

Abstract
Although the clinical manifestations of alcoholic liver disease are well-described, little is known about the molecular basis of liver injury. Recent studies have indicated that ethanol exposure induces global protein hyperacetylation. This reversible, post-translational modification on the epsilon-amino groups of lysine residues has been shown to modulate multiple, diverse cellular processes ranging from transcriptional activation to microtubule stability. Thus, alcohol-induced protein hyperacetylation likely leads to major physiological consequences that contribute to alcohol-induced hepatotoxicity. Lysine acetylation is controlled by the activities of two opposing enzymes, histone acetyltransferases and histone deacetylases. Currently, efforts are aimed at determining which enzymes are responsible for the increased acetylation of specific substrates. However, the greater challenge will be to determine the physiological ramifications of protein hyperacetylation and how they might contribute to the progression of liver disease. In this review, we will first list and discuss the proteins known to be hyperacetylated in the presence of ethanol. We will then describe what is known about the mechanisms leading to increased protein acetylation and how hyperacetylation may perturb hepatic function.
Keywords: Ethanol, Hepatotoxicity, Acetylation, Deacetylases, Acetyltransferases
INTRODUCTION
The liver is the major site of ethanol metabolism and thus sustains the most injury from chronic alcohol consumption. In the hepatocyte cytosol, alcohol dehydrogenase (ADH) converts ethanol to acetaldehyde, a highly reactive intermediate. The acetaldehyde is further metabolized in mitochondria to acetate by acetaldehyde dehydrogenase (ALDH). Alcohol is also metabolized by the resident ER enzyme, cytochrome P450 2E1 (CYP 2E1). CYP 2E1-mediated ethanol metabolism not only leads to the formation of acetaldehyde, but also to the formation of oxygen and hydroxyethyl radicals that in turn promote the formation of other highly reactive intermediates[1]. All of these reactive metabolites can readily and covalently modify proteins, DNA and lipids[2–7]. More recently, it has become apparent that alcohol exposure induces protein covalent-modifications that are part of the natural repertoire. To date, these post-translational modifications include increased methylation, phosphorylation and acetylation[8–14]. In particular, numerous proteins have been identified that are hyperacetylated upon ethanol exposure, and this list is expanding rapidly. As for adduct formation, it is not clear how increased acetylation is related to the progression of alcohol-induced hepatotoxicity. In this review, we will first list and discuss hepatic proteins known to be hyperacetylated by ethanol exposure. We will then describe our current understanding of the mechanisms and physiological consequences of protein hyperacetylation.
ETHANOL-INDUCED LYSINE HYPERACETYLATION
For over 40 years it has been recognized that proteins can be acetylated and that the modification comes in two forms[15]. One is the irreversible, co-translational N-terminal acetylation of α-amino groups of mainly serine and alanine, but also of threonine, methionine and glycine. The other form is the reversible, post-translational modification of epsilon-amino groups on lysine residues located within a polypeptide[15,16]. The reversibility of lysine acetylation and its presence on an ever expanding list of nuclear and nonnuclear proteins have led some to postulate that it might rival phosphorylation in its ability to regulate cellular processes[16]. Thus, alcohol-induced protein hyperacetylation likely results in major physiological consequences that contribute to the progression of hepatotoxicity. In this section, we will first discuss individual hepatic proteins that are known to be hyperacetylated upon ethanol exposure (Table 1). We will then comment on recent work that implicates many more candidates for alcohol-induced lysine acetylation (Table 2).
Table 1.
Ethanol-induced hyperacetylated proteins
| Protein | EtOH exposure | System | References |
| Histone H3 | Acute and chronic | Hepatocytes, rat liver, stellate cells | [8,17–21] |
| p53 | Chronic | Rat liver, VLA17 cells | [61] |
| PGC-1α | Chronic | Rat liver, mouse liver | [12,14] |
| SREPB-1c | Chronic | H4IIEC3 cells | [12] |
| α-tubulin | Chronic | WIF-B cells, rat liver | [11,39,75] |
| AceCS2 | Chronic | Rat Liver | [13] |
Table 2.
Newly identified ethanol-induced hyperacetylated proteins
| Liver subcellularlocation1 | EtOH exposure | Function | No. proteinsidentified |
| Cytosol | Chronic | AA metabolism | 7 |
| Carbohydrate metabolism | 4 | ||
| Other metabolic pathways | 3 | ||
| Oxidative stress | 3 | ||
| Other | 2 | ||
| Mitochondria | Chronic | Lipid metabolism | 12 |
| AA metabolism | 5 | ||
| Oxidative phosphorylation | 2 |
Our unpublished results.
Histone H3
Histones were the first proteins known to be acetylated[15,16], and histone H3 was the first protein known to be hyperacetylated after ethanol treatment. This effect has been observed in isolated hepatocytes and in rat liver after both acute and chronic ethanol exposure[8,17–21]. Although histone H3 encodes at least four acetylated lysines (lys 9, 14, 18 and 23), acetylation of lysine 9 is selectively increased after exposure to physiological ethanol concentrations[8]. Because histone H3 hyperacetylation was prevented by both 4-methylpyrazole (4-MP) and cyanamide, it was concluded that the modification requires alcohol metabolism and is likely mediated by the ethanol metabolite, acetate[8]. Interestingly, lysine 9 is also reversibly methylated, and in ethanol-treated hepatocytes, its methylation is decreased[9]. Furthermore, alcohol also induces hypermethylation of lysine 4 and phosphorylation of neighboring serine residues (ser 10 and ser 28)[10]. It will be important to consider all of these modifications as the functional consequences of histone H3 hyperacetylation are defined (see below).
p53
p53 is a tumor suppressor that is mutated in over half of all human cancers. It is activated by DNA damage and functions to stop cell cycle progression. For over a decade it has been known that p53 is reversibly lysine acetylated, and that this modification promotes enhanced DNA binding[15,16]. Very recently, p53 has been shown to be hyperacetylated upon chronic ethanol exposure in rat liver lysates[14]. Although multiple lysines are known to be acetylated, it is not yet known which residue(s) is hyperacetylated after ethanol consumption. Furthermore, it is not yet known whether hyperacetylation requires ethanol metabolism or whether it alters p53 DNA binding properties.
Sterol response element binding protein-1c (SREBP-1c)
SREBPs are a family of transcription factors that regulate lipid and cholesterol synthesis. SREBP-1c is the major form expressed in liver and is known to activate numerous lipogenic enzymes. In rat hepatoma H4IIEC3 cells, ethanol treatment for 24 h led to a dose-dependent increase in SREBP-1c acetylation[12]. This result was confirmed in livers from ethanol-fed mice indicating it has physiologic importance. It is not yet known whether hyperacetylation requires ethanol metabolism nor is it known which lysine is hyperacetylated. However, more is known about the mechanism by which SREBP-1c is acetylated/deacetylated and how that relates to SREBP-1c function in gene regulation and lipogenesis (see below).
Peroxisome proliferator-activated receptor γ coactivator α (PGC-1α)
The nuclear transcriptional coactivator, PGC-1α, is a key regulator of hepatic glucose homeostasis and lipid metabolism[22]. It has also been implicated in regulating mitochondrial biogenesis and respiration[22]. PGC-1α is known to be acetylated on many lysines, and in general, acetylation is correlated with decreased transcriptional activity resulting in decreased expression of genes involved in mitochondrial fatty acid oxidation and gluconeogenesis. Thus, it is notable that PGC-1α hyperacetylation is induced by ethanol consumption[12]. Currently, it is not known which lysine(s) is modified or whether PGC-1α hyperacetylation requires ethanol metabolism. However, as for SREBP-1c, there is more known about some likely functional consequences of PGC-1α hyperacetylation (see below).
Acetyl CoA synthetase 2 (AceCS2)
A recent proteomic survey of mammalian cell proteins identified nearly 200 lysine-acetylated nuclear and nonnuclear proteins[23]. Remarkably, this survey further revealed that more than 20% of mitochondrial proteins were lysine-acetylated[23]. Thus, it is not surprising that the acetylation of AceCS2, an enzyme involved in lipid metabolism, is increased upon alcohol exposure, probably on lysine 635[13,24]. Acetylation of this residue correlates with decreased AceCS2 catalytic activity[25], but whether its activity is decreased in ethanol-treated cells is not yet known. Interestingly, alcohol-induced acetylation of mitochondrial proteins, including AceCS2, was not observably altered in livers from CYP 2E1 knockout mice suggesting that the modification does not require CYP 2E1-mediated ethanol metabolism[13]. However, whether ethanol metabolism by ADH and ALDH is required for mitochondrial protein hyperacetylation is not yet known.
