

Abstract
Heat shock protein 90 has emerged as a promising target for the treatment of cancer and neurodegenerative diseases. This review summarizes recent advancements towards the development of natural products as they pertain to the biological and chemical understanding of this molecular chaperone.
Keywords: Hsp90, Inhibitors, Cancer, Neurodegenerative Diseases, Natural Products
Introduction
In recent years, molecular chaperones such as the 90 kDa heat-shock protein (Hsp90) have surfaced as promising targets for drug discovery [1-7]. Their role in the folding and maturation of various client proteins, as well as the rematuration of misfolded proteins [7-11], makes them potential targets for many diseases ranging from the disruption of multiple signaling pathways associated with cancer [1, 2, 4, 6, 12-17] to the clearance of protein aggregates in neurodegenerative diseases [4, 5, 18-23]. In fact, cytotoxic inhibitors of Hsp90 are the only cancer chemotherapeutic agents known to impact all six hallmarks of cancer simultaneously [6]. As defined by Hanahan and Weinberg, this includes 1) self-sufficiency in growth signals, 2) insensitivity to antigrowth signals, 3) evasion of apoptosis, 4) limitless replicative potential, 5) sustained angiogenesis, and 6) tissue invasion/metastasis [24]. Disruption of the Hsp90 protein folding machinery by non-cytotoxic agents promotes dissociation of heat shock factor 1 (HSF-1), which upregulates Hsp90 and facilitates the disaggregation of proteins responsible for several neurodegenerative diseases [21, 25].
Geldanamycin (GDA), a natural product isolated from the bacteria Streptomyces hygroscopicus (Fig. (1)), was the first identified Hsp90 inhibitor. Although it showed significant anti-proliferative activity against many cancer cell lines, its dose-limiting toxicity prevented successful completion of clinical trials. Since that time, a variety of natural product inhibitors of Hsp90 have emerged. Among these are herbimycin, radicicol, novobiocin, coumermycin A1, clorobiocin, epigallocatechin gallate (EGCG), taxol, pochonin, derrubone, gedunin, and celastrol.
Fig. (1).
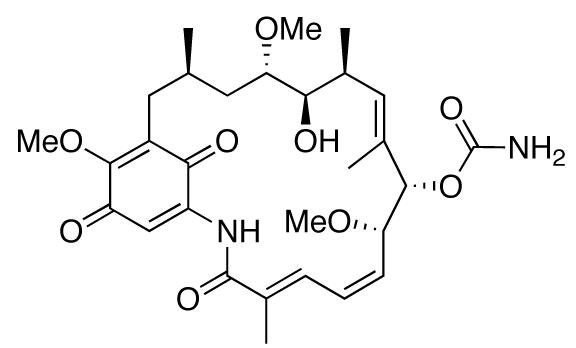
Structure of geldanamycin.
Properties of Hsp90
Heat-shock proteins (Hsps) act as molecular chaperones, guiding nascent polypeptides through the process of folding and maturation into three-dimensional structures [26, 27]. Chaperones are also responsible for refolding denatured proteins that result from cellular stresses such as nutrient deprivation, abnormal temperature or pH, malignancy, and exposure to various toxins and drugs [4, 28]. Heat-shock response is conserved across all species, from prokaryotes to eukaryotes, and provides a mechanism for general upkeep of intercellular processes, including protection against protein aggregation in the cytosol [29, 30].
Hsp90, the most prominent of the heat-shock proteins, makes up 1–2% of all cytosolic protein [8] and exists in four isoforms: Hsp90α, Hsp90β, glucose-regulated protein (GRP94), and Hsp75/tumor necrosis factor receptor associated protein 1 (TRAP-1). Hsp90α and Hsp90β can be found in the cytosol, and are the inducible and constitutive forms, respectively. GRP94 resides in the endoplasmic reticulum, while TRAP-1 is located in the mitochondrial matrix [31, 32]. To date, Hsp90 has been found to interact with over 200 client proteins, as well as ∼50 co-chaperones, making it a cornerstone in the cellular protein-folding machinery and an emerging target for the treatment of various disease states [33, 34].
