

Abstract
The majority of ethanol metabolism occurs in the liver. Consequently, this organ sustains the greatest damage from ethanol abuse. Ethanol consumption disturbs the delicate balance of protein homeostasis in the liver, causing intracellular protein accumulation due to a disruption of hepatic protein catabolism. Evidence indicates that ethanol or its metabolism impairs trafficking events in the liver, including the process of macroautophagy, which is the engulfment and degradation of cytoplasmic constituents by the lysosomal system. Autophagy is an essential, ongoing cellular process that is highly regulated by nutrients, endocrine factors and signaling pathways. A great number of the genes and gene products that govern the autophagic response have been characterized and the major metabolic and signaling pathways that activate or suppress autophagy have been identified. This review describes the process of autophagy, its regulation and the possible mechanisms by which ethanol disrupts the process of autophagic degradation. The implications of autophagic suppression are discussed in relation to the pathogenesis of alcohol-induced liver injury.
Keywords: Autophagy, Autophagosome, Ethanol metabolism, Hepatomegaly, Lysosomes, Signal transduction
INTRODUCTION
Lysosomes are the primary hydrolytic organelles of higher eukaryotic cells and have been appropriately named the cell’s “digestive system”. A large number of acid hydrolases inhabits these organelles. Together, these enzymes catalyze the breakdown of all forms of biopolymers, ranging from intracellular and extracellular proteins to storage carbohydrates (e.g. glycogen) to nucleic acids. Macromolecular catabolism takes place inside the lysosomal lumen at hydrogen ion concentrations that are 500-fold higher (pH about 4.7) than that in the cytoplasm (pH about 7.4)[1]. Maintenance of such an acidic lysosomal interior occurs through the continuous action of ATP-dependent proton transporters that reside on its membrane. Lysosomes exhibit morphological and functional plasticity; they undergo continuous change as they mature from larger primary vesicles that segregate and carry hydrolase precursors, to mature hydrolytic organelles that execute intracellular digestion. As the catabolic organelles for protein degradation, lysosomes differ from proteasomes in their size, their complexity, their intracellular locations and in the types of substrates they degrade. While proteasomes have essentially supplanted lysosomes as the primary system that degrades intracellular proteins in higher eukaryotes, the crucial task of breaking down protein substrates that are more difficult to digest, as well as hydrolyzing all other forms of polymeric substrates, is left to lysosomes. Certainly, compelling evidence of their importance is the existence of a number of human abnormalities known as lysosomal storage diseases, in which afflicted individuals possess genetically defective lysosomal hydrolases. Clinically, this group of diseases exhibits a spectrum of impaired function, from neurodegeneracy to premature death[2]. Furthermore, deficiencies in the pathways that participate in lysosome biogenesis or that have a crucial function in supplying lysosomes with their substrates, can be lethal to the organism[3]. Studies in the mammalian liver have provided a great deal of our current understanding of the lysosomal pathway, as this organ is highly responsive to changes in nutrient supply[4,5]. The liver is also the predominant organ that metabolizes ethanol and liver injury is the principal clinical complication of alcohol abuse[6]. This review will focus on the process of autophagy, its role in normal hepatic function and its alteration due to ethanol consumption. A portion of this review will consider how ethanol disrupts protein catabolism, which is believed to have a significant role in liver injury. The reader will note the citation of other excellent reviews that have been published on the process of autophagy and/or lysosomal proteolysis[3,7–9] The physiological and biomedical importance of autophagy is further underscored by the establishment of the international journal, Autophagy, the first issue of which was published in 2005.
FEEDING THE LYSOSOMAL APPARATUS; HETEROPHAGY AND AUTOPHAGY
In order for lysosomes to be “fed”, substrates of high molecular weight must gain entry to the interior of the organelle through a membrane that is normally impermeable to large molecules. Such access is accomplished by a number of mechanisms, each involving the active participation of membranes. During heterophagy extracellular materials are imported into the cell by either receptor-mediated or fluid phase endocytosis or by bulk phagocytosis of larger particles, including whole cells such as bacteria and apoptotic cells. All these are accomplished by invagination of the plasma membrane, thereby transporting the membrane-enclosed material, via endosomes to their final destinations, which, in many (but not all) cases is degradation in lysosomes. Heterophagy has a critical function in the normal turnover of senescent plasma proteins and in adaptive immunity, the latter involving antigen presentation through the action of endosomal proteases by the MHC class II pathway[10].
Autophagy is exclusively an intracellular lysosomal process, which literally means “self eating”. It was originally described in the 1950s and later systematically analyzed by the classical work of de Duve and Wattiaux in rat liver. Autophagy of intracellular proteins occurs by three distinct mechanisms: Macroautophagy is the vacuolar engulfment by membranes of large portions of cytoplasm and collateral organelles, forming a double membrane vesicle called an autophagosome or autophagic vacuole. The outer membrane of the autophagosome later fuses with existing lysosomes to form an autolysosome in which the contents are degraded. Microautophagy describes membrane uptake by existing lysosomes of smaller portions of cellular constituents by lysosomal membrane invagination followed by rapid hydrolysis of those molecules. Chaperone-mediated autophagy (CMA) is the singular uptake and degradation by lysosomes of specific protein substrates bearing a recognizable peptide motif, KFERQ. The latter process is mediated by molecular chaperones, the most prominent of which is a cytoplasmic form of heat shock protein 70 (HSC 70), which recognizes and binds the peptide motif of the substrate protein[11,12]. The CMA substrate/chaperone complex then moves to the lysosome, where a specific receptor, the lysosome-associated membrane protein type-2A (LAMP-2A) recognizes and binds the complex; the substrate protein is then unfolded and translocated across the lysosome membrane and degraded in the lysosomal lumen. CMA is enhanced by oxidative stress, and, in addition to the proteasome, thereby serves a quality control function by degrading modified proteins. CMA also declines with age, largely due to the depletion of LAMP-2A. Recent studies have demonstrated that CMA can be restored in livers of aged mice that are transgenic for LAMP-2A. These animals, compared with their wild type littermates, showed improved cellular quality control and hepatic function[13]. While microautophagy and CMA are largely ongoing constitutive processes, macroautophagy is more highly regulated. In general, when the term autophagy is used here, it will be used synonymously with macroautophagy.