Tubulin
Microtubules are one of the three major cytoskeletal systems of the cell. The polymer is made of repeating units of α- and β-tubulin heterodimers that form protofilaments, which in turn assemble into hollow tubes consisting of 13 protofilaments arranged in parallel. Microtubules exist as both dynamic and stable polymers. The latter population is characterized by a longer half-life, resistance to microtubule poisons (e.g. cold and nocodazole) and by specific post-translational modifications on the α-tubulin subunit[26]. These modifications include the removal of a carboxy-terminal tyrosine, polyglutamylation, polyglycylation and acetylation of lysine 40[26]. The functions of these modifications or whether they contribute to microtubule stability are still the subject of debate[27]. Recently, it was determined that chronic ethanol exposure enhanced α-tubulin acetylation at lysine 40 in polarized WIF-B cells and livers from ethanol-fed rats[11]. Increased acetylation correlated to increased stability suggesting that tubulin acetylation might in fact enhance microtubule stability. In WIF-B cells, increased tubulin acetylation and stability displayed both ethanol time- and dose-dependence[11]. Furthermore, tubulin hyperacetylation and stability was prevented by 4-MP and potentiated by cyanamide indicating that ethanol metabolism was required for the effects[11]. Thus, unlike acetate-mediated histone H3 hyperacetylation, tubulin hyperacetylation and increased stability are likely mediated by acetaldehyde. This disparity is likely to be due to the different mechanisms leading to enhanced acetylation of either substrate (see below).
The expanding list
With the growing number of known acetylated proteins and the large number of modifying enzymes, it is likely that numerous hepatic proteins are hyperacetylated in ethanol-treated cells. We initiated a proteomics approach to identify other hyperacetylated proteins from cytosolic and total membrane fractions prepared from livers from control and ethanol-fed rats (manuscript in preparation). So far, about 40 nonnuclear proteins have been identified (but not yet confirmed), half of which were from the cytosolic fraction and half from the total membranes. Remarkably, all the hyperacetylated proteins in the latter fraction were from mitochondria and most were metabolic enzymes (Table 2). Seven of these mitochondrial proteins were also identified in the proteomic survey for acetylated lysines described above[23] partially confirming our result. Also consistent with this finding is a recent study where purified mitochondria from alcohol-fed rats were immunblotted with anti-acetylated lysine antibodies[13]. Numerous immunoreactive species were observed (but not yet identified) suggesting massive mitochondrial protein hyperacetylation after ethanol exposure. Similarly, cytosolic fractions were highly hyperacetylated after ethanol exposure and the proteins identified varied widely in function, ranging from metabolic enzymes to proteins regulating oxidative stress to molecular chaperones. Efforts are currently underway to confirm the acetylation state of these proteins and to determine the functional consequences of their ethanol-induced hyperacetylation.
MECHANISMS OF ETHANOL-INDUCED LYSINE HYPERACETYLATION
Protein acetylation results from the coordinated activities of acetyltransferases and deacetylases[15,16]. Histones were the first proteins known to be acetylated, and accordingly the modifying enzymes were initially named histone acetyltransferases (HATs) and histone deacetylases (HDACs). Although the list of acetylated proteins has since grown to include numerous nonhistone substrates, their names have remained. In this section, we will briefly describe the two classes of enzymes and whether they are expressed in the liver. We will also discuss what is known about how these HATs and HDACs may be responsible for the alcohol-induced increase in lysine acetylation.
HDACs
To date, there are at least 18 known deacetylases that are categorized into four general classes based on sequence homology and cofactor/coenzyme dependence[28] (Table 3). Classes I, II and IV are closely related zinc-dependent enzymes whereas the class III HDACs are more distantly related, NAD+-dependent sirtuins[28,29]. Phylogenetic analysis of bacterial HDAC relatives indicates that the evolution of the class I, II and IV family members preceded the evolution of histones[29] suggesting that these HDACs have nonhistone substrates. This conclusion is consistent with the wide-range of mammalian HDAC substrates that have been identified (Table 3).
Table 3.
Deacetylases expressed in the liver
| Class | Enzyme | In liver? (methods) | Location | Substrates |
| I | HDAC1 | Yes (RT-PCR, IB, IHC, activity) | Nucleus | Histones, p53, retinoblastoma, STAT3, androgen receptor, estrogen receptor, Smad7, other transcription factors |
| HDAC2 | Yes (RT-PCR) | Nucleus | Histones, STAT3, other transcription factors | |
| HDAC3 | Yes (RT-PCR, IB, activity) | Nucleus | Histones, STAT3, Smad7, other transcription factors | |
| HDAC8 | Yes (RT-PCR) | Nucleus Cytoplasm | Histones, transcription factors, smooth muscle actin | |
| II | HDAC4 | Yes/No (RT-PCR) | Nucleus Cytoplasm | Transcription factors |
| HDAC5 | Yes (RT-PCR) | Nucleus Cytoplasm | Transcription factors | |
| HDAC6 | Yes (RT-PCR, IB, IF) | Cytoplasm | Tubulin, cortactin, HSP90 | |
| HDAC7 | No | Nucleus Cytoplasm | HIFα, other transcription factors | |
| HDAC9 | No | Nucleus Cytoplasm | Transcription factors | |
| HDAC10 | Yes (RT-PCR) | Nucleus Cytoplasm | Phosphatase pp1 | |
| III | SirT1 | Yes (RT-PCR, IB) | Nucleus | Histones, PGC-1α, LXR, p53, other transcription factors |
| SirT2 | Yes/No (RT-PCR, IB) | Cytoplasm | Histone H4, tubulin | |
| SirT3 | Yes (RT-PCR, IB) | Mitochondria | Acetyl CoA synthetase 2, glutamine dehydrogenase, ICDH2 | |
| SirT4 | Yes (RT-PCR) | Mitochondria | Glutamine dehydrogenase | |
| SirT5 | Yes (RT-PCR) | Mitochondria | Cytochrome c | |
| SirT6 | Yes (RT-PCR) | Nucleus | DNA polymerase β | |
| SirT7 | Yes (RT-PCR) | Nucleus | RNA polymerase I | |
| IV | HDAC11 | Unknown | Nucleus | Unknown |
IB: Immunoblotting; IHC: Immunohistochemistry; IF: Immunofluorescence; HIFα: Hypoxia-inducible factor α; LXR: Liver x receptor; ICHD2: Isocitrate dehydrogenase.
Mechanistically, classes I, II and IV all share a conserved catalytic core consisting of open α/β folds and a tubular active site pocket in which a zinc ion sits. The poised zinc mediates the nucleophilic attack of water on the acetylated lysine substrate resulting in the formation of a tetrahedral oxyanion intermediate. The nitrogen on the intermediate is then primed to accept a proton resulting in a charge relay system between histidine, aspartate, and tyrosine that results in the formation of acetate and the deacetylated lysine[29–31]. This catalytic mechanism was deduced with the help of HDAC inhibitors that displace the zinc ion thereby disrupting the charge relay system. Many of these inhibitors are proving to be promising anti-cancer therapies. The most potent inhibitor, trichostatin A (TSA), is a near perfect fit for the HDAC active site and is often used in studies to confirm the affect of protein acetylation on function (see below)[32].
The class I family members include HDACs 1, 2, 3 and 8 that are closely related to the yeast deacetylase, reduced potassium dependency 3 (RPD3)[32] (Table 3). They are all ubiquitously expressed (including in liver, Table 3), are found almost exclusively in the nucleus, are roughly the same length (370-480 aa) and encode one deacetylase domain[29,30,32,33]. These four HDACs have all been shown to deacetylate histones as well as a variety of transcription factors and other nuclear proteins including STATs and Smads[29,30,32,33] (Table 3). Recent reports suggest that class I enzymes may function together in multiprotein complexes that bind and deacetylate transcription factors[29,30]. Future work is needed to understand how the deacetylase activities are specifically coordinated leading to changes in gene expression.
Class II enzymes are much more variable in size (700-1200 aa) and exhibit more tissue-specific expression patterns with only HDACs 4, 5, 6 and 10 identified in the liver (Table 3). Like for class I enzymes, known class II substrates are mainly nuclear transcription factors[28–30,32–34]. However, the class II deacetylases encode both nuclear import and export signals allowing them to shuttle between the nucleus and cytoplasm where they bind to location-specific substrates[30] (Figure 1, bottom panel). Although interactions between these enzymes and their substrates are thought to regulate their activity and location[29,30], little is known how this process is regulated. The class II HDACs catalyze lysine deacetylation in much the same way as the class I enzymes but their active sites encode a glutamate instead of a glycine[29]. This larger side chain decreases active site size restricting substrate access thereby increasing enzyme specificity. Interestingly, HDAC6 and HDAC10 encode two deacetylase domains in tandem[28,34]. Whether the two sites deacetylate different substrates or whether both are needed for function is not yet clear[35–37]. Of particular interest to this review is HDAC6, the only exclusively cytosolic HDAC found in liver[38,39]. One of its known substrates is α-tubulin, one of the best characterized, nonnuclear, hyperacetylated proteins in ethanol exposed cells[11,39–42].
