with increasing sample loading, as described previously.[49,53] The amount of sample injected when the w1/2 is increased by 10% is defined as the maximum loading capacity. The loading capacity of the 2.5 m × 10 μm i.d. HILIC-PLOT column was determined to be roughly 40 fmol for 3-sialyl-tetrasaccharide (820 Da), which is comparable to value found for PS-DVB PLOT column [49].
The negative ESI-MS response of the 10 μm i.d. HILIC-PLOT column was evaluated using 3-sialyl-tetrasaccharide to determine the linear dynamic range. Various amounts of 3-sialyl-tetrasaccharide were split loaded on the HILIC-PLOT column, followed by a 30-min gradient separation. The peak area for 3-sialyl-tetrasaccharide increased linearly for sample loadings in the range of 0.5 – 300 fmol, and then began to level off. A linear dynamic range of approximately 3 orders of magnitude was achieved. Next, the detection level of the 10 μm i.d. HILIC-PLOT column was determined. A solution of sample containing 0.5 fmol of glycan was split loaded onto the column, and a signal to noise ratio of 5 was obtained in the full MS scan mode. It was estimated that the 10 μm i.d. HILIC-PLOT provided a detection limit (S/N=3) of ~0.3 fmol for 3-sialyl-tetrasaccharide.
3.6 NanoLC/ESI/MSn Characterization of Glycan Structure Using 10 μm i.d. HILIC-PLOT Column
Mass spectrometric analysis, especially MSn fragmentation, is very important for structure analysis of glycans. However, the sensitivity decreases 5 – 10 fold after each fragmentation step, and thus large amounts of sample are usually required for multistep fragmentation [54]. To illustrate the high sensitivity of the 10 μm i.d. HILIC-PLOT column for multiple CID fragmentation, 10 fmol of the standard glycans, one neutral and the other sialylated, were separated and subjected to fragmentation up to MS6 where in each step the highest intensity ion was selected for further fragmentation. Figure 5A shows the base peak chromatogram of the two glycans analyzed in the positive ion mode. It can be seen that, due to the electrostatic interaction of sialic acid with the positively charged amine-bonded stationary phase, the sialylated glycan was retained significantly longer. Figures 5B–G show the full MS and MS2 to MS6 spectra of the neutral glycan acquired in the data-dependant mode when the most intense ions was selected as a precursor for the next stage of fragmentation. Based on the known structure of the glycan, some of the high intensity fragments in MSn spectra were annotated; however, in the case of an unknown glycan, MSn spectra would be highly valuable in structure analysis. Importantly, in order to maximize the information content of the MSn spectra, fragmentation could be performed with additional ions besides the highest intensity ion. Finally, it should be noted that the MSn spectra of the sialylated glycan was also analyzed in a similar fashion (data not shown). For both glycans, it is important to emphasize that the multistep fragmentation was accomplished with only 10 fmol of analyte injected.
Figure 5.