Autophagy is clearly important for survival of the organism, particularly in times of nutrient deprivation when degradation of macromolecular constituents becomes necessary to recycle essential carbon sources to maintain viability. Autophagy was initially characterized morphometrically and biochemically in rat liver, one of the most sensitive organs to changes in nutrient supply. An illustration of this is when rats are fasted for 48 h, they exhibit a 40% loss of liver protein and a significant loss of liver mass, both of which are rapidly restored to normal levels 12 h after food is replenished. These respective changes correlate with an induction of autophagy by fasting followed by its nutrient-induced suppression after refeeding[4,14,15]. A similar suppression of both autophagy as well as lysosome biogenesis was reported 24 h after partial hepatectomy in regenerating rat liver[16]. Thus, autophagy rapidly responds to these changes and is an important component of cell adaptation. In fact, the half-life of the autophagosome is less than 10 min[5], which indicates that the molecular machinery required to form autophagic vacuoles is constitutive for rapid degradation of cellular constituents. Furthermore, like the proteasome, autophagy has a significant role in removing misfolded proteins from the hepatocyte. Such is the case in alpha-1-antitrypsin (A-1AT) deficiency, in which the cellular accumulation of the mutated unsecretable form of A-1AT, known as ATZ (the isoform most prone to aggregation), is directed to autophagic vacuoles for degradation[17]. Similarly, other aggregation-prone proteins are reported to have the same fate[18,19]. It was recently demonstrated that inhibition of proteasome activity by treatment of mice with bortezomib caused Mallory-Denk-(MD)-like body formation. Activation of autophagy with rapamycin treatment prevented the formation of these inclusions, indicating that proteasome inhibition causes the formation of M-D like bodies in livers of susceptible mice, while autophagosomes prevent M-D body accumulation by resorbing and degrading these insoluble inclusions[20]. Thus, autophagy, not only functions in cell survival but also provides an additional line of cellular defense, the latter by removing aggregated, potentially toxic proteins, presumably resistant to degradation by the proteasome.
REGULATION OF AUTOPHAGY
Hormonal and nutrient regulators of autophagy include food deprivation or glucagon treatment and both are reliable autophagy inducers. Conversely, treatment with growth factors such as insulin or amino acids are well-known methods of autophagic suppression. The molecular mechanisms by which these agents actually induce or suppress autophagy have been investigated more recently and this has led to investigations of the protein components and signaling mechanisms that regulate macroautophagy to form the autophagosome. To date, over 30 autophagy-related gene products (Atgs) are now known. There are extensive molecular interactions that occur between these gene products in the early stages of autophagosome formation. The most well known mechanism is that of the class I phosphoinositol-3-kinase (PI-3K) pathway, which initiates an upstream signaling cascade to regulate the activity of the mammalian target of rapamycin (mTOR), a kinase, which is a major regulatory anabolic protein, and a suppressor of autophagy. Activation of mTOR leads to inhibition of Atg1, a key signal in autophagy induction. If Atg1 is not inhibited by mTOR, it is free to recruit the autophagy proteins, Atg11, Atg13 and Atg17 to form a complex which signals the induction of autophagy. The cellular machinery that initiates the formation of the autophagosome occurs through the activation of Beclin-1 (Atg6), which interacts with the class III PI 3K pathway and complexes with Atg 14. A second complex involves the interaction of Atg12, Atg16, Atg5, and Atg 7 complex, which is critical for the recruitment of Atg8, (also known as LC31) to the phagophore, the isolation membrane at which the macroautophagic response is initiated and the precursor of the autophagosome. Autophagic induction leads to cleavage of Atg8 (LC3-I), which is covalently bound to phosphatidylethanolamine (PE) to form a distinct form of the protein, LC3-II. The latter is associated with and is a commonly used marker for autophagosome membranes. Atg8 is believed to be involved in autophagosome closure as well as its relative size to surround portions of the cytoplasm for degradation[21]. Eventual fusion of the autophagosome and the lysosome requires the participation of the LAMP-2 protein. Figure 1 depicts the major regulatory proteins and signaling factors in the autophagic response.
Figure 1.
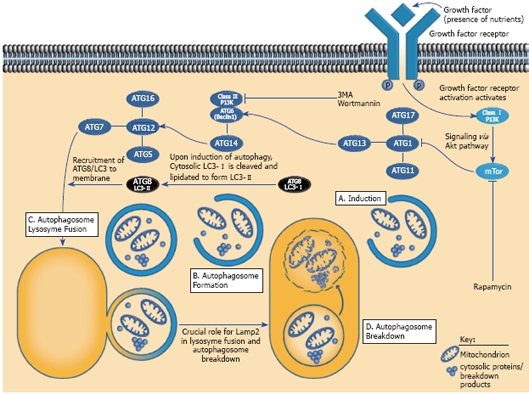
Interaction of gene products and pathways in the regulation of autophagy. Growth factors and nutrients activate the Class I P13K proteins, which, in turn, signal, via the AKT pathway to activate mTOR. This leads to inhibition of ATG1 - the primary signal for autophagy. Nutrient deprivation or inhibition of mTOR by rapamycin allows ATG1 to recruit ATG11, ATG13 and ATG 17 to form a complex to initiate formation of the autophagosome. This is dependent on the formation of 2 complexes: ATG 6 (Beclin1) which interacts with the Class III P13K protein complexes with ATG14. Another complex involves ATG12, ATG16, ATG5 and ATG7. The latter complex recruits ATG8 (LC3). Upon induction of autophagy, cytosolic LC3-I (ATG8) is cleaved and lipidated to form LC3-II. LC3-II is a marker for the autophagosome membrane. Fusion between the autophagosome and the lysosome and subsequent breakdown of the contents of the autophagic vacuole requires LAMP2 protein. Reproduced in modified form with permission (Abcam, Inc.) http://www.abcam.com/ps/pdf/cardiovascular/autophagic.pdf).