Figure 1.
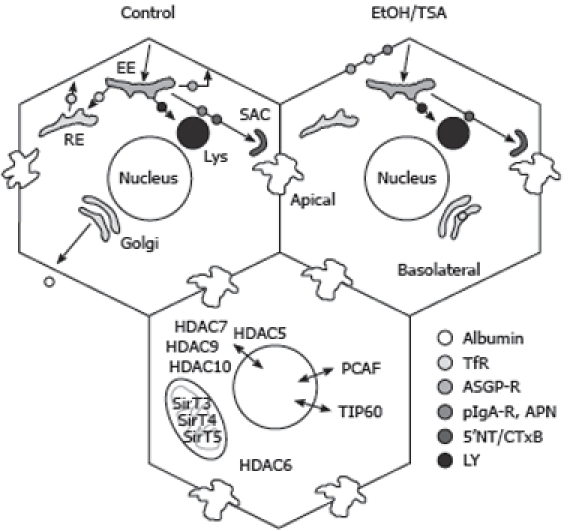
Alcohol-induced defects in protein trafficking can be explained by increased microtubule acetylation and stability. Cargo internalized from the basolateral plasma membrane is delivered to the early endosome (EE) and sorted into at least four different pathways. They are recycled directly back to the plasma membrane (e.g. ASGP-R), delivered to the recycling endosome (RE) before recycling back to the plasma membrane (e.g. Tf-R), delivered to lysosomes (lys) (e.g. LY) or to the apical plasma membrane via the sub-apical compartment (SAC) (e.g. APN, pIgA-R and 5’NT). Albumin secretion is also indicated. In ethanol (EtOH) or TSA treated cells, albumin secretion and the internalization of APN, pIgA-R, Tf-R and ASGP-R is impaired whereas the fluid-phase delivery of LY to lysosomes or the raft/caveolae-mediated internalization of 5’NT and CTxB are not changed. In the lower hepatocyte, the various deacetylases and acetyltransferases that may play a role in the acetylation of nonnuclear proteins are indicated.
Currently there is only one known class IV deacetylase, the recently identified HDAC11. As a result of its high sequence similarity to class I enzymes, HDAC11 is often considered a class I HDAC[30]. At present, little is known about HDAC11’s substrates, expression patterns or subcellular distributions. HDAC11 has been shown to act in a complex with other HDACs, most notably HDAC6, suggesting that it may not be an independent lysine deacetylase[29]. Clearly, more work is needed on this class IV enzyme before we can understand its role in protein acetylation.
Unlike the three classes of zinc-dependent HDACs, class III deacetylases are referred to collectively as sirtuins. Sirtuin activity requires NAD+, and thus each family member encodes a highly conserved, elongated catalytic core containing a Rossman fold characteristic of NAD+/NADH-binding proteins. Both the NAD+ and the acetylated lysine substrate bind to the “C pocket” which is located in the cleft between the Rossman fold and a smaller, more variable domain containing a zinc ion. This binding results in a nucleophilic substitution reaction where the NAD+ and acetylated lysine substrate are converted to nicotinamide, a deacetylated lysine and a novel metabolite, 2’-O-acetyl-adenosine diphosphate ribose[31]. In total, there are seven known sirtuins that all exhibit interesting expression patterns. SirT1 is a widely expressed, well characterized nuclear sirtuin whose long list of substrates includes transcription factors, histones, p53, and PGC-1α[42–44] (Table 3). SirT6 and 7 are two other nuclear sirtuins, and all three have been found in the liver by RT-PCR[44,45]. SirT2 is the lone cytoplasmic sirtuin, and like its counterpart, HDAC6, its main substrate is α-tubulin[46]. Although SirT2 was detected in liver homogenates by RT-PCR, the protein has not been detected at any level in liver lysates or WIF-B cells[39]. SirT3, 4 and 5 are all mitochondrial-specific sirtuins that are ubiquitously expressed (Figure 1, bottom panel). So far, only SirT3 and 5 have confirmed deacetylase activity[47,48]. Interestingly, recent reports suggest that SirT4 (and nuclear SirT6) are instead ADP ribosyltransferases that preferentially ADP-ribosylate acetylated substrates[49,50]. Further studies are needed to examine the effects of ethanol on this activity.
HATs
To date, there are 17 families of HATs that have been grouped according to sequence homology. In general, the N and C terminal domains of the acetyltransferases are structurally and functionally diverse while the catalytic domain is highly conserved. All HAT active sites contain a structurally conserved loop-β strand core domain that binds acetyl Co-A[51]. Also within this core domain is a glutamate that serves as a lysine docking site. The glutamate removes a proton from the docked lysine allowing acetyl transfer from acetyl CoA[51]. From the 17 families, three major groups have emerged: Gcn5/PCAF, p300/CBP and MYST (Table 4). Little is known about the mammalian members of the Gcn5/PCAF family, but in general these HATs have been detected in liver, reside in the nucleus and have mainly nuclear substrates. Only PCAF has been found to distribute to both the nucleus and cytoplasm[52–55] allowing for more varied substrates including histones, p53, and the actin binding protein, cortactin[56–58]. The p300/CBP family consists of p300 and CBP that are nearly structurally identical and are referred to interchangeably[57,58]. They also reside in the nucleus and are expressed in the liver. The least characterized MYST family consists of the mammalian enzymes, TIP60, HBO, MOZ, and MORF[57,58]. Although each of these HATs have been shown to acetylate histones H3 and H4, much still needs to be learned about other possible substrates and whether they function alone or in large multi-HAT complexes.
Table 4.
Histone acetyltransferases expressed in the liver
| Family | HAT | In liver? (methods) | Location | Substrates | References |
| Gcn5/PCAF | PCAF | Yes (IB) | Nucleus Cytoplasm | Histones, p53, PGC-1a, other transcription factors, cortactin | [52–58] |
| p300/CBP | CBP | Yes (IB) | Nucleus | Histones, E1A, p53, SRC-1, TIF2, ACTR, SREBP, other transcription factors | [54,57,58] |
| p300 | Yes (IB) | Nucleus | Histones, E1A, p53, SRC-1,TIF2, TAT, ACTR, SREBP, other transcription factors | [18,54,55,57,58] | |
| MYST | TIP60 | Yes (IB) | Nucleus Cytoplasm | Histones H3 and H4, androgen receptor | [54,57] |
| HBO | Unknown | Nucleus | Histones H3 and H4 | [57,58] | |
| MOZ | No | Nucleus | Histones H3 and H4 | [100] | |
| MORF | Yes (RT-PCR) | Nucleus | Histones H3 and H4 | [101,102] |
E1A: Adenovirus early region 1A; SRC-1: Steroid receptor coactivator; TIF2: Transcriptional intermediary factor 2.
General roles for HATs and HDACs in ethanol-induced protein acetylation
Changes in both HAT and HDAC expression have been correlated with global changes in protein acetylation. Thus, a simple explanation for ethanol-induced protein acetylation may be altered enzyme levels. The prediction is that either a decrease in HDAC expression or an increase in HAT levels will lead to increased protein acetylation. In addition, changes in enzyme activity or subcellular distribution may lead to hyperacetylation. For example, either a decrease in HDAC activity or a loss of nuclear import could enhance nuclear protein acetylation. Additionally, the sirtuin deacetylases are NAD+-dependent enzymes. Because ethanol metabolism is NAD+-depleting, sirtuin activity may be impaired thereby leading to increased protein acetylation. Below, we summarize what is known about how alterations in HAT or HDAC activity/function contribute to ethanol-induced protein hyperacetylation.
Histone H3 hyperacetylation and p300
At present, the mechanisms responsible for ethanol-induced hyperacetylation of histone H3 are not known, but recent studies have provided some clues. In livers of ethanol-fed rats, p300 protein levels were increased about 3 fold, which correlated with increases in overall nuclear HAT activity[17,18,21]. Acetate treatment also led to enhanced nuclear HAT activity and histone H3 hyperacetylation indicating ethanol metabolism is required for this modification[17]. Interestingly, the protein expression of the closely related p300 homologue, CBP, was not altered[18] indicating that ethanol-induced histone H3 acetylation may be specific to p300. These studies further suggest that these two HATs may not be completely interchangeable. To date, it is not known which (if any) HDAC is involved in histone H3 hyperacetylation in ethanol-treated cells. However, total nuclear HDAC activity was significantly decreased in ethanol-treated WIF-B cells, and that decreased activity correlated with increased histone H3 acetylation[39]. In contrast, total nuclear HDAC activity was not altered in ethanol-treated isolated hepatocytes where enhanced histone H3 acetylation was observed[17,21]. The reasons for these disparate results are not known but may be explained by differences in the cell systems used, acute vs. chronic alcohol-treatment, or differences in the deacetylase assay used. Further research is needed to elucidate this further.
