

Abstract
We have developed a lentiviral vector system for human factor IX (hFIX) gene transfer in hematopoietic stem cells (HSCs) that provides erythroid cell-derived systemic protein delivery following nonmyeloablative conditioning and in vivo methylguanine methyltransferase (MGMT) drug selection. After bone marrow transplantation under moderate Busulfan conditioning, the initial hFIX expression in the chimeras was minimally detectable. However, the hFIX levels rose sharply following in vivo MGMT-drug selection and eventually reached a level that is considered curative in hemophilia B therapy (>500 ng/ml). The rise of hFIX levels was proportional to the increase in vector copy (VC) number in peripheral blood cells. High levels of hFIX expression were maintained in serially engrafted mice chimeras for 18 months. Importantly, high-level hFIX expression by erythroid cells did not result in anemia or adversely affect red blood cell counts. The prospect of combining reduced intensity conditioning, a presumably lowered risk of insertional mutagenesis due to low VC number requirement and erythroid-restricted transgene expression, as well as long-term protein expression at high level, strongly supports the potential applicability of adult stem cell-based gene therapy in nonlethal blood or metabolic disorders, as demonstrated here for hemophilia.
INTRODUCTION
The safe and effective gene modification of hematopoietic stem cells (HSCs) is one of the essential goals of gene therapy. HSCs are very promising target cells for developing gene therapies to treat various hematological as well as nonhematological diseases because of their self-renewal capacity and ability to generate all blood lineages.1-3 However, the broad use of genetically modified HSCs is currently limited by the toxicity associated with the use of conditioning regimens needed to achieve good engraftment of transduced cells, suboptimal transgene expression as a result of poor vector design, low gene transfer in HSCs and gene silencing after vector integration, as well as the risk of insertional mutagenesis associated with the use of long terminal repeat-driven γ retroviral vectors.
Using an erythroid-specific lentiviral vector and lethal irradiation as conditioning, we previously demonstrated the feasibility of directing long-term secretion of a clotting factor, human factor IX (hFIX), from HSC-derived erythroid cells. Therapeutic levels (250-350 ng/ml) of plasmic hFIX were obtained in peripheral blood of radiation chimeras harboring 0.2-0.6 vector copies (VCs) per cell.4 Having developed a dual-gene expressing lentiviral vector encoding hFIX and methylguanine methyltransferase (MGMT) mutant P140K, we investigate here whether high-level therapeutic gene expression can be achieved in HSC-based gene therapy under low toxicity nonmyeloablative conditioning and subsequent O6-benzylguanine (BG) and 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) in vivo MGMT-drug selections, which has been demonstrated previously to be an effective in vivo drug selection strategy.5-7
High levels of hFIX expression were achieved in primary hematopoietic chimeras conditioned with a nonlethal dose of Busulfan and BG/BCNU. These therapeutic levels of hFIX expression were sustained over long term in the primary, secondary, and tertiary chimeras (total 18 months). We therefore conclude that this nonmyeloablative approach using the dual-gene expressing lentiviral vector system warrants further investigation as a potentially safe and efficient HSC-based systemic protein delivery strategy.
RESULTS
High-level hFIX expression achieved by efficient MGMT in vivo selection
We first constructed a dual-gene expressing lentiviral vector that encodes hFIX and MGMT mutant P140K. hFIX is under the transcriptional control of the human β-globin promoter, enhancers and locus control regions (Figure 1a). This erythroid-specific gene expression system has been demonstrated to confer a robust transgene expression in erythroid lineage.8 In addition, this vector expresses the MGMT mutant P140K9 under the transcriptional control of the human phosphoglycerate kinase promoter.
Figure 1. Schematic representation of the self-inactivating lentiviral vector and experimental design.
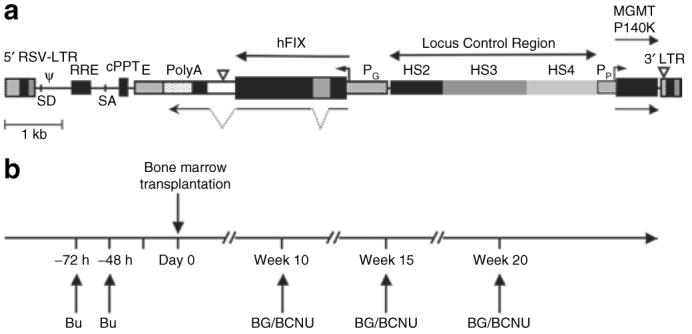
(a) RT9-hFIX-SI-MGMT vector encodes human factor IX (hFIX) cDNA with a truncated hFIX intron placed between the first and second exon sequences of hFIX under the transcriptional control of the β-globin promoter (PG), the 3′ β-globin enhancer (E) and β-globin locus control region (LCR); and the human phosphoglycerate kinase promoter (PP) and methylguanine methyltransferase (MGMT) P140K mutant gene is also included. (b) Experimental design for nonmyeloablative hematopoietic stem cell-based gene transfer and in vivo drug selection. Basically, C57BL/6, FIX-/- recipient mice were conditioned by administration of Busulfex, intraperitoneally 20 mg/kg, twice at 72 and 48 hours before engraftment. Bone marrow cells from syngeneic donor mice were then transduced overnight using RT9-hFIX-SI-MGMT vector. Three MGMT selections were performed using BG/BCNU at indicated time intervals. BCNU, 1,3-bis(2-chloroethyl)-1-nitrosourea; BG, O6-benzylguanine; cPPT, central polypurine tract; LTR, long terminal repeat.
We transduced bone marrow cells of hemophilia B mice, transplanted them in syngeneic recipients after nonmyeloablative conditioning using Busulfan. In vivo MGMT selection was initiated after transplantation as shown in Figure 1b. After engraftment, low levels of gene transfer and hFIX expression were obtained initially in Busulfan-conditioned recipients (Figure 2a and c), as well as in control mice treated with lethal irradiation (Figure 2b and c). After first BG/BCNU selection, the plasma levels of hFIX expression were significantly increased in Busulfan-conditioned mice (195.0 ± 85.6 ng/ml, week 14 post-transplantation) (Figure 2a). Following second BG/BCNU selection, hFIX levels in this group of mice were further increased to 487.5 ± 191.9 ng/ml with corresponding increase of the VC number in the peripheral blood (0.73 ± 0.13 VC per cell, week 20 post-transplantation) (Figure 2a and c). The hFIX levels in these mice continued to rise following third BG/BCNU selection and eventually reached 1,669.8 ± 441.4 ng/ml at 2.29 ± 0.16 VC per cell (week 27 post-transplantation) (Figure 2a and c).
Figure 2. Efficient selection of methylguanine methyltransferase (MGMT) P140K expressing hematopoietic stem cells leads to high-level and sustained hFIX expression in primary and secondary mice.

hFIX plasma levels monitored over 27 weeks in (a) Busulfan treatment group (n = 4) and (b) irradiation control group (n = 5). Three BG/BCNU selections (30 and 5 mg/kg, respectively) were performed in Busulfan-conditioned mice only (a, c, d) (indicated by black arrows). The 5 and 10% levels in a and b corresponding to human physiological levels of plasma FIX are indicated. These levels are considered therapeutic and curative, respectively, in human therapy. (c) Peripheral blood vector copy number (VCN) was determined by Taqman quantitative PCR analysis and was monitored over the same period. (d) hFIX expression in Busulfan-conditioned mice normalized by peripheral blood VCN. (e) The hFIX production and corresponding proportion of Ter119/hFIX double positive cells in total Ter119 positive cells analyzed by flow cytometry in three long-term chimeras (S1.1, S1.2, T2.2). (f) High levels of hFIX expression was sustained in secondary chimeras over 30 weeks (n = 4). Statistical analyses were done for a-d, and f. The probability of a statistically significant difference between the mean values of two data sets was determined by unpaired t-test with Welch's correction using GraphPad Prism version 4.0 (GraphPad software). In a and c, data sets marked with asterisk symbol indicates that there was a statistical significance when compared the present data set with the data set from the previous time point. No statistical significance in data sets between each time point was found in b, d and f. BCNU, 1,3-bis(2-chloroethyl)-1-nitrosourea; BG, O6-benzylguanine.