ETHANOL CONSUMPTION AND THE HEPATIC UTOPHAGIC/LYSOSOMAL SYSTEM
The majority of ethanol is metabolized in the liver and individuals who abuse alcohol by routinely drinking 50-60 g (about 4 to 5 drinks) of ethanol per day are at risk for developing alcoholic liver disease[6]. The pathogenesis of liver disease from alcohol abuse comes from the interaction of several factors, including the generation of oxidants and reactive metabolites from ethanol oxidation, which, in turn, causes other metabolic derangements. In addition, both acute and chronic ethanol administration cause enhanced formation of cytokines, especially TNF-alpha by hepatic Kupffer cells, which have a significant role in liver injury. This latter aspect is reviewed and described elsewhere[22–24]. Besides the development of fatty liver (steatosis), another early sign of excessive ethanol consumption is liver enlargement and protein accumulation, both of which are common findings in alcoholics and heavy drinkers. Baraona et al[25,26] originally described this in alcohol-fed rats. Later investigations sought to determine the origin of ethanol-elicited protein accumulation. They showed that chronic ethanol consumption slowed down the catabolism of long-lived proteins in rat liver[27] and also depressed hepatic protein synthesis[28]. Other studies confirmed that rats subjected to chronic ethanol feeding exhibited lower hepatic proteolysis than control rats. This was associated with reduced volume densities of autophagosomes and autolysosomes, as determined morphometrically[29]. Our later studies, using isolated lysosomes from chronically ethanol-fed rats also revealed a reduced capacity of these organelles for proteolysis of endogenous as well as exogenous (i.e. akin to CMA) substrates in vitro. Furthermore, there was an enhanced tendency of lysosomes from ethanol-fed rats to leak their contents, but the intact organelles still exhibited lower levels of cathepsins, B and L compared with those from pair-fed control rats. A later report showed that lysosomes from ethanol-fed rats exhibited altered sedimentation properties on density gradients. Pulse-chase analyses, using isolated hepatocytes demonstrated that the reduced lysosomal content of cathepsin L was the result of delayed trafficking of its nascent precursor and impaired processing to its mature catalytic form in cells from ethanol-fed rats[16,30,31]. Both flow cytometric and microscopy studies showed that vesicular movement and acidification of lysosomes in hepatocytes are both impeded after ethanol exposure[32,33].
MECHANISMS OF ETHANOL-ELICITED SUPPRESSION OF AUTOPHAGY
The exact mechanisms responsible for the aforementioned ethanol-induced changes in hepatic autophagy and lysosomal proteolysis are not clear but some cogent explanations have been advanced. One is that oxidants (e.g. peroxynitrite) and reactive species (e.g. acetaldehyde and malondialdehyde-acetaldehyde)[34] derived from ethanol metabolism may impair autophagy, similar to that which occurs with the proteasome[35–37]. Some of the current evidence for this is circumstantial, but nevertheless compelling as there is evidence of lysosomal damage, as judged by enhanced lysosomal fragility, which could result from either altered lipid metabolism, oxidative stress or both[15]. By analogy, other instances of lysosome fragility by iron-induced oxidative stress are documented[38,39]. However, ethanol administration has little to no effect on the activities of lysosomal hydrolases[31] and, because a significant number of cathepsins rely on a reduced sulfhydryl group for their catalytic activity in their active centers, the alteration of lysosomal glutathione or cysteine content by ethanol seems unlikely. It is worth noting that lysosomes and proteasomes seem to exhibit differential sensitivity to ethanol levels in the serum. Animal experiments have revealed that proteasome activity declines in animals that have higher (i.e. > 40 mmol/L) serum ethanol concentrations[40]. The lysosomal system, on the other hand, appears to be impaired by lower serum ethanol levels, as recently reported in livers of female ethanol-fed rats[41].
Ethanol-induced suppression of autophagy may result from alterations in hepatic amino acid pool sizes, especially those of leucine, phenylalanine, methionine, histidine, tryptophan, glutamine, proline, and tyrosine, which have been deemed regulatory amino acids and suppressors of macroautophagy[5]. L-leucine appears to be one of the strongest autophagic suppressors in this group. It is noteworthy that reports indicate that chronic ethanol administration in rats increases the intrahepatic levels of leucine by 1.4 to 1.8-fold over pair-fed controls[27,42]. Thus, the association of an ethanol-induced reduction in autophagy with higher levels of intrahepatic leucine may partially explain autophagic suppression in the ethanol-fed state. However, it remains to be conclusively demonstrated whether this association represents a causal relationship. It is also paradoxical that ethanol feeding results in higher levels of leucine during a slowdown in protein catabolism when one would expect the opposite situation. Still, leucine accumulation could reflect a reduced ability of the liver to synthesize proteins, which indeed occurs in ethanol-fed animals[42,43].
A third likely mechanism of autophagic suppression by ethanol is its well-documented ability to disrupt protein trafficking in the liver. Autophagy requires the action of cytoskeletal elements, including the microtubules and microfilaments. Both are necessary for autophagosome formation and their fusion with other vesicular bodies, as demonstrated by blockage of these processes with specific inhibitors, including nocadazole and vinblastine (microtubules) and the cytochalasins (microfilaments)[44,45]. Disruption of vesicular movement within the hepatocyte by ethanol treatment occurs by mechanisms that are independent of the molecular motors, dyenin and kinesin, although there is evidence for alterations in the protein, dynamin[32]. Trafficking of exogenous proteins into the hepatocyte by endocytosis[46,47] and the intracellular delivery of proteases to lysosomes[31] are both inhibited by ethanol consumption. Furthermore, the anti-secretory properties of ethanol in the liver are well documented. Studies with liver slices and cultured cells indicate that ethanol metabolism is required for disruption of these protein trafficking events[48–51]. In vitro investigations also revealed that acetaldehyde, the initial product of ethanol oxidation, inhibits the polymerization of tubulin to form microtubules, indicating that the reactive metabolite may impair protein trafficking by forming adducts with tubulin subunits, thereby blocking their polymerization into microtubules[52]. Further, the finding that acetaldehyde can undergo secondary reactions with malondialdehyde (MDA) to form more bulky substituents on proteins, known as malondialdehyde acetaldehyde adducts (MAA) with proteins, makes this mechanism of autophagic suppression an attractive hypothesis.