SREBP-1c hyperacetylation and SirT1 and p300
As for histone H3, the mechanisms responsible for ethanol-induced hyperacetylation of SREBP-1c are not known, but work has provided some hints. From studies performed in H4IIEC3 cells, overexpressing SirT1 and p300 were found to deacetylate or acetylate SREBP-1c, respectively, confirming that SREBP-1c is a substrate for both enzymes[12]. Increasing ethanol concentrations enhanced p300-mediated SREBP acetylation in these cells while a concomitant decrease in SirT1 protein levels was observed[12,14]. Thus, SREBP-1c hyperacetylation was the result of changes in both types of modifying enzymes. As for SREBP-1c, PGC-1α hyperacetylation was also correlated with decreased SirT1 protein expression[12,14]. At present, it is not known if changes in SirT1 or p300 activity levels correlate with changes in protein levels or whether SREBP-1c and PGC-1α hyperacetylation requires ethanol metabolism. Future work is clearly needed to understand this fully.
Tubulin hyperacetylation and HDAC6
Since the specific microtubule acetyltransferase has not yet been identified, studies thus far have focused on the known hepatic tubulin deacetylase, HDAC6 (Table 3). Although ethanol treatment did not alter HDAC6 subcellular distributions in polarized WIF-B cells, it led to a 25% decrease in HDAC6 protein levels[39]. HDAC6 binding to endogenous microtubules was also found to be significantly impaired by about 70% in ethanol-treated cells and this impairment partially required ethanol metabolism. Measuring HDAC6 tubulin deacetylase activity by two methods further revealed that ethanol did not impair HDAC6’s ability to bind or deacetylate exogenous tubulin. This suggests that tubulin from ethanol-treated cells was modified, thereby preventing HDAC6 binding[39].
Although decreased HDAC6 protein levels are a simple explanation for increased tubulin acetylation, it is likely that the impaired microtubule binding has more impact. HDAC6 is abundant in the liver[56,59] such that a 25% decrease in levels may not likely have profound effects on tubulin acetylation. Rather, the 70% impairment in HDAC6 binding to microtubules may have a more dramatic effect; much less of the available enzyme can bind its substrate leading to decreased deacetylation. At present, the nature of the alcohol-induced tubulin modification is not known, but there are some interesting possibilities. Both impaired HDAC6 binding to microtubules and alcohol-induced tubulin hyperacetylation require ethanol metabolism, and from studies using 4-MP, both events are likely mediated by acetaldehyde[11,39]. Because this highly reactive ethanol metabolite can readily and covalently modify a highly reactive lysine in α-tubulin in vitro[60], one provocative possibility is that tubulin acetaldehyde adducts impede HDAC6 binding. Because decreased HDAC6 binding to microtubules was only partially prevented by 4-MP, it is also possible that other reactive ethanol metabolites form detrimental tubulin adducts. Future studies are needed to elucidate this and other details.
Mitochondrial protein hyperacetylation and sirtuins
The ever-expanding list of ethanol-induced hyperacetylated mitochondrial proteins is likely to generate much interest in the mitochondrial-specific sirtuins, but so far, only protein levels of SirT3 and 5 have been examined. Although SirT3 is considered the predominant mitochondrial deacetylase, its expression levels were not changed in livers from ethanol-fed rats[13]. In contrast, SirT5 protein levels were significantly decreased[61]. To date, nothing is known about the subcellular distributions, activities or substrate specificities of these deacetylases in ethanol-treated cells. Furthermore, virtually nothing is known about the HATs required for mitochondrial protein acetylation. Clearly, this is a fertile area of investigation for many researchers.
Cytosolic protein hyperacetylation and nonnuclear HATs and HDACs
Recent proteomics studies have revealed that many cytosolic proteins are hyperacetylated upon ethanol exposure (Table 2). At present, very little is known about the HATs or HDACs responsible for these modifications. However, attention is being turned to those modifying enzymes that are exclusively cytosolic (HDAC6) or those that shuttle between the nucleus and cytoplasm (PCAF, TIP60 and HDACs 5, 7, 9 and 10) (Figure 1, bottom panel). Not only will future studies likely provide a better understanding of alcohol-induced protein acetylation and the progression of hepatotoxicity, they will also undoubtedly identify new substrates for these enzymes. As for studies on the mitochondrial modifying enzymes, this area of research promises to be fruitful.
CONSEQUENCES OF ETHANOL-INDUCED LYSINE HYPERACETYLATION
Although there is an ever-expanding list of proteins that are known to be hyperacetylated upon ethanol exposure, little is known about the functional consequences of this modification. In general, the added acetyl group likely neutralizes the positive charge on lysine while increasing the overall size and hydrophobicity of the side chain. Such changes may result in protein conformational changes that alter function, albeit to a much lesser extent than the addition of a large, highly charged phosphoryl group. Also, lysine acetylation sites have been identified that overlap with nuclear localization signals[23] such that the modification may induce altered protein subcellular distributions. Not only can lysine residues be acetylated, they can also be methylated, sumoylated and ubiquitinylated such that ethanol-induced hyperacetylation may displace other modifications further altering protein function. In fact, p300 acetylation has been shown to prevent its sumoylation thereby repressing its activity[62]. Clearly, mechanistic studies are required to not only understand the functional consequences of acetylation in the normal liver, but also how alcohol-induced hyperacetylation alters hepatic function in the alcoholic liver. In this section, we will describe some recent advances in our understanding of how ethanol-induced acetylation impairs hepatic function. Although it is too early for specific mechanistic details, the results provide an exciting framework for continued investigation.
Histone H3 hyperacetylation alters transcriptional regulation
Alcohol consumption has long been known to lead to changes in gene expression. Many genes are up-regulated including those encoding for enzymes involved in alcohol metabolism, lipogenesis and the regulation of oxidative stress[63,64]. Many genes are also down-regulated, but fewer seem to functionally group together[63,64]. The specific mechanisms responsible for changes in alcohol-induced gene expression are not well defined. Recent studies have been aimed at understanding the role of histone modifications in transcriptional regulation. The amino-termini of histones are characterized by at least six different modifications (acetylation, ubiquitinylation, methylation, phosphorylation, sumoylation and ADP-ribosylation) occurring on lysines, arginines, serines, threonines and histidines[65]. In general, these reversible modifications are thought to change the net negative charge of the amino-terminal domain leading to altered DNA binding and changes in gene expression[66]. Thus, the simple hypothesis is that alcohol-induced lysine 9 hyperacetylation will decrease the amino-terminal net negative charge thereby loosening histone H3 associations with DNA. The relaxed DNA becomes more accessible to the transcriptional machinery, leading to enhanced transcription.
This prediction was tested in ethanol-treated hepatocytes using a series of chromatin immunoprecipitations. In general, histone H3 with acetylated lysine 9 residues was found to be more highly associated with the promoters of genes known to be up-regulated by ethanol exposure (ADH and glutathione S-transferase)[9], consistent with this hypothesis. Also consistent with this hypothesis is that histone H3 with methylated lysine 9 residues (this modification is associated with gene silencing) was more highly associated with promoters of genes known to be down-regulated by ethanol (L-serine dehydratase and CYP 2C11)[9]. Somewhat surprisingly, histone H3 containing methylated lysine 4 residues was found to be highly associated with ADH and glutathione S-transferase promoters, not with promoters of down-regulated genes[9]. These associations suggest that lysine 4 hyperacetylation and lysine 4 methylation enhance gene expression while lysine 9 methylation represses transcription.
At present, the site-specific differences in promoter associations cannot be fully explained. One implication is that promoter regions are characterized by specific microenvironments that are differentially accessible to histone modifying enzymes and by extension, are differentially affected by ethanol exposure. In order to fully understand these site-specific differences, it will be necessary to fully account for all six possible histone H3 modifications and the modifying enzymes in control and ethanol-treated cells. Also, to determine how each modification or combination of modifications regulates gene expression, assays that directly monitor transcription activation (rather than promoter associations) will need to be developed. Furthermore, recent reports demonstrate that histone hyperacetylation may also lead to nucleosome instability allowing transcription at cryptic promoters resulting in aberrant gene product expression[67,68]. This emerging hypothesis must be considered as the transcriptional consequences of ethanol-induced histone acetylation as elucidated.