To assess the efficiency of hFIX production by this erythroid-specific lentiviral vector in chimeras following MGMT selection, the hFIX plasma levels were normalized against peripheral blood VC numbers. The average hFIX level reached 771.8 ± 112.6 ng/ml per VC at week 27 post-transplantation (Figure 2d). Three long-term chimeras were further analyzed to determine the fraction of hFIX-positive cells in total erythroid (Ter119 positive) lineage cells (Figure 2e). The average hFIX-fluorescein isothiocyanate (FITC) staining positive cells was 70.1 ± 10.8% as determined by fluorescence-activated cell sorting analysis, corresponding to plasma hFIX level of 773.3 ± 55.2 ng/ml as assayed by enzyme-linked immunosorbent assay. In contrast, hFIX expression in the irradiation control mice without MGMT selection remained at very low level over the same period (Figure 2b). These data indicate that therapeutic levels of hFIX expression can be achieved by incorporating single VC per cell using this erythroid-specific vector system in large population of erythroid cells, rather than multiple VC insertion per cell using other vector systems.
Therapeutic levels of hFIX were sustained in serially engrafted mice
High levels of hFIX expression were sustained in the secondary (728.8 ± 145.3 ng/ml, week 30 post secondary transplantation) (Figure 2f) and tertiary engrafted mice (671.5 ± 115.8 ng/ml, week 15 post tertiary transplantation) (data not shown) up to 18 months after the initial primary bone marrow engraftment. The overall high level and sustained hFIX expression in the primary, secondary, and tertiary chimeras indicates the successful gene transfer and subsequent enrichment of vector-integrated multipotent progenitor cells in the primary engrafted mice after MGMT selection, as well as the healthy self-renewal and propagation of these progenitor cells in the subsequent engraftments.
RT9-hFIX/PGK-MGMT dual-gene expression system allows erythroid-restricted hFIX and nonlineage-specific MGMT expression
To demonstrate the dual-gene expression capability of this vector system, fluorescence-activated cell sorting analysis was performed on hematopoietic cells collected from long-term chimeras. hFIX was exclusively expressed in erythroid compartments (Ter119 and CD71 positive cells) by RT9-hFIX-SI-MGMT chimeras, but not in other lineages of blood cells (Figures 3a and 4a). The fact that hFIX (+) splenocytes were mostly Ter119 positive (34.3% positive, 0.81% negative), but only partially CD71 positive (27.3% positive, 16.1% negative) indicates the presence of hFIX protein in more mature erythroid cells, as expected from the developmental stage specifically of the β-globin promoter. Ter119 surface antigen is a pan-erythroid marker, whereas CD71 is present only up to the orthochromatic erythroblast stage. The population of hFIX-positive mature erythrocytes is thus present in the CD71 negative quadrant (Figure 3a). In contrast, MGMT P140K gene expression was detected in all lymphoid and myeloid lineage cells (Figures 3b and 4b). The exclusive erythroid lineage-restricted expression of hFIX is in agreement with our previous finding using a monogenic erythroid-specific lentiviral vector.4 These results demonstrate that we have established a dual-gene expression vector system that is capable of erythroid-specific delivery of therapeutic protein in the presence of a nontissue-specific drug-resistant gene MGMT P140K expression.
Figure 3. Erythroid-restricted expression of human factor IX (hFIX) in Ter119 and CD71 positive cells and nonlineage specific expression of methylguanine methyltransferase (MGMT) P140K.
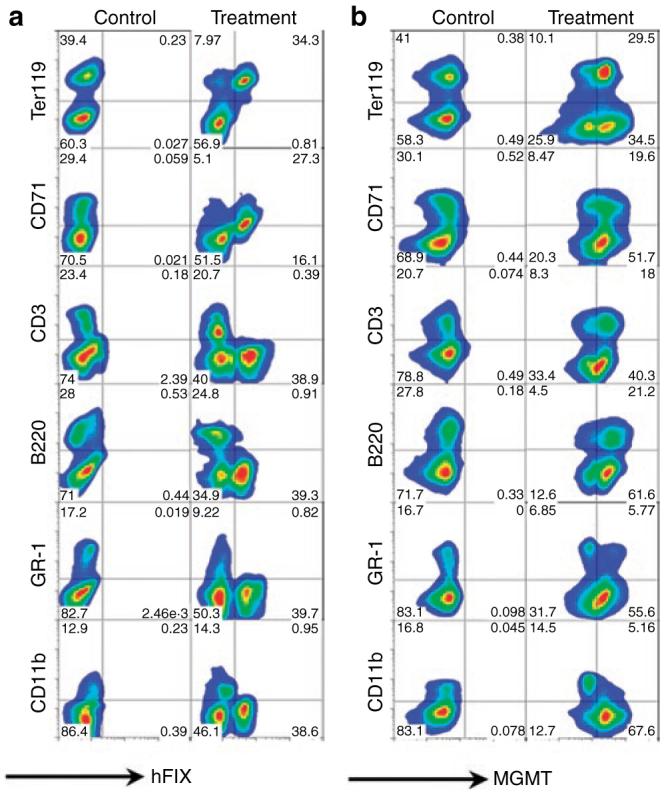
Splenocytes from a control C57BL/6-FIX-/- mouse (control) and a RT9-hFIX-SIMGMT transduced chimera (treatment) were stained for cell surface blood lineage markers Ter119, CD71, CD3, B220, GR-1, and CD11b; as well as intracellularly stained by (a) goat anti-hFIX-FITC or (b) biotin-labeled anti-MGMT and followed by streptavidin-FITC. FITC, fluorescein isothiocyanate.
Figure 4. Summary of fluorescence-activated cell sorting (FACS) analysis.