Illustration of the nanoLC/ESI/MSn analysis of a neutral and a sialylated glycans in the positive ion mode. (A) Base peak chromatogram of 10 fmol of each glycan injected on a 2.5 m × 10 μm i.d. HILIC-PLOT column. (B) – (G) MS to MS6 of the first eluted neutral glycan. Highest intensity ion was selected for next fragmentation step. For other conditions, see Figure 4, except a 20 min gradient was used.
3.7 Comprehensive Analysis of N-Linked Glycans Released from Ovalbumin
The combination of HILIC nanoLC separation with MS/MS analysis can be a powerful approach for glycan characterization in complex samples. In this study, a model glycoprotein, ovalbumin, was used to evaluate the performance of the HILIC-PLOT/ESI/MS/MS. Although ovalbumin has only a single N-linked glycosylation site, more than 30 glycan structures have been reported and extensively characterized [55]. Ovalbumin glycans were enzymatically released using PNGase F, and roughly 3 ng of the ovalbumin digest was split loaded onto the HILIC-PLOT column without any sample cleanup. During the sample loading, the deglycosylated ovalbumin, PNGase F, as well as digestion buffer, were not retained on the HILIC column and thus did not interfere with the subsequent analysis. After washing with mobile phase A (90% acetonitrile), the glycans were gradient eluted and analyzed using LC-MS in the data dependent mode. Figure 6A shows the base peak chromatogram of nanoLC/ESI/MS/MS analysis of 3 ng ovalbumin PNGase F digest using a 2.5 m × 10 μm i.d. HILIC-PLOT column operating at a flow rate of 20 nL/min. High quality MS spectra were obtained and manually interpreted to assign glycan compositions. Overall, 28 high mannose type glycans were found in a single analysis, and the glycan structures are listed in Table 2. It should be emphasized that the high-mannose type glycans are generally very difficult to be detected because of their low chargeability and high hydrophobicity [13,56]. It is worth noting that 3 of the 28 high mannose glycans, specifically, Man5GlcNac2, Man6GlcNac2, and Man7GlcNac2, were also identified based on their high quality MS/MS spectra from only 3 ng of ovalbumin PNGase F digest in this study. The MS/MS spectra of the identified structures are shown in Figure 6B–D. The nanoLC/ESI/MS/MS system using 10 μm i.d. HILIC-PLOT column demonstrates the high potential to be used for comprehensive analysis of complex glycans sample at trace level.
Figure 6.

Illustration of the nanoLC/ESI/MS/MS analysis of ovalbumin PNGase F digest. (A) Base peak chromatogram of N-linked glycans released from 3 ng of ovalbumin on a 2.5 m × 10 μm i.d. HILIC-PLOT column. (B) – (D) MS/MS spectra of the identified high mannose glycans, Man5GlcNac2, Man6GlcNac2, and Man7GlcNac2.
Table 2.
Structures and masses of the N-glycans cleaved from ovalbumin
| Mass |
Charge State | Structure | |
|---|---|---|---|
| Calca | Exp | ||
| 910.3 | 910.1 | 1 |
|
| 1072.4 | 1072.4 | 1 |
|
| 1113.4 | 1113.4 | 1 |
|
| 1234.4 | 1234.5 | 1 |

|
| 1275.5 | 1276.2 | 1 |
|
| 1316.5 | 1316.4 | 1 |
|
| 1396.5 | 1395.2 | 1 |

|
| 1437.5 | 1435.7 | 2 |
|
| 1478.5 | 1478.1 | 2 |
|
| 1519.6 | 1518.9 | 2 |

|
| 1558.5 | 1559.5 | 1 |

|
| 1640.6 | 1639.9 | 2 |

|
| 1681.6 | 1680.8 | 2 |

|
| 1722.6 | 1721.5 | 2 |
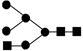
 or or 
|
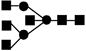
| 1843.7 | 1844.5 | 2 |
 or or 
|
| 1884.7 | 1883.9 | 2 |
 or or 
|
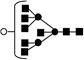
| 1925.7 | 1924.6 | 2 |
 or or 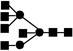
|
| 2087.8 | 2087.0 | 2 |
 or or 
|
| 2128.8 | 2128.0 | 2 |