Structure
Since the first reported crystal structure by Prodromou and co-workers in 1996 [35], it has been determined that Hsp90 is comprised of three distinct structural domains: a 10 kDa C-terminus, a 55 kDa middle domain, and a 25 kDa N-terminus [36, 37]. In its biologically active form, Hsp90 exists as a homodimer bound in a quaternary helix bundle formed by overlapping and antiparallel pairs of helices from each of the C-terminus domains [38-41]. C-Terminal crystal structures of bacterial HtpG [42] and eukaryotic Hsp90 [43] were solved in 2004 and 2006, respectively. Although rumors of its existence have surfaced in industry, a co-crystal structure of the C-terminal bound to an inhibitor has not been published. Csermely et al. first reported this binding site in 1998 [44], and in 2000 Neckers and co-workers were able to show that inhibition of Hsp90 at the C-terminus interrupts activity in a non-ATP competitive fashion [45, 46]. This discovery makes the C-terminus of Hsp90 a promising target for drug development, and highlights the importance of utilizing a co-crystal structure to further understand this process.
The 55 kDa middle domain of Hsp90 is the most variable region across species, but nonetheless is intimately involved in the binding and maturation of client proteins [9, 38]. The 25 kDa N-terminal domain is similar in composition to DNA gyrase B, histidine kinase, and MutL – together forming the GHKL (ATPase/kinase) superfamily [47]. This homology was determined through domain-specific human [48] and yeast [49] crystal structures and eventually led to elucidation of the ATP-binding site at the N-terminus. A co-crystal structure with ATP bound in a bent conformation, characteristic of the GHKL superfamily, was reported soon after [50]. These structures have played a critical role in the design of new and more potent Hsp90 inhibitors [51].
Hsp90 Folding Mechanism
Under normal physiological conditions, HSF-1 is tightly bound to and regulated by Hsp90 in its inactive state (2a, Fig. (2)). Upon activation, Hsp90 releases HSF-1, allowing translocation to the nucleus and induction of Hsps by binding to the heat shock response element [52]. These newly formed molecular chaperones are then responsible for governing the folding and maturation of nascent and denatured polypeptides into biologically active structures. It should be noted that the following description of this process has been simplified for the purpose of this review. A gamut of proteins have been linked to this folding mechanism, but only key interactions are highlighted herein.
Fig. (2).
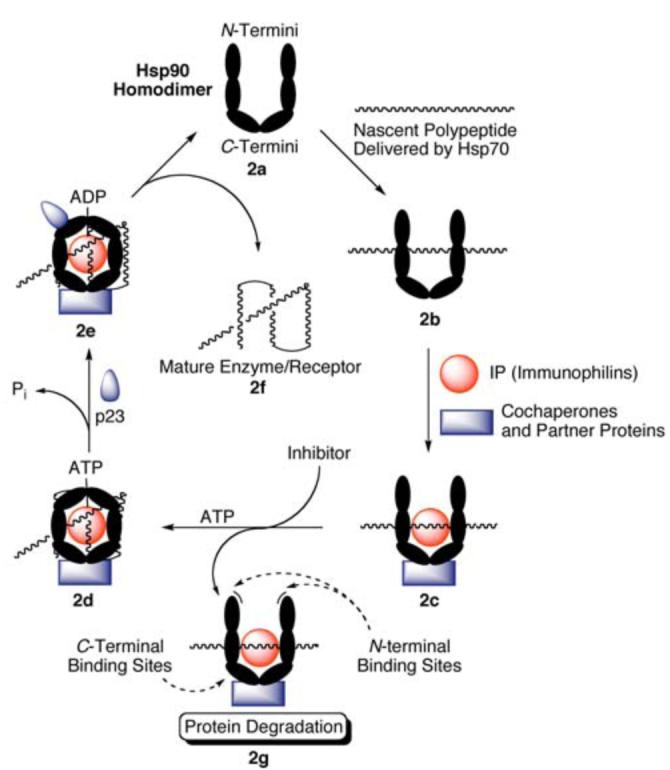
The protein folding mechanism of Hsp90.