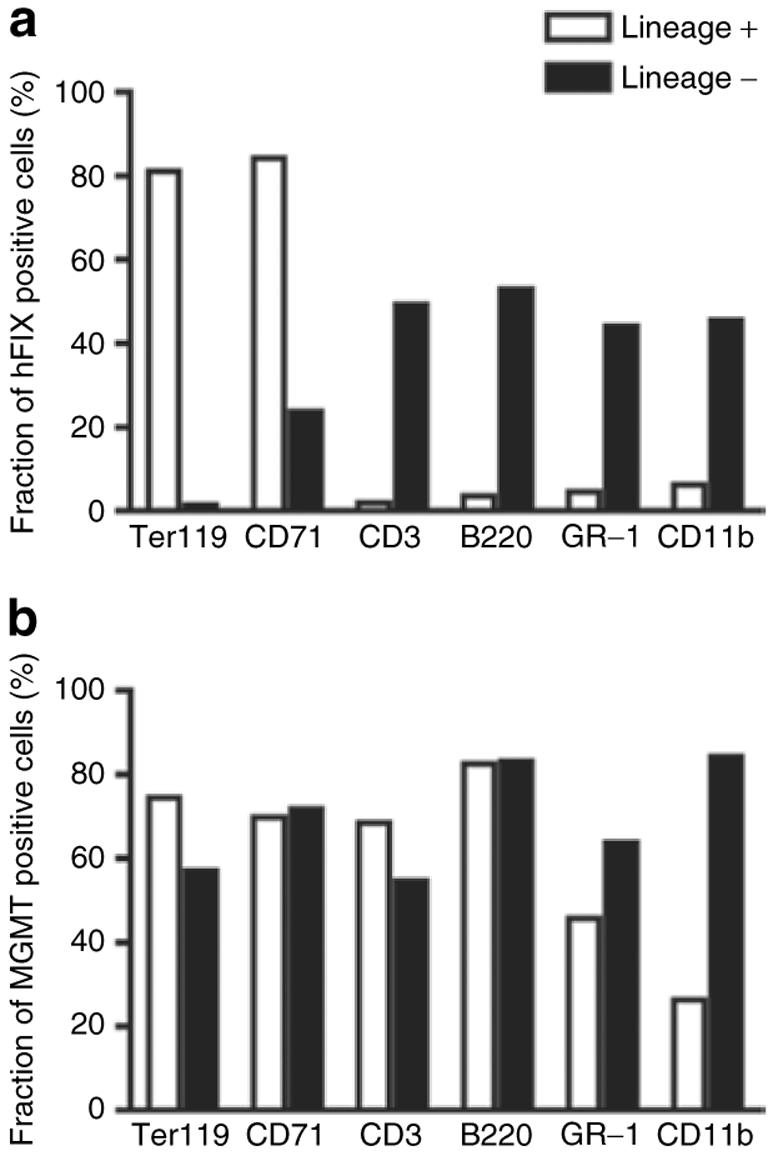
Fractions of (a) human factor IX (hFIX) or (b) methylguanine methyltransferase (MGMT) positive cells in lineage positive or negative cells. Representative data derived from FACS analysis (Figure 3).
Therapeutic protein delivery by erythroid cells does not adversely affect red blood cell homeostasis
We next investigated whether expression of a secreted exogenous protein by erythroid cells would affect hemoglobin synthesis and red blood cell homeostasis. By linear regression analysis of the correlation between plasma hFIX expression levels measured by enzyme-linked immunosorbent assay and the hemoglobin concentration levels and erythrocyte counts, it was concluded that hFIX expression levels in the chimeras did not significantly interfere with the plasma hemoglobin levels, nor did they affect the erythrocyte counts (Figure 5). Therefore, this analysis indicates that high-level expression of hFIX directed by erythroid cells does not result in anemia or adversely affect red blood cell counts. Compared with Busulfan-treated mice (Figure 5a and b), the overall hemoglobin concentration levels and erythrocyte counts were low in mice conditioned by lethal irradiation (Figure 5c and d). The slower recovery of hematopoiesis in these mice is likely due to the more toxic treatment as a result of the myeloablative irradiation conditioning.
Figure 5. High level of human factor IX (hFIX) expression by erythroid cells does not disrupt hemoglobin (Hb) synthesis or red blood cell (RBC) homeostasis.
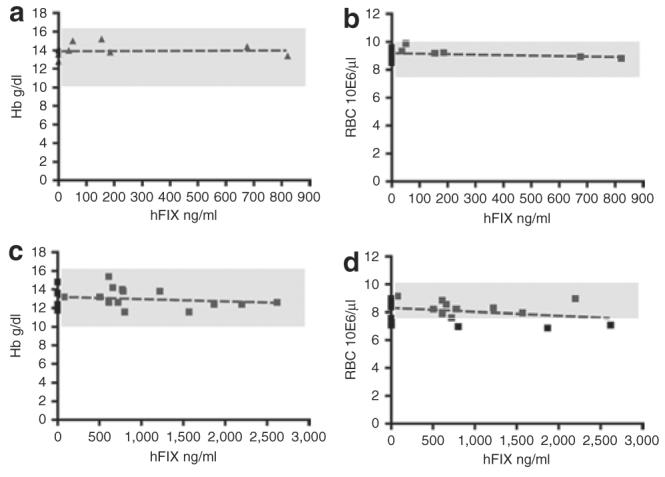
(a,b) Heparinized peripheral blood samples from Busulfan-conditioned mice (9 mice were hFIX negative, 6 mice were hFIX positive) and (c,d) lethally irradiation conditioned mice (6 mice were hFIX negative, 15 mice were hFIX positive) were analyzed for Hb levels (a,c) and RBC counts (b,d) in RT9-hFIX-SI-MGMT chimeras expressing various levels of hFIX. The normal ranges of RBC counts and Hb levels were depicted in the shaded areas. No significant correlation between plasma hFIX levels and RBC counts or Hb concentration has been found, according to linear regression analysis (Prism version 4; GraphPad Software). P values: 0.8790 (a), 0.4333 (b), 0.4320 (c), and 0.1769 (d), respectively.
To further assess the effect of exogenous hFIX expression on morphological development of red blood cells, confocal microscopy was performed using peripheral blood collected from RT9-hFIX-SI-MGMT chimeras. Based on our previous observation that residue hFIX protein trapped in mature erythrocytes after hFIX expression in late stage of erythroblast can be immunologically detected,4 red blood cells collected from peripheral blood of RT9-hFIX-SI-MGMT chimeras were double stained for surface erythroid marker Ter119 and intracellular hFIX. Erythrocytes expressing hFIX were specifically identified by the FITC-labeled anti-hFIX antibody, as compared to the control mouse samples (Figure 6a and b). The morphologic phenotype of hFIX-expressing erythrocytes does not appear to be different from red blood cells collected from control mice (Figure 6a and b), in contrast to that of thalassemic mice (Hbbth3/+) (data not shown). Altogether, these results suggest that high-level hFIX expression by erythroid cells does not adversely affect the development and morphology of erythrocytes.
Figure 6. Ectopically expressed human factor IX (hFIX) does not alter the morphological phenotype of erythrocytes.
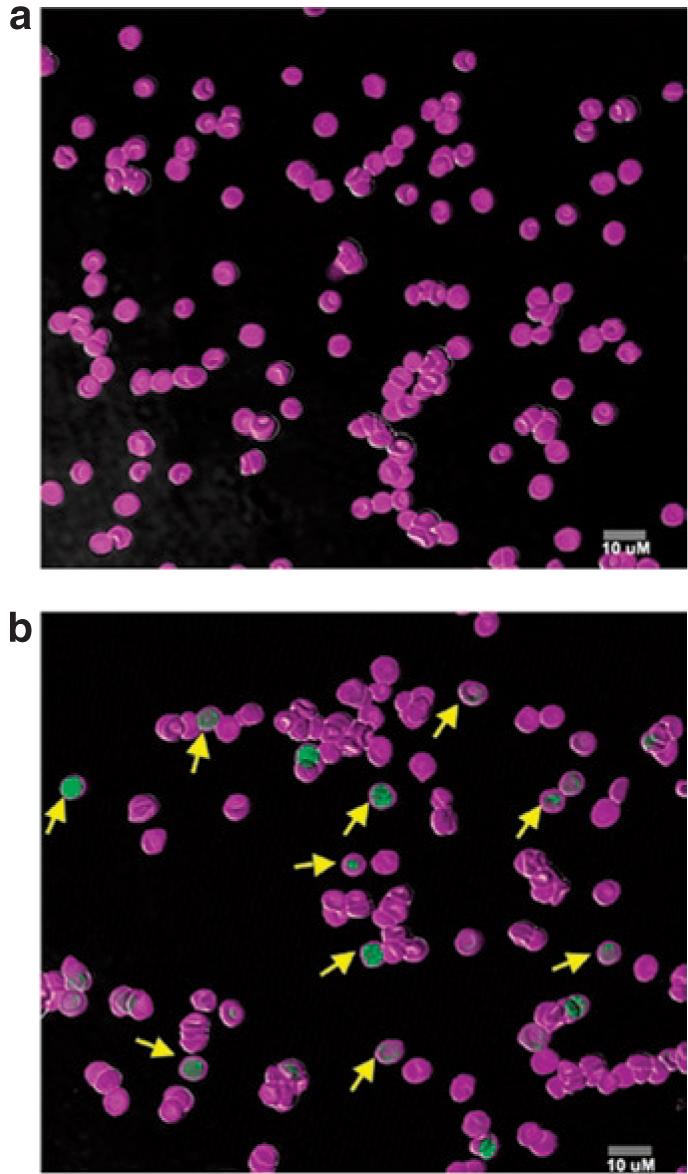
Confocal microscopy of peripheral blood erythrocytes from a (a) control C57BL/6, FIX-/- mouse and a C57BL/6, FIX-/- mouse engrafted with bone marrow cells from a (b) primary RT9-hFIX-SI-MGMT chimera. Cells were double stained by Ter119-Alexa and hFIX-FITC. Yellow arrows in b indicate typical hFIX-positive cells. MGMT, methylguanine methyltransferase.