ETHANOL-INDUCED ALTERATIONS IN CELLULAR SIGNALING: REGULATION OF AMP KINASE
While the foregoing mechanisms of autophagic suppression by ethanol are plausible, it is likely that ethanol and/or its metabolites impact signaling events that govern the autophagic response. There have been significant advances in our understanding of these processes at the molecular level, as recently reviewed[3]. Because autophagy is an energy-dependent process and requires ATP[53], it follows that ATP depletion naturally suppresses autophagy. Ethanol consumption has been reported to inhibit ATP production by mitochondria[54], in part by enhancing oxidative modification and inactivation of proteins within these organelles[55]. However, others have reported an increased efficiency of mitochondrial oxidative phosphorylation in livers of ethanol-fed animals[56], Thus, reduced ATP availability from mitochondria may not fully explain such downstream effects on autophagy. Adenosine monophosphate-activated protein kinase (AMPK) is a heterotrimeric protein that is, itself, activated by elevated ratios of AMP/ATP and is a significant regulator of a variety of metabolic and signal transduction pathways, including autophagy. Elevated AMP/ATP ratios are indicators of low intracellular energy charge. Therefore, when AMP activates the AMPK it generally down-regulates energy requiring pathways and stimulates catabolic pathways. Interestingly, however, inhibition of AMPK also suppresses autophagy[57]. These findings are consistent with the suppression of autophagy by ethanol consumption, which significantly reduces AMPK activity in liver[58]. Such a decline in AMPK activity would, in turn cause the release of suppression of mTOR activity by AMPK[59], thereby permitting mTOR to suppress proteolysis. Conversely, AMPK activation suppresses mTOR, thereby allowing autophagy to proceed. Recent work with cultured hepatoma cells determined that ethanol metabolism is required for suppression of AMPK phosphorylation. This occurs by an inactivation of two upstream kinases, LKB-1 and protein kinase C-zeta. Simultaneously, ethanol exposure activates protein phosphatase 2A (PP2A), which causes dephosphorylation (and inactivation) of AMPK[60]. The latter findings seem paradoxical, as AMPK is generally activated by reactive oxygen and nitrogen species[22,61]. Thus, while ethanol metabolism similarly generates oxidants and reactive species, including acetaldehyde, these molecules down-regulate upstream kinases and up-regulate the downstream phosphatase, PP2A, resulting in AMPK inactivation, which, in turn, can cause autophagic suppression. AMPK also has an important role in regulating lipid metabolism and AMPK suppression by ethanol allows activation of rate-limiting enzymes involved in lipid biosynthesis, which contribute to ethanol-induced fatty liver[62,63]. Figure 2 depicts the putative mechanisms of autophagic suppression.
Figure 2.
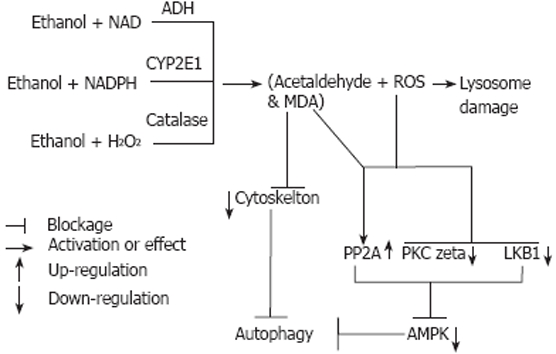
Putatative mechanisms of autophagy suppression by ethanol. Ethanol metabolism by the three major pathways, alcohol dehydrogenase (ADH), cytochrome P450 2E1 (CYP2E1) and catalase generates acetaldehyde, which can undergo secondary reactions with malondialdehyde (MDA) to form MAA. In addition, reactive oxygen species (ROS) are generated by CYP2E1 catalysis. Reactive oxidants (ROS) may also contribute to lysosomal damage. The reactive aldehydes are known to form adducts with tubulin and other cytoskeletal elements to block trafficking and movement of autophagic vacuoles and their formation. The combined formation of ROS and acetaldehyde putatively cause upregulation of protein phosphatase 2A (PP2A) and a downregulation of LKB1 and PKC zeta. The latter changes cause inactivation of AMPK, which in turn suppresses autophagy due to up regulation of mTOR (not shown) as described in the text.
PATHOLOGICAL CONSEQUENCES OF AUTOPHAGIC SUPPRESSION
Protein accumulation due to ethanol-elicited decline in protein catabolism probably contributes to the formation of Mallory Denk (M-D) bodies in liver cells. These inclusions are prominent histological hallmarks of liver disease in alcoholics[64]. M-D bodies contain the cytokeratins 8 and 18 as well as ubiquitin, and p62, an adaptor protein. As pointed out earlier, M-D bodies comprise an aggresome, consisting of filamentous ubiquitylated misfolded proteins that are believed to represent a failed attempt by the proteasome to degrade them. Recent evidence indicates that autophagy can degrade these insoluble complexes[20]. However, clearance of such cellular debris is hampered by an ethanol-elicited suppression of autophagy, a situation that may have a perilous outcome if drinking continues.