Ethanol-induced acetylation of SREBP-1 and PGC-1α leads to altered lipid metabolism
One of the first clinical manifestations of alcohol consumption is the appearance of a fatty liver (steatosis). This is correlated with the up-regulation of many lipogenic enzymes that leads to the alcohol-induced synthesis of hepatic triglycerides and phospholipids[69]. An active area of steatosis research is aimed at identifying members of the transcriptional machinery that regulate gene expression of the enzymes involved in lipid metabolism. It has long been appreciated that SREBP-1 promotes the expression of many genes involved in lipogenesis that are up-regulated after alcohol consumption[69]. However, the exact mechanism by which SREBP-1 leads to enhanced expression is not well-defined. One emerging hypothesis is that SREBP-1 acetylation plays an important role. SREBP-1 is known to be acetylated in its DNA binding domain, and that when acetylated, DNA binding is enhanced. SREBP-1 is also known to be ubiquitinylated on the same lysine residues leading to its proteosomal degradation[70]. Thus, in alcohol-treated hepatocytes the prediction is that hyperacetylation prevents SREBP-1 proteosomal degradation by displacing the ubiquitin while enhancing its DNA binding which leads to increased transcription of lipogenic enzymes.
A similar scenario is emerging for the transcriptional activator, PGC-1α, but it is the deacetylated protein that up-regulates expression of genes regulating fatty acid β-oxidation[22]. Therefore in ethanol-treated hepatocytes, the prediction is that the hyperacetylated PGC-1α will be inactive. Based on this prediction and the one for SREBP-1, a straightforward scenario emerges. Alcohol-induced SREBP-1 hyperacetylation enhances lipid synthesis by activating lipogenic enzyme transcription while PGC-1α hyperacetylation impairs lipid catabolism by inhibiting transcription of enzymes involved in fatty acid oxidation. The altered transcriptional activation in either case leads to hepatic fatty acid accumulation that likely contributes to development of steatosis. Because many other components of the transcriptional machinery are known to be acetylated (Tables 3 and 4), it is likely that alcohol-induced hyperacetylation will have far- reaching effects on hepatic gene expression.
Ethanol-induced microtubule acetylation leads to impaired protein trafficking
Because microtubules are central to multiple cellular processes, changes in their dynamics will likely alter hepatic function. An active area of research has been aimed at understanding the relationship between protein trafficking and alterations in microtubule dynamics. Not only is protein trafficking microtubule-dependent, the trafficking of many hepatic proteins is also impaired by ethanol[71–74]. Two transport pathways appear to be affected: transport of newly-synthesized secretory or membrane proteins from the Golgi to the basolateral membrane and receptor-mediated endocytosis from the sinusoidal surface (Figure 1). One attractive hypothesis is that the alcohol-induced defects in secretion and endocytosis can be explained by increased microtubule acetylation and stability.
To test this hypothesis, recent studies have examined the trafficking of selected proteins in WIF-B cells treated with ethanol or TSA, a potent inhibitor of HDAC6, the major tubulin deacetylase in liver and WIF-B cells[39] (Table 3). Importantly, TSA induces increased microtubule acetylation and stability to the same extent as ethanol[75]. As shown previously in situ, the endocytic trafficking of asialoglycoprotein-receptor (ASGP-R) was impaired in ethanol-treated WIF-B cells[75] (Figure 1). This impairment required ethanol metabolism and was likely mediated by acetaldehyde[75]. TSA also impaired ASGP-R endocytic trafficking, but to a lesser extent. Similarly, both ethanol and TSA impaired transcytosis of a single spanning apical resident, aminopeptidase (APN). For both ASGP-R and APN, and for both treatments, the block in trafficking was internalization from the basolateral membrane. Interestingly, no changes in transcytosis of the GPI-anchored protein, 5’nucleotidase (5’NT) (Figure 1), were observed suggesting that increased microtubule acetylation and stability differentially regulate internalization. It was further determined that albumin secretion was impaired in both ethanol- and TSA-treated cells[75] indicating that increased microtubule acetylation and stability also disrupt this transport step. Thus, increased microtubule acetylation and stability explain, in part, the alcohol-induced defects in membrane trafficking.
There is evidence that suggests that different microtubule populations (and/or their modifications) support specific protein transport steps[76]. Of particular interest are studies performed in WIF-B cells that used a novel microtubule depolymerizing drug, 201-F[77]. This drug specifically depolymerizes dynamic microtubules leaving only stable, acetylated polymers behind. In 201-F-treated cells, both secretion and transcytosis were impaired[77]. Although the specific impaired transcytotic step was not identified, increased basolateral labeling of the apical proteins was observed. These results are remarkably consistent with the findings in ethanol or TSA treated cells where increased populations of stable microtubules were observed (presumably at the expense of dynamic microtubules) that correlated with impaired albumin secretion and basolateral internalization.
An unanswered question from these studies is why 5’NT distributions were not altered in treated cells. Furthermore, the internalization of cholera toxin B subunit (CTxB) (a known raft marker) was also not impaired by ethanol exposure (Figure 1). One possibility is that internalization mechanisms were differentially impaired by ethanol metabolism. There are at least three major internalization routes in mammalian cells: clathrin-mediated, caveolae/raft-mediated and non-clathrin/non-raft mediated[78] that are characterized by specific molecular players, cargoes and regulators. In general, the receptors that displayed impaired endocytosis in ethanol-treated hepatocytes in situ (ASGP-R, EGF-R, and to a lesser extent, insulin via its receptor)[71–73,79,80] and in WIF-B cells (ASGP-R, transferrin receptor (Tf-R) and polymeric IgA receptor (pIgA-R)[75] (Figure 1) are internalized via clathrin-mediated pathways. Interestingly, the non-clathrin/non-raft-mediated fluid phase uptake of Lucifer Yellow was not changed in livers of ethanol-fed rats or WIF-B cells suggesting this pathway is not affected by ethanol metabolism[81] (Figure 1). Thus, we propose that the molecular machinery that drives clathrin-mediated endocytosis is more prone to adduction (by acetaldehyde or other reactive metabolites) or covalent modification such that it is selectively impaired by alcohol treatment.
Another unanswered question from these studies is how the acetylation of lysine 40 specifically contributes to enhanced microtubule stability and trafficking defects. Although lysine 40 is thought to reside in the lumen of the microtubule[82], it is possible that its acetylation may lead to altered tubulin conformation such that interactions with microtubule associated proteins and motors are altered. This hypothesis is supported by the findings that kinesin, dynein and dynactin preferentially bound acetylated microtubules in neuronal cells[83–85]. This is further supported by the finding that vesicles recovered from livers of ethanol-fed rats have decreased motility in vitro[86]. Clearly, further studies are needed to fully understand these results.
Mitochondrial dysfunction and protein hyperacetylation
Despite the large number of mitochondrial proteins known to be acetylated, little is known about the functional consequences of the modification on mitochondrial function. So far, only glutamate dehydrogenase and AceCS2 activities have been related to their acetylation states, and in both cases, increased acetylation correlated with decreased activity[25]. Thus, the prediction is that in ethanol-treated hepatocytes, the hyperacetylated enzymes would be inactivated leading to altered lipid metabolism that contributes to the development of steatosis as described above. Also, as described above, acetylation may function as an on/off switch for these and other mitochondrial metabolic enzymes such that alcohol-induced changes in this modification may have a large impact on hepatic metabolism.
However, recent results from knockout mice are not consistent with this conclusion[48]. Striking levels of mitochondrial protein acetylation were observed in livers from SirT3 knockout mice whereas mitochondrial acetylation in SirT4 or SirT5 knockout mice was not changed. This suggests that SirT3 is the predominant mitochondrial deacetylase. Despite the high levels of mitochondrial protein acetylation, there was no discernible phenotype in SirT3 knockout mice. Specifically in liver, there was no change in morphology, no change in mitochondrial numbers and metabolism was not altered suggesting hyperacetylation does not affect mitochondrial function[48]. However, it is possible that hyperacetylation is only detrimental under stressed conditions such that in the alcoholic liver, it leads to mitochondrial dysfunction.
This conclusion is consistent with a recent hypothesis that mitochondrial protein acetylation functions as a sensor for the overall cellular energy status[23]. Kim et al[23] suggest that acetyl-CoA and NAD+ levels are the key indicators of energy status. Coincidentally, these two molecules serve as cofactors for HATs (acetyl-CoA) or sirtuins (NAD+). Furthermore, over 44% of mitochondrial dehydrogenases that require NAD+ for activity are known to be acetylated. Thus, one possibility is that lysine acetylation serves as a feedback mechanism for the regulation of dehydrogenases. For example, under NAD+-depleting conditions, sirtuins are less active resulting in higher protein acetylation and dehydrogenase activities. In contrast, when acetyl-CoA levels are limiting, HATs are inactivated leading to decreased protein acetylation and increased dehydrogenase activities. Thus, in the alcoholic liver where NAD+ is depleting, increased acetylation is predicted to correlate with impaired dehydrogenase activity and by extension, impaired mitochondrial function. Although it has been suggested that NAD+ levels recover after prolonged ethanol exposure, the finding that hyperacetylation remains long after chronic ethanol withdrawal[13] suggests that this mechanism may have physiologic relevance. Clearly, this exciting hypothesis needs to be rigorously tested.