The biological and immunological effects of hFIX production in hemophilia B mice
The biological activity of erythroid cell-expressed hFIX has been studied extensively before.4 It has been demonstrated to be biologically active as measured by activated partial thromboplastin testing, and is corresponding to 50-60% of what was expected on the basis of plasma hFIX concentrations determined by enzyme-linked immunosorbent assay.4 Here, we further analyzed the biological activity of erythroid cell-produced hFIX in mice treated by nonmyeloablative conditioning using Busulfan (Table 1). The results confirmed that erythroid cell-expressed hFIX is biologically active in mice either treated with nonmyeloablative Busulfan or with lethal irradiation conditioning, and the bioactivities are again in the 50-60% range even at high levels of expression. This level of bioactivity is consistent with a reduced level of γ-glutamyl carboxylase expression in hematopoietic cells in comparison to liver.10 We have previously demonstrated by tail-clipping assay that phenotypic correction of the hemophilia mice can be achieved in radiation chimeras producing 50 ng/ml hFIX or more.4 Here, we extended our previous findings and confirmed that phenotypic correction can also be achieved in mice treated by nonmyeloablative conditioning using Busulfan (Table 1). We have confirmed the absence of hFIX-specific antibodies in either nonmyeloablative Busulfan or radiation chimeras (data not shown). In addition, the majority of the mice expressing hFIX above a level of 37 ng/ml established immune tolerance against hFIX when challenged by Mononine and Titermax. These immune-tolerance effects induced by chimeras producing hFIX in their erythroid cells correlate well with our previous findings in lethal radiation chimeras.4
Table 1.
The biological and immunological effects of human factor IX (hFIX) production on hemophilia B mice
| Treatment | FIX (ng/ml) | Clotting activity (%) | Tail clip challenge | Tolerance |
|---|---|---|---|---|
| Bu | 105.7 | 1.25 | + | + |
| 26.3 | 0.28 | - | - | |
| 15.7 | 0.12 | - | - | |
| 0 | 0.00 | - | - | |
| 69.0 | 0.78 | + | + | |
| Bu/ATS | 600.7 | 7.33 | + | + |
| 10.3 | 0.10 | - | - | |
| 89.5 | 1.00 | + | + | |
| 37.3 | 0.37 | - | + | |
| 373.7 | 4.63 | + | + | |
| Irradiation | 603.4 | 8.57 | + | + |
| 538.3 | 5.81 | + | + | |
| 687.2 | 8.52 | + | + | |
| 851.6 | 9.37 | + | + | |
| 290.8 | 3.49 | + | + |
Bu, Busulfan-treated group [Busulfan, intraperitoneal (i.p.) 2 × 20mg/kg, -72 and -48 hours before engraftment]. Bu/ATS, Busulfan and antithymocyte serum treated group (Busulfan, i.p. 2 × 20mg/kg, -72 and -48 hours; antithymocyte serum, i.p. 2 × 30mg/kg, -24 and 2-4 hours before engraftment). Irradiation, irradiation-treated group (recipient mice were irradiated with 10.5 cGy). FIX concentration was measured by enzyme-linked immunosorbent assay, clotting activity by activated partial thromboplastin test. Survival was monitored for 6 hours and overnight after tail clipping. Mice survived the challenge is marked (+). Immune tolerance was assessed by immunization of mice with Mononine and Titermax, followed by testing for FIX-specific antibodies in the plasma samples 5 weeks after immunization. Mice established immune tolerance are defined as the mice that have plasma anti-FIX antibody level <100ng/ml, and are marked (+).
DISCUSSION
We have successfully developed a lentiviral vector system that yields high-level systemic protein delivery and also addresses some of the safety and efficiency concerns associated with transgene engineering of HSCs. This vector comprises two transcription units: one that overcomes silencing and expresses high level of hFIX at low VC number, and the other that allows for MGMT-based in vivo drug selection of transduced hemotopoietic cells. Using this approach, high-level hFIX expression has been achieved in chimeras after nonmyeloablative conditioning and in vivo MGMT-drug selection. Efficient hFIX transgene expression reached a level that is considered curative in hemophilia gene therapy (>500 ng/ml)11 in chimeras bearing in average a VC of 1-2 per cell. This is by far the highest level of therapeutic protein delivery achieved by HSC-based gene therapy approaches on a VC basis. The requirement for hFVIII expression in hemophilia A is considerably less (20-200 ng/ml).
This is a significant improvement over the erythroid-restricted therapeutic protein delivery platform we developed earlier,4 in which radiation chimeras with an average VC of 0.5 had levels of 250-350 ng/ml of hFIX expression. Although both vector systems adopted the powerful β-globin synthesis capability of erythroid cells for systemic therapeutic protein delivery, there have been major differences in conditioning regimens for bone marrow engraftment, the requirement of levels of gene transfer, and the option of enrichment of vector-integrated progenitor cells through in vivo drug selection. Combining the advantages of moderate conditioning for engraftment, lowered requirement for gene transfer, and enrichment of vector-integrated progenitor cells by MGMT-drug selection, we have extended our previous findings and established a clinically applicable erythroid-specific therapeutic protein delivery system. Importantly, hFIX levels in this study were increased from minimally detectable level to a very high plasma level (1,600 ng/ml) with corresponding VC increase after just three rounds of MGMT selection. This study thus demonstrates that under low gene transfer and mild conditioning regimens, it is possible to achieve therapeutic level of hFIX expression at a moderate level of VC number after MGMT-based in vivo drug selection.