|
| 2249.8 | 2249.6 | 2 |

|
| 2290.9 | 2291.2 | 2 |
|
Obtained from reference 45.● mannose; ▪ N-acetyl glucosamine; ○ galactose.
4. CONCLUSIONS
This paper has demonstrated the reproducible preparation and application of a high efficiency 2.5 m × 10 μm i.d. amine-bonded poly(vinylbenzyl chloride-divinylbenzene) HILIC-PLOT column. The polymer layer was covalently attached to the walls of the capillary, followed by a single step modification with ethylenediamine to create a hydrophilic interaction chromatographic stationary phase suitable for separation of glycans. Due to the open structure in the center of the capillary, the HILIC-PLOT column provided a flow rate of 20 nL/min at a back pressure of roughly 700 ps, even though the column was 2.5 m. At such a low flow rate, significant improvement in ESI/MS sensitivity was achieved along with reduced ion suppression, which is particularly important for the ESI/MS analysis of non-surface active compounds such as glycans. It was shown that the ionization efficiency of glycans increased more than 10-fold, and that the ratio of ionization efficiencies of angiotensin and glycan (i.e. ion suppression of glycan) decreased ~5-fold when the flow rate decreased from 1 μL/min to 20 nL/min. The high sensitivity of the nanoLC/ESI/MS system using 10 μm i.d. HILIC-PLOT column enabled comprehensive analysis of native N-linked glycans released from glycoproteins at the low ng level.
Work is continuing on the development and application of 10 μm i.d. HILIC-PLOT columns. We are implementing an on-line microSPE/PLOT/ESI/MS system using the 10 μm i.d. HILIC-PLOT column for ultratrace glycan analysis. In addition, we are preparing new HILIC columns suitable for glycans separation based on amide- or sulfoalkylbetaine-bonded HILIC phases. The combination of HILIC columns with different functionality will be very useful for the separation of highly complex samples. Finally, besides analysis of glycans, the HILIC columns could be used for the analysis of other polar analytes, such as metabolites [57] and phosphopeptides [58].
Acknowledgments
The authors thank NIH GM 15847 for support of this work. Contribution number 922 from the Barnett Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Packer NH, Von der Lieth CW, Aoki-Kinoshita KF, Lebrilla CB, Paulson JC, Raman R, Rudd P, Sasisekharan R, Taniguchi N, York WS. Proteomics. 2008;8:8. doi: 10.1002/pmic.200700917. [DOI] [PubMed] [Google Scholar]
- 2.Mechref Y, Novotny MV. Chem Rev. 2002;102:321. doi: 10.1021/cr0103017. [DOI] [PubMed] [Google Scholar]
- 3.Helenius A, Aebi M. Science. 2001;291:2364. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 4.Dell A, Morris HR. Science. 2001;291:2351. doi: 10.1126/science.1058890. [DOI] [PubMed] [Google Scholar]
- 5.Olajos M, Hajos P, Bonn G, Guttman A. Anal Chem. 2008;80:4241. doi: 10.1021/ac8002598. [DOI] [PubMed] [Google Scholar]
- 6.Pabst M, Bondili JS, Stadlmann J, Mach L, Altmann F. Anal Chem. 2007;79:5051. doi: 10.1021/ac070363i. [DOI] [PubMed] [Google Scholar]
- 7.Tang H, Mechref Y, Novotny MV. Bioinformatics. 2005;21:i431. doi: 10.1093/bioinformatics/bti1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campbell MP, Royle L, Radcliffe CM, Dwek RA, Rudd PM. Bioinformatics. 2008;24:1214. doi: 10.1093/bioinformatics/btn090. [DOI] [PubMed] [Google Scholar]