CONCLUSION
Chronic alcohol consumption leads to the hyperacetylation of numerous hepatic nuclear and nonnuclear proteins. Although many interesting and provocative mechanisms have been proposed that describe how hyperacetylation contributes to alcohol-induced hepatotoxicity, future work is clearly needed to test these hypotheses. New therapeutic strategies for treating patients with chronic liver disease may be aimed at reducing protein acetylation. Currently, specific SirT1 activators (e.g. resveratrol and SRT-501) are known to be well-tolerated in humans and are in clinical trials for treatment of various metabolic diseases including type 2 diabetes[87]. Furthermore, resveratrol has been shown to attenuate fatty liver in alcohol-exposed mice[88]. An exciting possibility is that this drug and other specific deacetylase activators or acetyltransferase inhibitors will be useful in treating alcoholic liver disease.
Footnotes
Supported by The National Institute of Alcohol Abuse and Alcoholism (R21 AA015683) awarded to P.L.T.
Peer reviewers: Gyongyi Szabo, MD, PhD, Director of Hepatology and Liver Center, University of Massachusetts Medical School, 364 Plantation Street, NRB Floor 2, Room 215, Worcester, Massachusetts 01605-2324, United States; Michael Trauner, Professor, Medical University Graz, Auenbruggerplatz 15, Graz A-8036, Austria
S- Editor Li LF L- Editor Kerr C E- Editor Zheng XM
References
- 1.Tuma DJ, Casey CA. Dangerous byproducts of alcohol breakdown--focus on adducts. Alcohol Res Health. 2003;27:285–290. [PMC free article] [PubMed] [Google Scholar]
- 2.Brooks PJ. DNA damage, DNA repair, and alcohol toxicity--a review. Alcohol Clin Exp Res. 1997;21:1073–1082. [PubMed] [Google Scholar]
- 3.Fraenkel-Conrat H, Singer B. Nucleoside adducts are formed by cooperative reaction of acetaldehyde and alcohols: possible mechanism for the role of ethanol in carcinogenesis. Proc Natl Acad Sci USA. 1988;85:3758–3761. doi: 10.1073/pnas.85.11.3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kenney WC. Acetaldehyde adducts of phospholipids. Alcohol Clin Exp Res. 1982;6:412–416. doi: 10.1111/j.1530-0277.1982.tb05000.x. [DOI] [PubMed] [Google Scholar]
- 5.Kenney WC. Formation of Schiff base adduct between acetaldehyde and rat liver microsomal phosphatidylethanolamine. Alcohol Clin Exp Res. 1984;8:551–555. doi: 10.1111/j.1530-0277.1984.tb05728.x. [DOI] [PubMed] [Google Scholar]
- 6.Ristow H, Obe G. Acetaldehyde induces cross-links in DNA and causes sister-chromatid exchanges in human cells. Mutat Res. 1978;58:115–119. doi: 10.1016/0165-1218(78)90103-9. [DOI] [PubMed] [Google Scholar]
- 7.Wehr H, Rodo M, Lieber CS, Baraona E. Acetaldehyde adducts and autoantibodies against VLDL and LDL in alcoholics. J Lipid Res. 1993;34:1237–1244. [PubMed] [Google Scholar]
- 8.Park PH, Miller R, Shukla SD. Acetylation of histone H3 at lysine 9 by ethanol in rat hepatocytes. Biochem Biophys Res Commun. 2003;306:501–504. doi: 10.1016/s0006-291x(03)01040-4. [DOI] [PubMed] [Google Scholar]
- 9.Pal-Bhadra M, Bhadra U, Jackson DE, Mamatha L, Park PH, Shukla SD. Distinct methylation patterns in histone H3 at Lys-4 and Lys-9 correlate with up- & down-regulation of genes by ethanol in hepatocytes. Life Sci. 2007;81:979–987. doi: 10.1016/j.lfs.2007.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee YJ, Shukla SD. Histone H3 phosphorylation at serine 10 and serine 28 is mediated by p38 MAPK in rat hepatocytes exposed to ethanol and acetaldehyde. Eur J Pharmacol. 2007;573:29–38. doi: 10.1016/j.ejphar.2007.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kannarkat GT, Tuma DJ, Tuma PL. Microtubules are more stable and more highly acetylated in ethanol-treated hepatic cells. J Hepatol. 2006;44:963–970. doi: 10.1016/j.jhep.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 12.You M, Liang X, Ajmo JM, Ness GC. Involvement of mammalian sirtuin 1 in the action of ethanol in the liver. Am J Physiol Gastrointest Liver Physiol. 2008;294:G892–G898. doi: 10.1152/ajpgi.00575.2007. [DOI] [PubMed] [Google Scholar]
- 13.Picklo MJ Sr. Ethanol intoxication increases hepatic N-lysyl protein acetylation. Biochem Biophys Res Commun. 2008;376:615–619. doi: 10.1016/j.bbrc.2008.09.039. [DOI] [PubMed] [Google Scholar]
- 14.Lieber CS, Leo MA, Wang X, Decarli LM. Effect of chronic alcohol consumption on Hepatic SIRT1 and PGC-1alpha in rats. Biochem Biophys Res Commun. 2008;370:44–48. doi: 10.1016/j.bbrc.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 15.Polevoda B, Sherman F. The diversity of acetylated proteins. Genome Biol. 2002;3:reviews0006. doi: 10.1186/gb-2002-3-5-reviews0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kouzarides T. Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 2000;19:1176–1179. doi: 10.1093/emboj/19.6.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park PH, Lim RW, Shukla SD. Involvement of histone acetyltransferase (HAT) in ethanol-induced acetylation of histone H3 in hepatocytes: potential mechanism for gene expression. Am J Physiol Gastrointest Liver Physiol. 2005;289:G1124–G1136. doi: 10.1152/ajpgi.00091.2005. [DOI] [PubMed] [Google Scholar]
- 18.Bardag-Gorce F, French BA, Joyce M, Baires M, Montgomery RO, Li J, French S. Histone acetyltransferase p300 modulates gene expression in an epigenetic manner at high blood alcohol levels. Exp Mol Pathol. 2007;82:197–202. doi: 10.1016/j.yexmp.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim JS, Shukla SD. Histone h3 modifications in rat hepatic stellate cells by ethanol. Alcohol Alcohol. 2005;40:367–372. doi: 10.1093/alcalc/agh170. [DOI] [PubMed] [Google Scholar]
- 20.Kim JS, Shukla SD. Acute in vivo effect of ethanol (binge drinking) on histone H3 modifications in rat tissues. Alcohol Alcohol. 2006;41:126–132. doi: 10.1093/alcalc/agh248. [DOI] [PubMed] [Google Scholar]
- 21.Choudhury M, Shukla SD. Surrogate alcohols and their metabolites modify histone H3 acetylation: involvement of histone acetyl transferase and histone deacetylase. Alcohol Clin Exp Res. 2008;32:829–839. doi: 10.1111/j.1530-0277.2008.00630.x. [DOI] [PubMed] [Google Scholar]
- 22.Rodgers JT, Lerin C, Gerhart-Hines Z, Puigserver P. Metabolic adaptations through the PGC-1 alpha and SIRT1 pathways. FEBS Lett. 2008;582:46–53. doi: 10.1016/j.febslet.2007.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006;23:607–618. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 24.Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci USA. 2006;103:10230–10235. doi: 10.1073/pnas.0604392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci USA. 2006;103:10224–10229. doi: 10.1073/pnas.0603968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Westermann S, Weber K. Post-translational modifications regulate microtubule function. Nat Rev Mol Cell Biol. 2003;4:938–947. doi: 10.1038/nrm1260. [DOI] [PubMed] [Google Scholar]
- 27.Palazzo A, Ackerman B, Gundersen GG. Cell biology: Tubulin acetylation and cell motility. Nature. 2003;421:230. doi: 10.1038/421230a. [DOI] [PubMed] [Google Scholar]
- 28.Yang XJ, Grégoire S. Class II histone deacetylases: from sequence to function, regulation, and clinical implication. Mol Cell Biol. 2005;25:2873–2884. doi: 10.1128/MCB.25.8.2873-2884.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hildmann C, Riester D, Schwienhorst A. Histone deacetylases--an important class of cellular regulators with a variety of functions. Appl Microbiol Biotechnol. 2007;75:487–497. doi: 10.1007/s00253-007-0911-2. [DOI] [PubMed] [Google Scholar]
- 30.Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9:206–218. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hodawadekar SC, Marmorstein R. Chemistry of acetyl transfer by histone modifying enzymes: structure, mechanism and implications for effector design. Oncogene. 2007;26:5528–5540. doi: 10.1038/sj.onc.1210619. [DOI] [PubMed] [Google Scholar]