The high level of gene expression conferred by this vector system should be mainly credited to the use of erythroid-directed transgene expression utilizing multiple optimized regulatory elements, which we have previously shown to overcome gene silencing and position effect to drive efficient expression of human β-globin chain in thalassemic mice,8 as well as hFIX in hemophilia B mice.4 This tissue-specific and regulated expression approach is a significant advance over previous studies using nontissue-specific promoters, which either produced undetectable or very low levels of the clotting factors,12-15 or only reached therapeutic levels when multiple VCs were incorporated per cell using a strong nontissue-specific SFFV U3 promoter.16 The later practice may potentially increase the risk of integration-dependent genotoxicity.17-19
To overcome the limitation of lethal irradiation treatment traditionally used for bone marrow transplantation and to make HSC-based gene therapy more applicable for development of therapeutics of nonhematological diseases, we have developed a combination protocol of cell engineering and Busulfan-nonmyeloablative conditioning, followed by in vivo MGMT-drug selection. Busulfan is an alkylating chemotherapeutic agent that preferentially exerts more toxicity on quiescent HSCs, which makes space for more effective engraftment. High doses of Busulfan have been extensively used in preparatory regimen for allogeneic HSC transplantation. There is a species difference in the pharmacokinetics of Busulfan:20,21 the myeloablative dose in humans is 20 mg/kg, whereas in mice it is 150 mg/kg.22 Recently, low doses of Busulfan have been used to enhance engraftment of autologous CD34+ cells in patients with adenosine deaminase-defective severe combined immunodeficiency patients (4 mg/kg)23 and in patients with chronic granulomatous disease (8 mg/kg).24 The dose of 8 mg/kg of Busulfan has also been recently approved for the first US thalassemia clinical trial by RAC.25 The Busulfan dose used in this study is approximately one-fourth of the myeloablative dose used in mice and is considered nonmyeloablative.26 Although effective as an alkylating agent, Busulfan should be studied further in the future as it is presently unknown whether it may have long-term undesirable side effects in patients.
To enrich the gene-modified cells after engraftment of bone marrow cells transduced under low multiplicity of infection, MGMT drug resistance gene selection was used.5-7,27,28 The MGMT mutant P140K encoded by the vector was under the transcriptional control of a relatively weak human phosphoglycerate kinase promoter,29-33 in order to avoid possible cellular toxicity when MGMT is expressed at high levels (D.A. Williams, Cincinnati Children's Hospital Medical Center, personal communication, 23 November 2007). We demonstrated here that MGMT selection offered the selective advantage for enrichment of a small population of vector-integrated stem cells and achieved an average 1-2 VCs per cell in the peripheral blood of chimeras. Furthermore, these MGMT-selected stem cells were capable of expanding without loss of repopulating ability as demonstrated by sustained and high levels of transgene expression in the primary, secondary, and tertiary engrafted mice for a total of 18 months. It is still unclear at this time whether drug selection, which leads to clonal expansion of transduced progenitor cells, may increase the probability of insertional mutagenesis. However, the major advantage of having a powerful tissue-specific promoter shown here is that successful therapy could be achieved with relatively low overall VC number.
One potential concern of ectopically expressing hFIX in erythroid cells is whether it will affect hemoglobin synthesis and red blood cell development. We demonstrated here that production of hFIX at high level did not seem to disrupt hemoglobin synthesis, nor did it disturb red blood cell development or the morphologic phenotype. This indicates that therapeutic protein delivery from erythroid cells can be achieved without sacrificing the normal development and homeostasis of red blood cells due to the enormous capacity of hemoglobin synthesis in the erythroid cells (7.2 g daily output of hemoglobin in a healthy adult).33 FIX is a secreted vitamin K-dependent glycoprotein normally synthesized in the liver as a precursor molecule with a signal peptide which is proteolytically cleaved before secretion.34,35 Similar to other secreted proteins, it goes through several post-translational modifications before its secretion in the endoplasmic reticulum and Golgi apparatus, including γ-carboxylation, glycosylation, sulfation, phosphorylation, and hydroxylation.36 Although detailed mechanisms remain to be elucidated, the production of secreted hFIX by erythroid cells is not expected to be different. hFIX transcription should be initiated in early erythroblast (basophilic) cells, and expression and secretion should be maximized in the late erythroblast (polychromatic and orthochromic) stage, prior to the ejection of the nucleus. We know from the cell association of hFIX in our fluorescence-activated cell sorting analysis and confocal microscopy study that hFIX secretion by erythroid cells is not absolute. There is still some residue of FIX in the fully matured erythrocytes. However, it is important to know that most of the FIX produced is secreted and the residue cell-associated hFIX is not harmful to the red blood cells, assessing from the high levels of circulating FIX, the normal levels of hemoglobin and red blood cell counts, as well as normal erythrocyte morphology. Furthermore, judging from the high levels of FIX expression normalized to VC number (600-800 ng/ml per VC number), and in comparison with one of the strongest nontissue-specific gene expression systems4 that yielded low initial FIX levels and eventually silenced over 5-7 months after engraftment, our erythroid cell specific FIX expression system is perhaps the most efficient HSC-based gene expression system so far characterized in literature.
Recently, advances have been made in the preclinical studies of HSC-based hemophilia A gene therapy by several groups.26,37-39 Hemophilia A is a clinically more prevalent disease than hemophilia B, with more severe inhibitor problems in patients receiving clotting factor treatment.40 The physiological level of the clotting factor VIII (200 ng/ml), which is deficient in hemophilia A, is 25-fold less than that of the FIX (5,000 ng/ml).33 Therefore, the requirement for gene expression in hemophilia A gene therapy is much less demanding. Using murine stem cell virus-based vector and nontissue-specific gene expression, therapeutic levels of FVIII have been achieved in hemophilia A mice under nonmyeloablative doses of irradiation, Busulfan, or Busulfan with antithymocyte serum.26,37 In addition, megakaryocyte/platelet-directed FVIII gene expression has been attempted using either transgenics or lentiviral gene delivery into HSC.41,42 Although these promoters are relatively weaker than our erythroid-specific delivery system, the potential advantage of this platelet-directed FVIII delivery is that FVIII can be therapeutic even in the presence of high titer anti-FVIII inhibitory antibodies, although the degree of improved protection in the presence of inhibitors is limited, as demonstrated in the transgenic model.41 In comparison with our erythroid delivery system, platelet-directed FVIII gene delivery, which is confined to platelets and therefore not detectable in circulation, is more restricted for potential management of hemostasis and megakaryocyte-platelet related diseases.
We report here a nonmyeloablative conditioning and MGMT selection protocol to address in part the safety and efficiency issues associated with HSC-based gene therapy. To translate this research finding to hemophilia treatment, we plan to first study this therapeutic strategy in canine model of hemophilia. Recent clinical studies aiming to treat hemophilia B by genetic approaches have been hampered by limited gene transfer in the muscle or transient and immunogenic gene transfer in the liver.40 The HSC-based gene therapy approach described here, as well as other progenitor cell and systemic genetic approaches43,44 are therefore relevant therapeutic strategies for future hemophilia treatment. Furthermore, this powerful dual-gene expression vector system can be used as a paradigm for tissue-specific therapeutic protein delivery in vivo (Figure 7). The high-level hFIX expression afforded using our erythroid-specific protein expression approach can be extended to a number of diseases that require high-level and systemic protein delivery.33 These diseases include congenital or acquired deficiencies of clotting factors (factors VIII and IX), various enzymes (lysosomal storage diseases, adenosine deaminase, and ApoE), as well as antiangiogenic factors or humanized antibodies for cancer treatment.
Figure 7. The RT9-hFIX/PGK-MGMT hematopoietic stem cell-based gene therapy paradigm.