In addition to the accumulation of potentially toxic proteins, ethanol also causes mitochondrial damage. Changes ranging from morphologically detectable mitochondrial swelling, to DNA fragmentation, to depolarization of the inner membrane, (commonly known as the mitochondrial permeability transition or MPT) to disruption of mitochondrial gene products as well as decreases in glutathione occur in livers of ethanol-fed animals[55,65–72]. It is crucial that such damaged organelles be removed from the cell and there is evidence that selective engulfment of mitochondria (called mitophagy) into autophagic vacuoles occurs in cells as a quality control device[73–75]. Similarly, there appears to be rather selective autophagy of ER components (ER-phagy) as well as peroxisomes (pexophagy) in cells under conditions where there is an accumulation of these organelles[76,77]. It remains to be demonstrated whether such selective autophagy represents actual recognition of damaged organelles or simply occurs because of mass action due to their accumulation, however, reports show that the permeability transition may act as a signal for autophagic destruction of the mitochondrion[74]. While there is no firm evidence of ethanol-elicited suppression of mitophagy, the detection of increased numbers of damaged mitochondria in livers of ethanol-fed animals provides circumstantial evidence of mitophagy inhibition. In this regard it is also worth noting that mitochondrial damage also occurs by non-oxidative products of ethanol metabolism, namely fatty acid ethyl esters (FAEE). These condensation products between ethanol and fatty acids are generated enzymatically and have been shown to cause mitochondrial damage[78]. Thus a mechanism for ethanol-induced injury in nonhepatic tissue such as pancreas, heart and brain could occur by the generation of FAEE.
CONCLUSION
This review has summarized the evidence for the ethanol-elicited suppression of autophagy as a mechanism by which liver cells can accumulate damaged proteins and organelles. Suppression of proteolysis in general can be lethal to cells, as normal turnover is disrupted and the removal of potentially toxic proteins is prevented. As an organ that is sensitive to nutrients as well as toxins, the liver is highly active in autophagy. The inability to respond to depletion of nutrient supply in the alcoholic state is a potentially perilous condition. Still, while there have been numerous investigations of hepatic autophagy, the actual mechanism(s) by which ethanol may influence this process remain(s) to be conclusively determined. From this summary of results from our laboratory and from the literature, we hypothesize that ethanol consumption probably influences pathways upstream of the autophagic process, including inhibition of AMPK, but this awaits experimental confirmation. Finally, while this review has specifically focused on autophagy in hepatic tissue, a recent report indicates that ethanol exposure also slows autophagy in neural cells[79], indicating that this effect of ethanol is not exclusively liver-specific. In conclusion, while autophagy has been recognized for decades as an indispensable means of macromolecular disposal, its roles in other cellular processes from differentiation to cell death are now being documented, thus rekindling new interest in this fascinating field of investigation.
Footnotes
Supported by Development funds from the Section of Gastroenterology/Hepatology, University of Nebraska Medical Center; Bridge Research Grant from the University of Nebraska Medical Center and Medical Research Funds from the Department of Veterans Affairs, United States of America
Peer reviewer: Minoti V Apte, Associate Professor, Pancreatic Research Group, South Western Sydney Clinical School, The University of New South Wales, Liverpool, NSW 2170, Australia
S- Editor Li LF L- Editor Webster JR E- Editor Yin DH
References
- 1.Kharbanda KK, McVicker DL, Zetterman RK, MacDonald RG, Donohue TM Jr. Flow cytometric analysis of vesicular pH in rat hepatocytes after ethanol administration. Hepatology. 1997;26:929–934. doi: 10.1002/hep.510260419. [DOI] [PubMed] [Google Scholar]
- 2.Kuranda MJ, Aronson NN Jr. Receptor-mediated endocytosis and lysosomal degradation of asialoglycoproteins by the liver. In: Glaumann H, Ballard FJ, editors. Lysosomes: Their Role in Protein Breakdown. London: Academic Press; 1987. pp. 241–282. [Google Scholar]
- 3.Yin XM, Ding WX, Gao W. Autophagy in the liver. Hepatology. 2008;47:1773–1785. doi: 10.1002/hep.22146. [DOI] [PubMed] [Google Scholar]
- 4.Mortimore GE, Ward WF. Internalization of cytoplasmic protein by hepatic lysosomes in basal and deprivation-induced proteolytic states. J Biol Chem. 1981;256:7659–7665. [PubMed] [Google Scholar]
- 5.Mortimore GE, Pösö AR. Intracellular protein catabolism and its control during nutrient deprivation and supply. Annu Rev Nutr. 1987;7:539–564. doi: 10.1146/annurev.nu.07.070187.002543. [DOI] [PubMed] [Google Scholar]
- 6.Zakhari S, Li TK. Determinants of alcohol use and abuse: Impact of quantity and frequency patterns on liver disease. Hepatology. 2007;46:2032–2039. doi: 10.1002/hep.22010. [DOI] [PubMed] [Google Scholar]
- 7.Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol. 2004;14:70–77. doi: 10.1016/j.tcb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 8.Cuervo AM, Dice JF. How do intracellular proteolytic systems change with age? Front Biosci. 1998;3:d25–d43. doi: 10.2741/a264. [DOI] [PubMed] [Google Scholar]