- 32.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26:5310–5318. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- 34.Bertos NR, Wang AH, Yang XJ. Class II histone deacetylases: structure, function, and regulation. Biochem Cell Biol. 2001;79:243–252. [PubMed] [Google Scholar]
- 35.Zhang Y, Gilquin B, Khochbin S, Matthias P. Two catalytic domains are required for protein deacetylation. J Biol Chem. 2006;281:2401–2404. doi: 10.1074/jbc.C500241200. [DOI] [PubMed] [Google Scholar]
- 36.Zou H, Wu Y, Navre M, Sang BC. Characterization of the two catalytic domains in histone deacetylase 6. Biochem Biophys Res Commun. 2006;341:45–50. doi: 10.1016/j.bbrc.2005.12.144. [DOI] [PubMed] [Google Scholar]
- 37.Haggarty SJ, Koeller KM, Kau TR, Silver PA, Roberge M, Schreiber SL. Small molecule modulation of the human chromatid decatenation checkpoint. Chem Biol. 2003;10:1267–1279. doi: 10.1016/j.chembiol.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 38.Wang AG, Kim SU, Lee SH, Kim SK, Seo SB, Yu DY, Lee DS. Histone deacetylase 1 contributes to cell cycle and apoptosis. Biol Pharm Bull. 2005;28:1966–1970. doi: 10.1248/bpb.28.1966. [DOI] [PubMed] [Google Scholar]
- 39.Shepard BD, Joseph RA, Kannarkat GT, Rutledge TM, Tuma DJ, Tuma PL. Alcohol-induced alterations in hepatic microtubule dynamics can be explained by impaired histone deacetylase 6 function. Hepatology. 2008;48:1671–1679. doi: 10.1002/hep.22481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y, Kwon S, Yamaguchi T, Cubizolles F, Rousseaux S, Kneissel M, Cao C, Li N, Cheng HL, Chua K, et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol Cell Biol. 2008;28:1688–1701. doi: 10.1128/MCB.01154-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Li N, Caron C, Matthias G, Hess D, Khochbin S, Matthias P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003;22:1168–1179. doi: 10.1093/emboj/cdg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 43.Frye RA. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem Biophys Res Commun. 1999;260:273–279. doi: 10.1006/bbrc.1999.0897. [DOI] [PubMed] [Google Scholar]
- 44.Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell. 2005;16:4623–4635. doi: 10.1091/mbc.E05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamamoto H, Schoonjans K, Auwerx J. Sirtuin functions in health and disease. Mol Endocrinol. 2007;21:1745–1755. doi: 10.1210/me.2007-0079. [DOI] [PubMed] [Google Scholar]
- 46.North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003;11:437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 47.Schlicker C, Gertz M, Papatheodorou P, Kachholz B, Becker CF, Steegborn C. Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J Mol Biol. 2008;382:790–801. doi: 10.1016/j.jmb.2008.07.048. [DOI] [PubMed] [Google Scholar]
- 48.Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol. 2007;27:8807–8814. doi: 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liszt G, Ford E, Kurtev M, Guarente L. Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J Biol Chem. 2005;280:21313–21320. doi: 10.1074/jbc.M413296200. [DOI] [PubMed] [Google Scholar]
- 50.Ahuja N, Schwer B, Carobbio S, Waltregny D, North BJ, Castronovo V, Maechler P, Verdin E. Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J Biol Chem. 2007;282:33583–33592. doi: 10.1074/jbc.M705488200. [DOI] [PubMed] [Google Scholar]
- 51.Marmorstein R. Structure and function of histone acetyltransferases. Cell Mol Life Sci. 2001;58:693–703. doi: 10.1007/PL00000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamauchi T, Yamauchi J, Kuwata T, Tamura T, Yamashita T, Bae N, Westphal H, Ozato K, Nakatani Y. Distinct but overlapping roles of histone acetylase PCAF and of the closely related PCAF-B/GCN5 in mouse embryogenesis. Proc Natl Acad Sci USA. 2000;97:11303–11306. doi: 10.1073/pnas.97.21.11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wong K, Zhang J, Awasthi S, Sharma A, Rogers L, Matlock EF, Van Lint C, Karpova T, McNally J, Harrod R. Nerve growth factor receptor signaling induces histone acetyltransferase domain-dependent nuclear translocation of p300/CREB-binding protein-associated factor and hGCN5 acetyltransferases. J Biol Chem. 2004;279:55667–55674. doi: 10.1074/jbc.M408174200. [DOI] [PubMed] [Google Scholar]
- 54.Ohta K, Ohigashi M, Naganawa A, Ikeda H, Sakai M, Nishikawa J, Imagawa M, Osada S, Nishihara T. Histone acetyltransferase MOZ acts as a co-activator of Nrf2-MafK and induces tumour marker gene expression during hepatocarcinogenesis. Biochem J. 2007;402:559–566. doi: 10.1042/BJ20061194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Osada S, Nishikawa J, Nakanishi T, Tanaka K, Nishihara T. Some organotin compounds enhance histone acetyltransferase activity. Toxicol Lett. 2005;155:329–335. doi: 10.1016/j.toxlet.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 56.Zhang X, Yuan Z, Zhang Y, Yong S, Salas-Burgos A, Koomen J, Olashaw N, Parsons JT, Yang XJ, Dent SR, et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol Cell. 2007;27:197–213. doi: 10.1016/j.molcel.2007.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang XJ. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res. 2004;32:959–976. doi: 10.1093/nar/gkh252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev. 2000;64:435–459. doi: 10.1128/mmbr.64.2.435-459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grozinger CM, Hassig CA, Schreiber SL. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc Natl Acad Sci USA. 1999;96:4868–4873. doi: 10.1073/pnas.96.9.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tuma DJ, Smith SL, Sorrell MF. Acetaldehyde and microtubules. Ann N Y Acad Sci. 1991;625:786–792. doi: 10.1111/j.1749-6632.1991.tb33920.x. [DOI] [PubMed] [Google Scholar]
- 61.Lieber CS, Leo MA, Wang X, Decarli LM. Alcohol alters hepatic FoxO1, p53, and mitochondrial SIRT5 deacetylation function. Biochem Biophys Res Commun. 2008;373:246–252. doi: 10.1016/j.bbrc.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 62.Bouras T, Fu M, Sauve AA, Wang F, Quong AA, Perkins ND, Hay RT, Gu W, Pestell RG. SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J Biol Chem. 2005;280:10264–10276. doi: 10.1074/jbc.M408748200. [DOI] [PubMed] [Google Scholar]
- 63.Deaciuc IV, Arteel GE, Peng X, Hill DB, McClain CJ. Gene expression in the liver of rats fed alcohol by means of intragastric infusion. Alcohol. 2004;33:17–30. doi: 10.1016/j.alcohol.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 64.Deaciuc IV, Doherty DE, Burikhanov R, Lee EY, Stromberg AJ, Peng X, de Villiers WJ. Large-scale gene profiling of the liver in a mouse model of chronic, intragastric ethanol infusion. J Hepatol. 2004;40:219–227. doi: 10.1016/j.jhep.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 65.Sims RJ 3rd, Reinberg D. Is there a code embedded in proteins that is based on post-translational modifications? Nat Rev Mol Cell Biol. 2008;9:815–820. doi: 10.1038/nrm2502. [DOI] [PubMed] [Google Scholar]
- 66.Eberharter A, Becker PB. Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep. 2002;3:224–229. doi: 10.1093/embo-reports/kvf053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Clayton AL, Hazzalin CA, Mahadevan LC. Enhanced histone acetylation and transcription: a dynamic perspective. Mol Cell. 2006;23:289–296. doi: 10.1016/j.molcel.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 68.Mellor J. Dynamic nucleosomes and gene transcription. Trends Genet. 2006;22:320–329. doi: 10.1016/j.tig.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 69.Donohue TM Jr. Alcohol-induced steatosis in liver cells. World J Gastroenterol. 2007;13:4974–4978. doi: 10.3748/wjg.v13.i37.4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Giandomenico V, Simonsson M, Grönroos E, Ericsson J. Coactivator-dependent acetylation stabilizes members of the SREBP family of transcription factors. Mol Cell Biol. 2003;23:2587–2599. doi: 10.1128/MCB.23.7.2587-2599.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tuma DJ, Casey CA, Sorrell MF. Effects of ethanol on hepatic protein trafficking: impairment of receptor-mediated endocytosis. Alcohol Alcohol. 1990;25:117–125. doi: 10.1093/oxfordjournals.alcalc.a044986. [DOI] [PubMed] [Google Scholar]