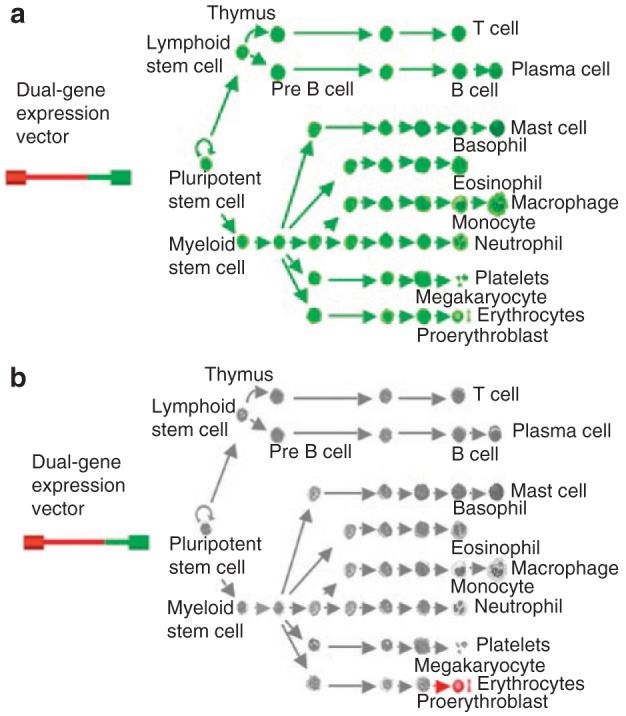
Dual-gene expressing lentiviral vector system allows nonlineage specific methylguanine methyltransferase (MGMT) P140K drug-resistant gene expression driven by a (a) relatively weak human phosphoglycerate kinase promoter in all cells and (b) as well as high-level protein delivery in late stage erythroid cells.
MATERIALS AND METHODS
Vector design and production
The RT9-hFIX-SI-MGMT construct (Figure 1a) was derived from RT9-hFIX-SI4 that encoded the hFIX cDNA with a short intron under the transcriptional control of the β-globin promoter, enhancers and locus control region. The human phosphoglycerate kinase promoter and MGMT P140K cDNA (a kind gift from Dr. Nachimuthu Chinnasamy)7 were cloned to the 3′-end of the vector. Viral stocks were generated by triple transfection of the recombinant vectors, pCMVΔR8.91, and pMD.G into 293T cells and concentrated as described.4
Animal procedures
All animal work was approved by the Institutional Animal Care and Use Committee at Memorial Sloan-Kettering Cancer Center. Donor bone marrow was collected from 8- to 16-week-old male C57BL/6 FIX-/- mice45 (kind gift of Dr. Inder Verma) that had been injected intravenously 4-6 days earlier with 5-fluorouracil (Pharmacia, Piscataway, NJ) (150 mg/kg body weight). Bone marrow cells were resuspended in X-VIVO 15 (BioWhittaker, Cambrex Bio Sci), supplemented with stem cell factor (100 ng/ml) and thrombopoietin (50 ng/ml) (R&D Systems), β-mercaptoethanol (0.5 mmol/l; Sigma), L-glutamine (200 mmol/l), penicillin [100 International units (IU)/ml], and streptomycin (100 μg/ml), and cultured overnight (8-12 hours). For nonmyeloablative conditioning, recipient mice (8- to 16-week-old female C57BL/6 FIX-/- mice) were injected with Busulfan (Busulfex; ESP Pharma) (diluted to 2 mg/ml in saline) at 20 mg/kg intraperitoneally, 72 and 48 hours before transplant. Bone marrow cells were transduced in serum-free medium on Retro-Nectin-coated 6-well plate (15 μg/ml; TAKARA Shuzo, Otsu, Japan) at low multiplicity of infection (1.1:1) for 8 hours. The transduced cells were then administered intravenously to each of the irradiated recipients (5 × 105 to 1 × 106 bone marrow cells per mouse). To establish secondary and tertiary bone marrow chimeras, 1 × 107 to 2 × 107 bone marrow cells from primary donors were injected into lethally irradiated recipient mice. At indicated time, mice for MGMT-based in vivo drug selection were given BG (Sigma) intraperitoneally at 30 mg/kg, which was followed by Carmustin (BCNU; Bristol-Myers Squibb) intraperitoneally at 5 mg/kg after 1 hour (Figure 1b).
Immunoassay to determine concentrations of hFIX
Concentrations of hFIX in mouse plasma samples were determined using enzyme-linked immunosorbent assay as described.4 Briefly, 96-well plate was coated with a monoclonal anti-hFIX antibody (clone HIX-1; Sigma) in a dilution of 1:800. The wells were blocked by phosphate-buffered saline with 5% fetal calf serum. Mouse plasma samples were diluted in phosphate-buffered saline with 2% fetal calf serum to concentrations within the linear range of the standard curve. A polyclonal goat anti-hFIX antibody conjugated with horseradish peroxidase (Affinity Biologicals, Hamilton, Ontario, Canada) was used as the secondary antibody in a dilution of 1:800. After applying ABTS substrate (Zymed, South San Francisco, CA), the 96-well plate was read at a single wavelength of 415 nm.
Determination of VC number using Taqman PCR
DNA was extracted from retro-orbitally collected mouse blood samples using a DNAeasy kit (Qiagen). The real-time Taqman PCR was performed and analyzed on a ABI 7500 PCR System (Applied Biosystems, Foster City, CA). For multiplex PCR, two sets of primer-probe combinations were used. The primer-probe set for the detection of the lentiviral vector was as follows: the forward primer, GAG-F (5′-GGAGCTAGAACGATTCGCAGTTA-3′); the reverse primer, GAG-R (5′-GGTTGTAGCTGTCCCAGTATTTGTC-3′); and the probe, GAG-P (5′-6FAM-ACAGCCTTCTGATGTTTCTAACAGGCCAGGTAMRA-3′).46 The primer-probe set for the endogenous control was a mouse β-actin endogenous control from Applied Biosystems (20×, VIC dye/MGB probe). The VC number per cell was determined by a relative quantification analysis45 over VC control NIH 3T3 cells.
Flow cytometric analysis of cell surface blood lineage markers and intracellular staining of hFIX and MGMT expression
Mouse splenocyte samples were collected from mouse by grinding the cut spleen against a 40 μmol/l Mesh filter (Fisher Scientific). The cells were then surface stained by erythroid cell markers Ter119, CD71, markers for myeloid (GR-1, CD11b), or lymphoid (CD3, B220), using antibodies conjugated with phycoerythrin (all from BD Biosciences). Following surface staining with indicated lineage markers, the cells were fixed and permeablized with the FIX & PERM kit (Caltag Laboratories) according to the manufacturer's instructions. For intracellular staining of hFIX, the affinity purified polyclonal goat anti-hFIX antibody conjugated with FITC (Affinity Biologicals, Hamilton, Ontario, Canada) was used. For intracellular staining of MGMT, a biotin-labeled mouse anti-MGMT monoclonal antibody (NeoMarkers, Fremont, CA) was used, which was followed by streptavidin-FITC (BD Biosciences). All flow cytometry analysis was done on a FACScan cytometer (BD Biosciences) and analyzed on FlowJo software, Version 6.2.1 (TreeStar).
Confocal microscopy
Mouse red blood cell samples collected from retro-orbital bleeding were fixed and permealized as described.4 Briefly, blood samples were first surface stained by Alexa Flur 700 labeled anti-mouse Ter119 (Biolegend, San Diego, CA) and fixed in 1 ml cold 0.05% glutaraldehyde in phosphate-buffered saline for 10 minutes. Surface-stained cells were then permeablized by 0.1% Triton X-100 for 10 minutes at room temperature, blocked by phosphate-buffered saline with 5% normal goat serum, and intracellularly stained with goat anti-hFIX conjugated with FITC (Affinity Biologicals, Ontario, Canada). Cells were then washed and transferred onto poly-L-lysine coated glass slides by cytospin (500 rpm for 3 minutes). Mobilized cells were mounted in ProLong Gold antifade reagent (Invitrogen) and visualized after a 24-hour curing period with a Leica SP2 confocal microscope. Images were analyzed using the MetaMorph offline software (Universal Imaging) and the Adobe Photoshop (Adobe).