- 9.Haslam SM, North SJ, Dell A. Curr Opin Struct Biol. 2006;16:584. doi: 10.1016/j.sbi.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 10.Harvey DJ. Expert Rev Proteomics. 2005;2:87. doi: 10.1586/14789450.2.1.87. [DOI] [PubMed] [Google Scholar]
- 11.Zaia J. Mass Spectrom Rev. 2004;23:161. doi: 10.1002/mas.10073. [DOI] [PubMed] [Google Scholar]
- 12.Kang P, Mechref Y, Klouckova I, Novotny MV. Rapid Commun Mass Spectrom. 2005;19:3421. doi: 10.1002/rcm.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mechref Y, Klouckova I, Novotny MV. Anal Chem. 1998;70:455. doi: 10.1021/ac970947s. [DOI] [PubMed] [Google Scholar]
- 14.Ciucanu I, Costello CE. J Am Chem Soc. 2003;125:16213. doi: 10.1021/ja035660t. [DOI] [PubMed] [Google Scholar]
- 15.Estrella RP, Whitelock JM, Packer NH, Karlsson NG. Anal Chem. 2007;79:3597. doi: 10.1021/ac0622227. [DOI] [PubMed] [Google Scholar]
- 16.Zhao J, Qiu W, Simeone DM, Lubman DM. J Proteome Res. 2007;6:1126. doi: 10.1021/pr0604458. [DOI] [PubMed] [Google Scholar]
- 17.Kamoda S, Kakehi K. Electrophoresis. 2006;27:2495. doi: 10.1002/elps.200500853. [DOI] [PubMed] [Google Scholar]
- 18.Zamfir A, Peter-Katalinic J. Electrophoresis. 2004;25:1949. doi: 10.1002/elps.200405825. [DOI] [PubMed] [Google Scholar]
- 19.Kawasaki N, Itoh S, Ohta M, Hayakawa T. Anal Biochem. 2003;316:15. doi: 10.1016/s0003-2697(03)00031-9. [DOI] [PubMed] [Google Scholar]
- 20.Barroso B, Dijkstra R, Geerts M, Lagerwerf F, van Veelen P. A de Ru Rapid Commun Mass Spectrom. 2002;16:1320. doi: 10.1002/rcm.723. [DOI] [PubMed] [Google Scholar]
- 21.Thomsson KA, Karlsson H, Hansson GC. Anal Chem. 2000;72:4543. doi: 10.1021/ac000631b. [DOI] [PubMed] [Google Scholar]
- 22.Wuuhrer M, Deelder AM, Hokke CH. J Chromatogr B. 2005;825:124. doi: 10.1016/j.jchromb.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 23.Hemstrom P. K Irgum J Sep Sci. 2006;29:1784. doi: 10.1002/jssc.200600199. [DOI] [PubMed] [Google Scholar]
- 24.Guile GR, Rudd PM, Wing DR, Prime SB, Dwek RA. Anal Biochem. 1996;240:210. doi: 10.1006/abio.1996.0351. [DOI] [PubMed] [Google Scholar]
- 25.Takegawa Y, Deguchi K, Ito H, Keira T, Nakagawa H, Nishimura S. J Sep Sci. 2006;29:2533. doi: 10.1002/jssc.200600133. [DOI] [PubMed] [Google Scholar]
- 26.Alpert AJ. J Chromatogr A. 1990;499:177. doi: 10.1016/s0021-9673(00)96972-3. [DOI] [PubMed] [Google Scholar]
- 27.Tomiya N, Awaya J, Kurono M, Endo S, Arata Y, Takahashi N. Anal Biochem. 1988;171:73. doi: 10.1016/0003-2697(88)90126-1. [DOI] [PubMed] [Google Scholar]
- 28.Takahashi N. J Chromatogr A. 1996;720:217. doi: 10.1016/0021-9673(95)00328-2. [DOI] [PubMed] [Google Scholar]
- 29.Makino Y, Omichi K, Hase S. Anal Biochem. 1998;264:172. doi: 10.1006/abio.1998.2848. [DOI] [PubMed] [Google Scholar]
- 30.Charlwood J, Birrell H, Bouvier ESP, Langridge J, Camilleri P. Anal Chem. 2000;72:1469. doi: 10.1021/ac991267n. [DOI] [PubMed] [Google Scholar]
- 31.Karas M, Bahr U, Dulcks T. Fresenius J Anal Chem. 2000;366:669. doi: 10.1007/s002160051561. [DOI] [PubMed] [Google Scholar]
- 32.Schmidt A, Karas M, Dulcks T. J Am Soc Mass Spectrom. 2003;14:492. doi: 10.1016/S1044-0305(03)00128-4. [DOI] [PubMed] [Google Scholar]
- 33.Wilm M, Mann M. Anal Chem. 1996;68:1. doi: 10.1021/ac9509519. [DOI] [PubMed] [Google Scholar]