- 9.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 10.Chapman HA. Endosomal proteases in antigen presentation. Curr Opin Immunol. 2006;18:78–84. doi: 10.1016/j.coi.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 11.Terlecky SR, Dice JF. Polypeptide import and degradation by isolated lysosomes. J Biol Chem. 1993;268:23490–23495. [PubMed] [Google Scholar]
- 12.Agarraberes FA, Terlecky SR, Dice JF. An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J Cell Biol. 1997;137:825–834. doi: 10.1083/jcb.137.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med. 2008;14:959–965. doi: 10.1038/nm.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mortimore GE, Surmacz CA. Liver perfusion: an in vitro technique for the study of intracellular protein turnover and its regulation in vivo. Proc Nutr Soc. 1984;43:161–177. doi: 10.1079/pns19840039. [DOI] [PubMed] [Google Scholar]
- 15.Donohue TM Jr, McVicker DL, Kharbanda KK, Chaisson ML, Zetterman RK. Ethanol administration alters the proteolytic activity of hepatic lysosomes. Alcohol Clin Exp Res. 1994;18:536–541. doi: 10.1111/j.1530-0277.1994.tb00906.x. [DOI] [PubMed] [Google Scholar]
- 16.Watanabe K, Ishidoh K, Ueno T, Sato N, Kominami E. Suppression of lysosomal proteolysis at three different steps in regenerating rat liver. J Biochem. 1998;124:947–956. doi: 10.1093/oxfordjournals.jbchem.a022212. [DOI] [PubMed] [Google Scholar]
- 17.Perlmutter DH. The role of autophagy in alpha-1-antitrypsin deficiency: a specific cellular response in genetic diseases associated with aggregation-prone proteins. Autophagy. 2006;2:258–263. doi: 10.4161/auto.2882. [DOI] [PubMed] [Google Scholar]
- 18.Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007;14:230–239. doi: 10.1038/sj.cdd.4401984. [DOI] [PubMed] [Google Scholar]
- 19.Kruse KB, Dear A, Kaltenbrun ER, Crum BE, George PM, Brennan SO, McCracken AA. Mutant fibrinogen cleared from the endoplasmic reticulum via endoplasmic reticulum-associated protein degradation and autophagy: an explanation for liver disease. Am J Pathol. 2006;168:1299–1308; quiz 1404-1405. doi: 10.2353/ajpath.2006.051097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harada M, Hanada S, Toivola DM, Ghori N, Omary MB. Autophagy activation by rapamycin eliminates mouse Mallory-Denk bodies and blocks their proteasome inhibitor-mediated formation. Hepatology. 2008;47:2026–2035. doi: 10.1002/hep.22294. [DOI] [PubMed] [Google Scholar]
- 21.Xie Z, Nair U, Klionsky DJ. Dissecting autophagosome formation: the missing pieces. Autophagy. 2008;4:920–922. doi: 10.4161/auto.6692. [DOI] [PubMed] [Google Scholar]
- 22.Zhou Z, Wang L, Song Z, Lambert JC, McClain CJ, Kang YJ. A critical involvement of oxidative stress in acute alcohol-induced hepatic TNF-alpha production. Am J Pathol. 2003;163:1137–1146. doi: 10.1016/s0002-9440(10)63473-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thurman RG. II. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am J Physiol. 1998;275:G605–G611. doi: 10.1152/ajpgi.1998.275.4.G605. [DOI] [PubMed] [Google Scholar]
- 24.Tsukamoto H, Takei Y, McClain CJ, Joshi-Barve S, Hill D, Schmidt J, Deaciuc I, Barve S, Colell A, Garcia-Ruiz C, et al. How is the liver primed or sensitized for alcoholic liver disease? Alcohol Clin Exp Res. 2001;25:171S–181S. doi: 10.1097/00000374-200105051-00029. [DOI] [PubMed] [Google Scholar]
- 25.Baraona E, Leo MA, Borowsky SA, Lieber CS. Alcoholic hepatomegaly: accumulation of protein in the liver. Science. 1975;190:794–795. doi: 10.1126/science.1198096. [DOI] [PubMed] [Google Scholar]
- 26.Baraona E, Leo MA, Borowsky SA, Lieber CS. Pathogenesis of alcohol-induced accumulation of protein in the liver. J Clin Invest. 1977;60:546–554. doi: 10.1172/JCI108806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Donohue TM Jr, Zetterman RK, Tuma DJ. Effect of chronic ethanol administration on protein catabolism in rat liver. Alcohol Clin Exp Res. 1989;13:49–57. doi: 10.1111/j.1530-0277.1989.tb00283.x. [DOI] [PubMed] [Google Scholar]
- 28.Donohue TM Jr, Sorrell JH, Sorrell MF, Tuma DJ. Measurement of protein synthetic activity by determination of peptidyl[3H]puromycin formation in liver slices after ethanol administration. Alcohol Clin Exp Res. 1985;9:526–530. doi: 10.1111/j.1530-0277.1985.tb05597.x. [DOI] [PubMed] [Google Scholar]
- 29.Pösö AR, Hirsimäki P. Inhibition of proteolysis in the liver by chronic ethanol feeding. Biochem J. 1991;273:149–152. doi: 10.1042/bj2730149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kharbanda KK, McVicker DL, Zetterman RK, Donohue TM Jr. Ethanol consumption reduces the proteolytic capacity and protease activities of hepatic lysosomes. Biochim Biophys Acta. 1995;1245:421–429. doi: 10.1016/0304-4165(95)00121-2. [DOI] [PubMed] [Google Scholar]
- 31.Kharbanda KK, McVicker DL, Zetterman RK, Donohue TM Jr. Ethanol consumption alters trafficking of lysosomal enzymes and affects the processing of procathepsin L in rat liver. Biochim Biophys Acta. 1996;1291:45–52. doi: 10.1016/0304-4165(96)00043-8. [DOI] [PubMed] [Google Scholar]
- 32.Török N, Marks D, Hsiao K, Oswald BJ, McNiven MA. Vesicle movement in rat hepatocytes is reduced by ethanol exposure: alterations in microtubule-based motor enzymes. Gastroenterology. 1997;113:1938–1948. doi: 10.1016/s0016-5085(97)70014-3. [DOI] [PubMed] [Google Scholar]