Red cell hematologic studies
Blood samples were obtained by retro-orbital puncture under anesthesia. Total hemoglobin levels and red cell counts were measured on a blood cell count analyzer (Coulter ACT diff Analyzer, Beckman Coulter).
Statistical methods
Linear regression analysis was used to test whether there was any correlation between plasma hFIX levels and red blood cell counts or hemoglobin concentration using Prism version 4 (GraphPad Software).
ACKNOWLEDGMENTS
We thank Inder Verma for providing the C57BL/6-FIX-/- mice and Nachimuthu Chinnasamy for the MGMT P140K cDNA. We also acknowledge Garfield Springer and Jerry Chow, from Pharmacy/Memorial Sloan-Kettering Cancer Center for assistance in obtaining chemotherapy drugs used for this study, and Arthur J. Chang for assistance in editing this manuscript. This work was supported by National Institutes of Health grants HL57612, CA08748, and the Starr Foundation. The authors declared no competing financial interests.
REFERENCES
- 1.Dunbar CE. Gene transfer to hematopoietic stem cells: implications for gene therapy of human disease. Annu Rev Med. 1996;47:11–20. doi: 10.1146/annurev.med.47.1.11. [DOI] [PubMed] [Google Scholar]
- 2.Ballas CB, Zielske SP, Gerson SL. Adult bone marrow stem cells for cell and gene therapies: implications for greater use. J Cell Biochem Suppl. 2002;38:20–28. doi: 10.1002/jcb.10127. [DOI] [PubMed] [Google Scholar]
- 3.Engel BC, Kohn DB. Gene therapy for inborn and acquired immune deficiency disorders. Acta Haematol. 2003;110:60–70. doi: 10.1159/000072455. [DOI] [PubMed] [Google Scholar]
- 4.Chang AH, Stephan MT, Sadelain M. Stem cell-derived erythroid cells mediate long-term systemic protein delivery. Nat Biotechnol. 2006;24:1017–1021. doi: 10.1038/nbt1227. [DOI] [PubMed] [Google Scholar]
- 5.Bowman JE, Reese JS, Lingas KT, Gerson SL. Myeloablation is not required to select and maintain expression of the drug-resistance gene, mutant MGMT, in primary and secondary recipients. Mol Ther. 2003;8:42–50. doi: 10.1016/s1525-0016(03)00141-2. [DOI] [PubMed] [Google Scholar]
- 6.Persons DA, Allay ER, Sawai N, Hargrove PW, Brent TP, Hanawa H, et al. Successful treatment of murine beta-thalassemia using in vivo selection of genetically modified, drug-resistant hematopoietic stem cells. Blood. 2003;102:506–513. doi: 10.1182/blood-2003-03-0677. [DOI] [PubMed] [Google Scholar]
- 7.Chinnasamy D, Fairbairn LJ, Neuenfeldt J, Treisman JS, Hanson JP, Jr., Margison GP, et al. Lentivirus-mediated expression of mutant MGMTP140K protects human CD34+ cells against the combined toxicity of O6-benzylguanine and 1,3-bis(2-chloroethyl)-nitrosourea or temozolomide. Hum Gene Ther. 2004;15:758–769. doi: 10.1089/1043034041648417. [DOI] [PubMed] [Google Scholar]
- 8.May C, Rivella S, Callegari J, Heller G, Gaensler KM, Luzzatto L, et al. Therapeutic haemoglobin synthesis in beta-thalassaemic mice expressing lentivirus-encoded human beta-globin. Nature. 2000;406:82–86. doi: 10.1038/35017565. [DOI] [PubMed] [Google Scholar]
- 9.Xu-Welliver M, Kanugula S, Pegg AE. Isolation of human O6-alkylguanine-DNA alkyltransferase mutants highly resistant to inactivation by O6-benzylguanine. Cancer Res. 1998;58:1936–1945. [PubMed] [Google Scholar]
- 10.Liu Y, Nelson AN, Lipsky JJ. Vitamin K-dependent carboxylase: mRNA distribution and effects of vitamin K-deficiency and warfarin treatment. Biochem Biophys Res Commun. 1996;224:549–554. doi: 10.1006/bbrc.1996.1063. [DOI] [PubMed] [Google Scholar]
- 11.High KA. Gene transfer as an approach to treating hemophilia. Circ Res. 2001;88:137–144. doi: 10.1161/01.res.88.2.137. [DOI] [PubMed] [Google Scholar]
- 12.Hoeben RC, Einerhand MP, Briët E, van Ormondt H, Valerio D, van der Eb AJ. Toward gene therapy in haemophilia A: retrovirus-mediated transfer of a factor VIII gene into murine haematopoietic progenitor cells. Thromb Haemost. 1992;67:341–345. [PubMed] [Google Scholar]
- 13.Evans GL, Morgan RA. Genetic induction of immune tolerance to human clotting factor VIII in a mouse model for hemophilia A. Proc Natl Acad Sci USA. 1998;95:5734–5739. doi: 10.1073/pnas.95.10.5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tiede A, Eder M, von Depka M, Battmer K, Luther S, Kiem HP, et al. Recombinant factor VIII expression in hematopoietic cells following lentiviral transduction. Gene Ther. 2003;10:1917–1925. doi: 10.1038/sj.gt.3302093. [DOI] [PubMed] [Google Scholar]
- 15.Kootstra NA, Matsumura R, Verma IM. Efficient production of human FVIII in hemophilic mice using lentiviral vectors. Mol Ther. 2003;7:623–631. doi: 10.1016/s1525-0016(03)00073-x. [DOI] [PubMed] [Google Scholar]
- 16.Bigger BW, Siapati EK, Mistry A, Waddington SN, Nivsarkar MS, Jacobs L, et al. Permanent partial phenotypic correction and tolerance in a mouse model of hemophilia B by stem cell gene delivery of human factor IX. Gene Ther. 2006;13:117–126. doi: 10.1038/sj.gt.3302638. [DOI] [PubMed] [Google Scholar]
- 17.Baum C, Düllmann J, Li Z, Fehse B, Meyer J, Williams DA, et al. Side effects of retroviral gene transfer into hematopoietic stem cells. Blood. 2003;101:2099–2114. doi: 10.1182/blood-2002-07-2314. [DOI] [PubMed] [Google Scholar]
- 18.Sadelain M. Insertional oncogenesis in gene therapy: how much of a risk? Gene Ther. 2004;11:569–573. doi: 10.1038/sj.gt.3302243. [DOI] [PubMed] [Google Scholar]
- 19.Biffi A, Naldini L. Gene therapy of storage disorders by retroviral and lentiviral vectors. Hum Gene Ther. 2005;16:1133–1142. doi: 10.1089/hum.2005.16.1133. [DOI] [PubMed] [Google Scholar]
- 20.Kuramoto K, Follman D, Hematti P, Sellers S, Laukkanen MO, Seggewiss R, et al. The impact of low-dose busulfan on clonal dynamics in nonhuman primates. Blood. 2004;104:1273–1280. doi: 10.1182/blood-2003-08-2935. [DOI] [PubMed] [Google Scholar]
- 21.Kang EM, Hsieh MM, Metzger M, Krouse A, Donahue RE, Sadelain M, et al. Busulfan pharmacokinetics, toxicity, and low-dose conditioning for autologous transplantation of genetically modified hematopoietic stem cells in the rhesus macaque model. Exp Hematol. 2006;34:132–139. doi: 10.1016/j.exphem.2005.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mauch P, Down JD, Warhol M, Hellman S. Recipient preparation for bone marrow transplantation. I. Efficacy of total-body irradiation and busulfan. Transplantation. 1988;46:205–210. doi: 10.1097/00007890-198808000-00004. [DOI] [PubMed] [Google Scholar]