- 34.Andrews CL, Li F, Yang E, Yu CP, Vouros P. J Mass Spectrom. 2006;41:43. doi: 10.1002/jms.944. [DOI] [PubMed] [Google Scholar]
- 35.Cech NB, Enke CG. Mass Spectrom Rev. 2001;20:362. doi: 10.1002/mas.10008. [DOI] [PubMed] [Google Scholar]
- 36.Smith RD, Shen Y, Tang K. Accounts Chem Res. 2004;37:269. doi: 10.1021/ar0301330. [DOI] [PubMed] [Google Scholar]
- 37.Ivanov AR, Zang L, Karger BL. Anal Chem. 2003;75:5306. doi: 10.1021/ac030163g. [DOI] [PubMed] [Google Scholar]
- 38.Shen Y, Moore RJ, Zhao R, Blonder J, Auberry DL, Masselon C, Pasa-Tolic L, Hixson KK, Auberry KJ, Smith RD. Anal Chem. 2003;75:3596. doi: 10.1021/ac0300690. [DOI] [PubMed] [Google Scholar]
- 39.Luo Q, Shen Y, Hixson KK, Zhao R, Yang F, Moore RJ, Mottaz HM, Smith RD. Anal Chem. 2005;77:5028. doi: 10.1021/ac050454k. [DOI] [PubMed] [Google Scholar]
- 40.Andre CE, Mosier AR. Anal Chem. 1973;45:1971. doi: 10.1021/ac60324a047. [DOI] [PubMed] [Google Scholar]
- 41.Kennedy RT, Jorgenson JW. Science. 1989;246:75. doi: 10.1126/science.2675314. [DOI] [PubMed] [Google Scholar]
- 42.Guiochon G. Anal Chem. 1981;53:1318. [Google Scholar]
- 43.Swart R, Kraak JC, Poppe H. J Chromatogr A. 1995;689:177. [Google Scholar]
- 44.Guo Y, Colon LA. Anal Chem. 1995;67:2511. [Google Scholar]
- 45.Yang L, Guihen E, Holmes JD, Loughran M, O’Sullivan GP, Glennon JD. Anal Chem. 2005;77:1840. doi: 10.1021/ac048544x. [DOI] [PubMed] [Google Scholar]
- 46.Huang X, Zhang J, Horvath C. J Chromatogr A. 1999;858:91. doi: 10.1016/s0021-9673(99)00795-5. [DOI] [PubMed] [Google Scholar]
- 47.Shen T. J Chromatogr Sci. 1992;30:239. doi: 10.1093/chromsci/30.6.239. [DOI] [PubMed] [Google Scholar]
- 48.Eeltink S, Svec F, Frechet JMJ. Electrophoresis. 2006;27:4249. doi: 10.1002/elps.200600259. [DOI] [PubMed] [Google Scholar]
- 49.Yue G, Luo Q, Zhang J, Wu S, Karger BL. Anal Chem. 2007;79:938. doi: 10.1021/ac061411m. [DOI] [PubMed] [Google Scholar]
- 50.Luo Q, Yue G, Valaskovic GA, Gu Y, Wu S, Karger BL. Anal Chem. 2007;79:6174. doi: 10.1021/ac070583w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luo Q, Gu Y, Wu S, Rejtar T, Karger BL. Electrophoresis. 2008;29:1604. doi: 10.1002/elps.200700741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harvey DJ. J Am Soc Mass Spectrom. 2005;26:622. doi: 10.1016/j.jasms.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 53.Premstaller A, Oberacher H, Walcher W, Timperio AM, Zolla L, Chervet JP, Cavusoglu N, Van Dorsselaer A, Huber CG. Anal Chem. 2001;73:2390. doi: 10.1021/ac010046q. [DOI] [PubMed] [Google Scholar]
- 54.Ashline DJ, Lapadula AJ, Liu YH, Lin M, Grace M, Pramanik B, Reinhold VN. Anal Chem. 2007;79:3830. doi: 10.1021/ac062383a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harvey DJ, Wing DR, Kuster B, Wilson IBH. J Am Soc Mass Spectrom. 2000;111:564. doi: 10.1016/S1044-0305(00)00122-7. [DOI] [PubMed] [Google Scholar]
- 56.Jitsuhara Y, Toyoda T, Itai T, Yamaguchi H. J Biochem. 2002;132:803. doi: 10.1093/oxfordjournals.jbchem.a003290. [DOI] [PubMed] [Google Scholar]
- 57.Cubbon S, Bradbury T, Wilson J, Thomas-Oates J. Anal Chem. 2007;79:8911. doi: 10.1021/ac071008v. [DOI] [PubMed] [Google Scholar]
- 58.McNulty DE, Annan RS. Mol Cell Proteomics. 2008;7:971. doi: 10.1074/mcp.M700543-MCP200. [DOI] [PubMed] [Google Scholar]