- 33.Kharbanda KK, McVicker DL, Zetterman RK, MacDonald RG, Donohue TM Jr. Flow cytometric analysis of vesicular pH in rat hepatocytes after ethanol administration. Hepatology. 1997;26:929–934. doi: 10.1002/hep.510260419. [DOI] [PubMed] [Google Scholar]
- 34.Tuma DJ. Role of malondialdehyde-acetaldehyde adducts in liver injury. Free Radic Biol Med. 2002;32:303–308. doi: 10.1016/s0891-5849(01)00742-0. [DOI] [PubMed] [Google Scholar]
- 35.Bardag-Gorce F, French BA, Nan L, Song H, Nguyen SK, Yong H, Dede J, French SW. CYP2E1 induced by ethanol causes oxidative stress, proteasome inhibition and cytokeratin aggresome (Mallory body-like) formation. Exp Mol Pathol. 2006;81:191–201. doi: 10.1016/j.yexmp.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 36.Donohue TM Jr, Zetterman RK, Zhang-Gouillon ZQ, French SW. Peptidase activities of the multicatalytic protease in rat liver after voluntary and intragastric ethanol administration. Hepatology. 1998;28:486–491. doi: 10.1002/hep.510280228. [DOI] [PubMed] [Google Scholar]
- 37.Donohue TM Jr, Cederbaum AI, French SW, Barve S, Gao B, Osna NA. Role of the proteasome in ethanol-induced liver pathology. Alcohol Clin Exp Res. 2007;31:1446–1459. doi: 10.1111/j.1530-0277.2007.00454.x. [DOI] [PubMed] [Google Scholar]
- 38.Barreto R, Kawakita S, Tsuchiya J, Minelli E, Pavasuthipaisit K, Helmy A, Marotta F. Metal-induced oxidative damage in cultured hepatocytes and hepatic lysosomal fraction: beneficial effect of a curcumin/absinthium compound. Chin J Dig Dis. 2005;6:31–36. doi: 10.1111/j.1443-9573.2005.00184.x. [DOI] [PubMed] [Google Scholar]
- 39.Britton RS. Metal-induced hepatotoxicity. Semin Liver Dis. 1996;16:3–12. doi: 10.1055/s-2007-1007214. [DOI] [PubMed] [Google Scholar]
- 40.Donohue TM Jr, Curry-McCoy TV, Todero SL, White RL, Kharbanda KK, Nanji AA, Osna NA. L-Buthionine (S,R) sulfoximine depletes hepatic glutathione but protects against ethanol-induced liver injury. Alcohol Clin Exp Res. 2007;31:1053–1060. doi: 10.1111/j.1530-0277.2007.00393.x. [DOI] [PubMed] [Google Scholar]
- 41.Donohue TM, Curry-McCoy TV, Nanji AA, Kharbanda KK, Osna NA, Radio SJ, Todero SL, White RL, Casey CA. Lysosomal leakage and lack of adaptation of hepatoprotective enzyme contribute to enhanced susceptibility to ethanol-induced liver injury in female rats. Alcohol Clin Exp Res. 2007;31:1944–1952. doi: 10.1111/j.1530-0277.2007.00512.x. [DOI] [PubMed] [Google Scholar]
- 42.Bernal CA, Vazquez JA, Adibi SA. Leucine metabolism during chronic ethanol consumption. Metabolism. 1993;42:1084–1086. doi: 10.1016/0026-0495(93)90262-m. [DOI] [PubMed] [Google Scholar]
- 43.Donohue TM Jr, Sorrell MF, Tuma DJ. Hepatic protein synthetic activity in vivo after ethanol administration. Alcohol Clin Exp Res. 1987;11:80–86. doi: 10.1111/j.1530-0277.1987.tb01267.x. [DOI] [PubMed] [Google Scholar]
- 44.Aplin A, Jasionowski T, Tuttle DL, Lenk SE, Dunn WA Jr. Cytoskeletal elements are required for the formation and maturation of autophagic vacuoles. J Cell Physiol. 1992;152:458–466. doi: 10.1002/jcp.1041520304. [DOI] [PubMed] [Google Scholar]
- 45.Köchl R, Hu XW, Chan EY, Tooze SA. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic. 2006;7:129–145. doi: 10.1111/j.1600-0854.2005.00368.x. [DOI] [PubMed] [Google Scholar]
- 46.Casey CA, Kragskow SL, Sorrell MF, Tuma DJ. Zonal differences in ethanol-induced impairments in receptor-mediated endocytosis of asialoglycoproteins in isolated rat hepatocytes. Hepatology. 1991;13:260–266. [PubMed] [Google Scholar]
- 47.Casey CA, Kragskow SL, Sorrell MF, Tuma DJ. Chronic ethanol administration impairs the binding and endocytosis of asialo-orosomucoid in isolated hepatocytes. J Biol Chem. 1987;262:2704–2710. [PubMed] [Google Scholar]
- 48.Tuma DJ, Sorrell MF. Effects of ethanol on the secretion of glycoproteins by rat liver slices. Gastroenterology. 1981;80:273–278. [PubMed] [Google Scholar]
- 49.Tuma DJ, Sorrell MF. Effects of ethanol on protein trafficking in the liver. Semin Liver Dis. 1988;8:69–80. doi: 10.1055/s-2008-1040529. [DOI] [PubMed] [Google Scholar]
- 50.Clemens DL, Casey CA, Sorrell MF, Tuma DJ. Ethanol oxidation mediates impaired hepatic receptor-mediated enocytosis. Alcohol Clin Exp Res. 1998;22:778–779. doi: 10.1111/j.1530-0277.1998.tb03866.x. [DOI] [PubMed] [Google Scholar]
- 51.Clemens DL, Halgard CM, Cole JR, Miles RM, Sorrell MF, Tuma DJ. Impairment of the asialoglycoprotein receptor by ethanol oxidation. Biochem Pharmacol. 1996;52:1499–1505. doi: 10.1016/s0006-2952(96)00524-2. [DOI] [PubMed] [Google Scholar]
- 52.Smith SL, Jennett RB, Sorrell MF, Tuma DJ. Acetaldehyde substoichiometrically inhibits bovine neurotubulin polymerization. J Clin Invest. 1989;84:337–341. doi: 10.1172/JCI114159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cells. Int J Biochem Cell Biol. 2004;36:2445–2462. doi: 10.1016/j.biocel.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 54.Young TA, Bailey SM, Van Horn CG, Cunningham CC. Chronic ethanol consumption decreases mitochondrial and glycolytic production of ATP in liver. Alcohol Alcohol. 2006;41:254–260. doi: 10.1093/alcalc/agl017. [DOI] [PubMed] [Google Scholar]
- 55.Moon KH, Hood BL, Kim BJ, Hardwick JP, Conrads TP, Veenstra TD, Song BJ. Inactivation of oxidized and S-nitrosylated mitochondrial proteins in alcoholic fatty liver of rats. Hepatology. 2006;44:1218–1230. doi: 10.1002/hep.21372. [DOI] [PubMed] [Google Scholar]