- 23.Aiuti A, Slavin S, Aker M, Ficara F, Deola S, Mortellaro A, et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296:2410–2413. doi: 10.1126/science.1070104. [DOI] [PubMed] [Google Scholar]
- 24.Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12:401–409. doi: 10.1038/nm1393. [DOI] [PubMed] [Google Scholar]
- 25.Recombinant DNA Advisory Committee Discussion of Human Gene Transfer Protocol #0704-852: A Phase I Open-Label Clinical Trial for the Treatment of β-Thalassemia Major with Autologous CD34+ Hematopoietic Progenitor Cells Transduced with ThalagenTM, a Lentiviral Vector Encoding the Normal Human β-Globin Gene; Minutes of Meeting. June 19-21, 2007; 2007. Principal Investigator: Farid Boulad, M.D., Memorial Sloan-Kettering Cancer Center. [Google Scholar]
- 26.Ide LM, Gangadharan B, Chiang KY, Doering CB, Spencer HT. Hematopoietic stem-cell gene therapy of hemophilia A incorporating a porcine factor VIII transgene and nonmyeloablative conditioning regimens. Blood. 2007;110:2855–2863. doi: 10.1182/blood-2007-04-082602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ragg S, Xu-Welliver M, Bailey J, D'Souza M, Cooper R, Chandra S, et al. Direct reversal of DNA damage by mutant methyltransferase protein protects mice against dose-intensified chemotherapy and leads to in vivo selection of hematopoietic stem cells. Cancer Res. 2000;60:5187–5195. [PubMed] [Google Scholar]
- 28.Zielske SP, Reese JS, Lingas KT, Donze JR, Gerson SL. In vivo selection of MGMT(P140K) lentivirus-transduced human NOD/SCID repopulating cells without pretransplant irradiation conditioning. J Clin Invest. 2003;112:1561–1570. doi: 10.1172/JCI17922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyoshi H, Smith KA, Mosier DE, Verma IM, Torbett BE. Transduction of human CD34+ cells that mediate long-term engraftment of NOD/SCID mice by HIV vectors. Science. 1999;283:682–686. doi: 10.1126/science.283.5402.682. [DOI] [PubMed] [Google Scholar]
- 30.Woods NB, Fahlman C, Mikkola H, Hamaguchi I, Olsson K, Zufferey R, et al. Lentiviral gene transfer into primary and secondary NOD/SCID repopulating cells. Blood. 2000;96:3725–3733. [PubMed] [Google Scholar]
- 31.Salmon P, Kindler V, Ducrey O, Chapuis B, Zubler RH, Trono D. High-level transgene expression in human hematopoietic progenitors and differentiated blood lineages after transduction with improved lentiviral vectors. Blood. 2000;96:3392–3398. [PubMed] [Google Scholar]
- 32.Ramezani A, Hawley TS, Hawley RG. Lentiviral vectors for enhanced gene expression in human hematopoietic cells. Mol Ther. 2000;2:458–469. doi: 10.1006/mthe.2000.0190. [DOI] [PubMed] [Google Scholar]
- 33.Chang AH, Sadelain M. The genetic engineering of hematopoietic stem cells: the rise of lentiviral vectors, the conundrum of the LTR, and the promise of lineage-restricted vectors. Mol Ther. 2007;15:445–456. doi: 10.1038/sj.mt.6300060. [DOI] [PubMed] [Google Scholar]
- 34.Choo KH, Gould KG, Rees DJ, Brownlee GG. Molecular cloning of the gene for human anti-haemophilic factor IX. Nature. 1982;299:178–180. doi: 10.1038/299178a0. [DOI] [PubMed] [Google Scholar]
- 35.Kurachi K, Davie EW. Isolation and characterization of a cDNA coding for human factor IX. Proc Natl Acad Sci USA. 1982;79:6461–6464. doi: 10.1073/pnas.79.21.6461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Enjolras N, Plantier JL, Rodriguez MH, Rea M, Attali O, Vinciguerra C, et al. Two novel mutations in EGF-like domains of human factor IX dramatically impair intracellular processing and secretion. J Thromb Haemost. 2004;2:1143–1154. doi: 10.1111/j.1538-7836.2004.00756.x. [DOI] [PubMed] [Google Scholar]
- 37.Moayeri M, Hawley TS, Hawley RG. Correction of murine hemophilia A by hematopoietic stem cell gene therapy. Mol Ther. 2005;12:1034–1042. doi: 10.1016/j.ymthe.2005.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doering CB, Gangadharan B, Dukart HZ, Spencer HT. Hematopoietic stem cells encoding porcine factor VIII induce pro-coagulant activity in hemophilia A mice with pre-existing factor VIII immunity. Mol Ther. 2007;15:1093–1099. doi: 10.1038/sj.mt.6300146. [DOI] [PubMed] [Google Scholar]
- 39.Chuah M, Vandendriessche T. Gene therapy for hemophilia “A” and “B”: efficacy, safety and immune consequences. Verh K Acad Geneeskd Belg. 2007;69:315–334. [PubMed] [Google Scholar]
- 40.High K. Gene transfer for hemophilia: can therapeutic efficacy in large animals be safely translated to patients? J Thromb Haemost. 2005;3:1682–1691. doi: 10.1111/j.1538-7836.2005.01460.x. [DOI] [PubMed] [Google Scholar]
- 41.Gewirtz J, Thornton MA, Rauova L, Poncz M. Platelet-delivered factor VIII provides limited resistance to anti-factor VIII inhibitors. J Thromb Haemost. 2008;6:1160–1166. doi: 10.1111/j.1538-7836.2008.02992.x. [DOI] [PubMed] [Google Scholar]
- 42.Shi Q, Fahs SA, Wilcox DA, Kuether EL, Morateck PA, Mareno N, et al. Syngeneic transplantation of hematopoietic stem cells (HSC) that are genetically modified to express factor VIII (FVIII) in platelets restores hemostasis to hemophilia A mice with pre-existing FVIII immunity. Blood. 2008 doi: 10.1182/blood-2008-02-138214. (epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thorrez L, Vandenburgh H, Callewaert N, Mertens N, Shansky J, Wang L, et al. Angiogenesis enhances factor IX delivery and persistence from retrievable human bioengineered muscle implants. Mol Ther. 2006;14:442–451. doi: 10.1016/j.ymthe.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 44.Sinn PL, Goreham-Voss JD, Arias AC, Hickey MA, Maury W, Chikkanna-Gowda CP, et al. Enhanced gene expression conferred by stepwise modification of a nonprimate lentiviral vector. Hum Gene Ther. 2007;18:1244–1252. doi: 10.1089/hum.2006.127. [DOI] [PubMed] [Google Scholar]
- 45.Wang L, Zoppè M, Hackeng TM, Griffin JH, Lee KF, Verma IM. A factor IX-deficient mouse model for hemophilia B gene therapy. Proc Natl Acad Sci USA. 1997;94:11563–11566. doi: 10.1073/pnas.94.21.11563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salmon P, Trono D. Design and production of human immunodeficiency virus-derived vectors. In: Celis JE, Carter N, Simons K, Small JV, Hunter T, Shotton D, editors. Cell Biology: A Laboratory Handbook. 3rd edn. vol. 1. The Netherlands; Elsevier: Amsterdam: 2005. p. 425. [Google Scholar]