- 72.Tuma DJ, Casey CA, Sorrell MF. Effects of alcohol on hepatic protein metabolism and trafficking. Alcohol Alcohol Suppl. 1991;1:297–303. [PubMed] [Google Scholar]
- 73.Tuma DJ, Sorrell MF. Effects of ethanol on protein trafficking in the liver. Semin Liver Dis. 1988;8:69–80. doi: 10.1055/s-2008-1040529. [DOI] [PubMed] [Google Scholar]
- 74.McVicker BL, Casey CA. Effects of ethanol on receptor-mediated endocytosis in the liver. Alcohol. 1999;19:255–260. doi: 10.1016/s0741-8329(99)00043-9. [DOI] [PubMed] [Google Scholar]
- 75.Joseph RA, Shepard BD, Kannarkat GT, Rutledge TM, Tuma DJ, Tuma PL. Microtubule acetylation and stability may explain alcohol-induced alterations in hepatic protein trafficking. Hepatology. 2008;47:1745–1753. doi: 10.1002/hep.22014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mizuno M, Singer SJ. A possible role for stable microtubules in intracellular transport from the endoplasmic reticulum to the Golgi apparatus. J Cell Sci. 1994;107:1321–1331. doi: 10.1242/jcs.107.5.1321. [DOI] [PubMed] [Google Scholar]
- 77.Poüs C, Chabin K, Drechou A, Barbot L, Phung-Koskas T, Settegrana C, Bourguet-Kondracki ML, Maurice M, Cassio D, Guyot M, et al. Functional specialization of stable and dynamic microtubules in protein traffic in WIF-B cells. J Cell Biol. 1998;142:153–165. doi: 10.1083/jcb.142.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 79.Tuma DJ, Casey CA, Sorrell MF. Chronic ethanol-induced impairments in receptor-mediated endocytosis of insulin in rat hepatocytes. Alcohol Clin Exp Res. 1991;15:808–813. doi: 10.1111/j.1530-0277.1991.tb00606.x. [DOI] [PubMed] [Google Scholar]
- 80.Dalke DD, Sorrell MF, Casey CA, Tuma DJ. Chronic ethanol administration impairs receptor-mediated endocytosis of epidermal growth factor by rat hepatocytes. Hepatology. 1990;12:1085–1091. doi: 10.1002/hep.1840120502. [DOI] [PubMed] [Google Scholar]
- 81.Casey CA, Camacho KB, Tuma DJ. The effects of chronic ethanol administration on the rates of internalization of various ligands during hepatic endocytosis. Biochim Biophys Acta. 1992;1134:96–104. doi: 10.1016/0167-4889(92)90032-7. [DOI] [PubMed] [Google Scholar]
- 82.Nogales E, Whittaker M, Milligan RA, Downing KH. High-resolution model of the microtubule. Cell. 1999;96:79–88. doi: 10.1016/s0092-8674(00)80961-7. [DOI] [PubMed] [Google Scholar]
- 83.Liao G, Gundersen GG. Kinesin is a candidate for cross-bridging microtubules and intermediate filaments. Selective binding of kinesin to detyrosinated tubulin and vimentin. J Biol Chem. 1998;273:9797–9803. doi: 10.1074/jbc.273.16.9797. [DOI] [PubMed] [Google Scholar]
- 84.Reed NA, Cai D, Blasius TL, Jih GT, Meyhofer E, Gaertig J, Verhey KJ. Microtubule acetylation promotes kinesin-1 binding and transport. Curr Biol. 2006;16:2166–2172. doi: 10.1016/j.cub.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 85.Dompierre JP, Godin JD, Charrin BC, Cordelières FP, King SJ, Humbert S, Saudou F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J Neurosci. 2007;27:3571–3583. doi: 10.1523/JNEUROSCI.0037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Török N, Marks D, Hsiao K, Oswald BJ, McNiven MA. Vesicle movement in rat hepatocytes is reduced by ethanol exposure: alterations in microtubule-based motor enzymes. Gastroenterology. 1997;113:1938–1948. doi: 10.1016/s0016-5085(97)70014-3. [DOI] [PubMed] [Google Scholar]
- 87.Elliott PJ, Jirousek M. Sirtuins: novel targets for metabolic disease. Curr Opin Investig Drugs. 2008;9:371–378. [PubMed] [Google Scholar]
- 88.Ajmo JM, Liang X, Rogers CQ, Pennock B, You M. Resveratrol alleviates alcoholic fatty liver in mice. Am J Physiol Gastrointest Liver Physiol. 2008;295:G833–G842. doi: 10.1152/ajpgi.90358.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang GL, Salisbury E, Shi X, Timchenko L, Medrano EE, Timchenko NA. HDAC1 cooperates with C/EBPalpha in the inhibition of liver proliferation in old mice. J Biol Chem. 2008;283:26169–26178. doi: 10.1074/jbc.M803544200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yoo YG, Na TY, Seo HW, Seong JK, Park CK, Shin YK, Lee MO. Hepatitis B virus X protein induces the expression of MTA1 and HDAC1, which enhances hypoxia signaling in hepatocellular carcinoma cells. Oncogene. 2008;27:3405–3413. doi: 10.1038/sj.onc.1211000. [DOI] [PubMed] [Google Scholar]
- 91.Farooq M, Sulochana KN, Pan X, To J, Sheng D, Gong Z, Ge R. Histone deacetylase 3 (hdac3) is specifically required for liver development in zebrafish. Dev Biol. 2008;317:336–353. doi: 10.1016/j.ydbio.2008.02.034. [DOI] [PubMed] [Google Scholar]
- 92.Aagaard-Tillery KM, Grove K, Bishop J, Ke X, Fu Q, McKnight R, Lane RH. Developmental origins of disease and determinants of chromatin structure: maternal diet modifies the primate fetal epigenome. J Mol Endocrinol. 2008;41:91–102. doi: 10.1677/JME-08-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yin L, Lazar MA. The orphan nuclear receptor Rev-erbalpha recruits the N-CoR/histone deacetylase 3 corepressor to regulate the circadian Bmal1 gene. Mol Endocrinol. 2005;19:1452–1459. doi: 10.1210/me.2005-0057. [DOI] [PubMed] [Google Scholar]
- 94.Waltregny D, Glénisson W, Tran SL, North BJ, Verdin E, Colige A, Castronovo V. Histone deacetylase HDAC8 associates with smooth muscle alpha-actin and is essential for smooth muscle cell contractility. FASEB J. 2005;19:966–968. doi: 10.1096/fj.04-2303fje. [DOI] [PubMed] [Google Scholar]
- 95.Tong JJ, Liu J, Bertos NR, Yang XJ. Identification of HDAC10, a novel class II human histone deacetylase containing a leucine-rich domain. Nucleic Acids Res. 2002;30:1114–1123. doi: 10.1093/nar/30.5.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F, Mostoslavsky R, Alt FW, Schibler U. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell. 2008;134:317–328. doi: 10.1016/j.cell.2008.06.050. [DOI] [PubMed] [Google Scholar]
- 97.Bardag-Gorce F, Oliva J, Villegas J, Fraley S, Amidi F, Li J, Dedes J, French B, French SW. Epigenetic mechanisms regulate Mallory Denk body formation in the livers of drug-primed mice. Exp Mol Pathol. 2008;84:113–121. doi: 10.1016/j.yexmp.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hou X, Xu S, Maitland-Toolan KA, Sato K, Jiang B, Ido Y, Lan F, Walsh K, Wierzbicki M, Verbeuren TJ, et al. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J Biol Chem. 2008;283:20015–20026. doi: 10.1074/jbc.M802187200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.You M, Cao Q, Liang X, Ajmo JM, Ness GC. Mammalian sirtuin 1 is involved in the protective action of dietary saturated fat against alcoholic fatty liver in mice. J Nutr. 2008;138:497–501. doi: 10.1093/jn/138.3.497. [DOI] [PubMed] [Google Scholar]
- 100.Thomas T, Corcoran LM, Gugasyan R, Dixon MP, Brodnicki T, Nutt SL, Metcalf D, Voss AK. Monocytic leukemia zinc finger protein is essential for the development of long-term reconstituting hematopoietic stem cells. Genes Dev. 2006;20:1175–1186. doi: 10.1101/gad.1382606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang XJ, Ullah M. MOZ and MORF, two large MYSTic HATs in normal and cancer stem cells. Oncogene. 2007;26:5408–5419. doi: 10.1038/sj.onc.1210609. [DOI] [PubMed] [Google Scholar]
- 102.Champagne N, Bertos NR, Pelletier N, Wang AH, Vezmar M, Yang Y, Heng HH, Yang XJ. Identification of a human histone acetyltransferase related to monocytic leukemia zinc finger protein. J Biol Chem. 1999;274:28528–28536. doi: 10.1074/jbc.274.40.28528. [DOI] [PubMed] [Google Scholar]