- 56.Piquet MA, Nogueira V, Devin A, Sibille B, Filippi C, Fontaine E, Roulet M, Rigoulet M, Leverve XM. Chronic ethanol ingestion increases efficiency of oxidative phosphorylation in rat liver mitochondria. FEBS Lett. 2000;468:239–242. doi: 10.1016/s0014-5793(00)01225-4. [DOI] [PubMed] [Google Scholar]
- 57.Meley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, Meijer AJ. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem. 2006;281:34870–34879. doi: 10.1074/jbc.M605488200. [DOI] [PubMed] [Google Scholar]
- 58.You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology. 2004;127:1798–1808. doi: 10.1053/j.gastro.2004.09.049. [DOI] [PubMed] [Google Scholar]
- 59.Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA. 2005;102:8204–8209. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liangpunsakul S, Wou SE, Zeng Y, Ross RA, Jayaram HN, Crabb DW. Effect of ethanol on hydrogen peroxide-induced AMPK phosphorylation. Am J Physiol Gastrointest Liver Physiol. 2008;295:G1173–G1181. doi: 10.1152/ajpgi.90349.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xie Z, Dong Y, Zhang M, Cui MZ, Cohen RA, Riek U, Neumann D, Schlattner U, Zou MH. Activation of protein kinase C zeta by peroxynitrite regulates LKB1-dependent AMP-activated protein kinase in cultured endothelial cells. J Biol Chem. 2006;281:6366–6375. doi: 10.1074/jbc.M511178200. [DOI] [PubMed] [Google Scholar]
- 62.Choi HC, Song P, Xie Z, Wu Y, Xu J, Zhang M, Dong Y, Wang S, Lau K, Zou MH. Reactive nitrogen species is required for the activation of the AMP-activated protein kinase by statin in vivo. J Biol Chem. 2008;283:20186–20197. doi: 10.1074/jbc.M803020200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 63.Crabb DW, Liangpunsakul S. Alcohol and lipid metabolism. J Gastroenterol Hepatol. 2006;21 Suppl 3:S56–S60. doi: 10.1111/j.1440-1746.2006.04582.x. [DOI] [PubMed] [Google Scholar]
- 64.Jensen K, Gluud C. The Mallory body: theories on development and pathological significance (Part 2 of a literature survey) Hepatology. 1994;20:1330–1342. [PubMed] [Google Scholar]
- 65.Cahill A, Wang X, Hoek JB. Increased oxidative damage to mitochondrial DNA following chronic ethanol consumption. Biochem Biophys Res Commun. 1997;235:286–290. doi: 10.1006/bbrc.1997.6774. [DOI] [PubMed] [Google Scholar]
- 66.Fernández-Checa JC, Hirano T, Tsukamoto H, Kaplowitz N. Mitochondrial glutathione depletion in alcoholic liver disease. Alcohol. 1993;10:469–475. doi: 10.1016/0741-8329(93)90067-x. [DOI] [PubMed] [Google Scholar]
- 67.Mansouri A, Demeilliers C, Amsellem S, Pessayre D, Fromenty B. Acute ethanol administration oxidatively damages and depletes mitochondrial dna in mouse liver, brain, heart, and skeletal muscles: protective effects of antioxidants. J Pharmacol Exp Ther. 2001;298:737–743. [PubMed] [Google Scholar]
- 68.Kim JH, Kim JE, Kim HJ, Roh GS, Yoo JM, Kang SS, Cho YY, Cho GJ, Choi WS. Ethanol decreases the expression of mitochondrial cytochrome c oxidase mRNA in the rat. Neurosci Lett. 2001;305:107–110. doi: 10.1016/s0304-3940(01)01820-1. [DOI] [PubMed] [Google Scholar]
- 69.Bailey SM, Cunningham CC. Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Free Radic Biol Med. 2002;32:11–16. doi: 10.1016/s0891-5849(01)00769-9. [DOI] [PubMed] [Google Scholar]
- 70.Venkatraman A, Shiva S, Davis AJ, Bailey SM, Brookes PS, Darley-Usmar VM. Chronic alcohol consumption increases the sensitivity of rat liver mitochondrial respiration to inhibition by nitric oxide. Hepatology. 2003;38:141–147. doi: 10.1053/jhep.2003.50293. [DOI] [PubMed] [Google Scholar]
- 71.Bailey SM, Pietsch EC, Cunningham CC. Ethanol stimulates the production of reactive oxygen species at mitochondrial complexes I and III. Free Radic Biol Med. 1999;27:891–900. doi: 10.1016/s0891-5849(99)00138-0. [DOI] [PubMed] [Google Scholar]
- 72.Junge J, Horn T, Christoffersen P. Megamitochondria as a diagnostic marker for alcohol induced centrilobular and periportal fibrosis in the liver. Virchows Arch A Pathol Anat Histopathol. 1987;410:553–558. doi: 10.1007/BF00781692. [DOI] [PubMed] [Google Scholar]
- 73.Rodriguez-Enriquez S, Kim I, Currin RT, Lemasters JJ. Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy. 2006;2:39–46. doi: 10.4161/auto.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001;15:2286–2287. doi: 10.1096/fj.01-0206fje. [DOI] [PubMed] [Google Scholar]
- 76.Masaki R, Yamamoto A, Tashiro Y. Cytochrome P-450 and NADPH-cytochrome P-450 reductase are degraded in the autolysosomes in rat liver. J Cell Biol. 1987;104:1207–1215. doi: 10.1083/jcb.104.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Luiken JJ, van den Berg M, Heikoop JC, Meijer AJ. Autophagic degradation of peroxisomes in isolated rat hepatocytes. FEBS Lett. 1992;304:93–97. doi: 10.1016/0014-5793(92)80596-9. [DOI] [PubMed] [Google Scholar]
- 78.Beckemeier ME, Bora PS. Fatty acid ethyl esters: potentially toxic products of myocardial ethanol metabolism. J Mol Cell Cardiol. 1998;30:2487–2494. doi: 10.1006/jmcc.1998.0812. [DOI] [PubMed] [Google Scholar]
- 79.Prock TL, Miranda RC. Embryonic cerebral cortical progenitors are resistant to apoptosis, but increase expression of suicide receptor DISC-complex genes and suppress autophagy following ethanol exposure. Alcohol Clin Exp Res. 2007;31:694–703. doi: 10.1111/j.1530-0277.2007.00354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]