Hsp70 binds to nascent polypeptides emerging from the ribosome in an ATP and Hsp40-dependent fashion. This complex is stabilized by Hsp70 interacting protein (HIP), and can be dissociated by Bcl2-associated athanogene (BAG). Hsp70-Hsp90 organizing protein (HOP) contains tetratricopeptide repeats (TPRs) recognized by both molecular chaperones, and recruitment by the Hsp70 complex facilitates transfer of the client protein to the Hsp90 homodimer (2b, Fig. (2)) [53-55]. Next, several co-chaperones, partner proteins, and immunophilins (shown in Table 1) bind Hsp90 (2c, Fig. (2)), and form a stabilized heteroprotein complex capable of binding ATP at the N-terminus [56-58]. Upon ATP mediated dimerization of the N-termini the activated Hsp90 multiprotein complex takes on a closed “clamped” conformation, engulfing the bound client protein (2d, Fig. (2)) [59, 60]. Recruitment of p23 facilitates ATP hydrolysis (2e, Fig. (2)) and further stabilizes Hsp90 [61, 62], allowing for maturation and subsequent release of the client protein (2f, Fig. (2)) [63].
Table 1.
Co-chaperones, partner proteins, and immunophilins involved in the Hsp90 folding mechanism.
| Co-chaperones |
Partner Proteins |
Immunophilins |
|---|---|---|
| Hsp40 | HOP | FKBP51 |
| Hsp70 | Tom70 | FKBP52 |
| Cdc37 | PP5 | Cyclophilin-40 |
| Ahl | ARA9 | UNC-45 |
| p23 | CNS1 | |
| CHIP | Dpit47 | |
| Tpr2 | ||
| SGT1 | ||
| CRN | ||
| WISp39 | ||
| NASP | ||
| TAH1 | ||
| Rar1 |
Inhibition of Hsp90 prior to ATP-mediated dimerization (2g, Fig. (2)) can effectively destabilize the heteroprotein complex. ATP hydrolysis provides the energy necessary for conformational changes that facilitate folding and maturation of the client. By preventing the maturation process, the Hsp90 complex is essentially disabled, and becomes a substrate for ubiquination and subsequent proteasomal degradation [64, 65]. Application of this concept has proven very useful in the rematuration of heat denatured firefly luciferase. A representative example by Yonehara [66] has demonstrated that inhibition of Hsp90 reduces luciferase activity, and a 2007 publication by Galam and coworkers used this information to establish a high-throughput screening assay to identify both N- and C-terminal inhibitors of Hsp90 based on the rematuration of heat denatured firefly luciferase [67].
A Selective Target for Cancer Treatment
Cancer is often referred to as a multifaceted class of diseases [68], dependent upon satisfaction of each of the six hallmarks as defined by Hanahan and Weinberg [24]. Although many cancer chemotherapeutics have successfully targeted proteins associated with multiple hallmarks, none have been able to simultaneously affect all six. Of the numerous client proteins dependent upon Hsp90 for folding and maturation, many are deemed essential for malignant progression. Over the last ten years, Hsp90 client proteins have been linked to all six hallmarks of cancer (Table 2) [6, 14], making Hsp90 inhibition an exciting new chemotherapeutic target. Whiteshell and Lindquist reviewed this concept in 2005 [13], and several studies and clinical trials have verified Hsp90 as a viable cancer target [69-71].
Table 2.
The six hallmarks of cancer.
| Hallmark | Hsp90 Client Proteins |
|---|---|
| Self-sufficiency in growth signals |
Raf-1, AKT, Her-2, MEK, Bcr-Abl, FLT-3, EGFR, IGF-1R, FGFR, KDR |
| Insensitivity to antigrowth signals |
Wee 1, Myt 1, CDK4, CDK6, Plk |
| Evasion of apoptosis | RIP, AKT, mutant p53, c-MET, Apaf-1, Survivin |
| Limitless replicative potential | Telomerase (h-TERT) |
| Sustained angiogenesis | FAK, AKT, HIF-1α, VEGFR, FLT-3 |
| Tissue invasion/metastasis | c-MET, MMP |
Inhibitors of Hsp90 have shown as high as a 200-fold differential selectivity toward malignant versus normal cells. Several mechanisms have been suggested to explain this high selectivity. First, Hsp90 is significantly upregulated in malignant cells to compensate for their dependency on the overexpression of client proteins, including ErbB2, Her-2, c-Met, Raf-1, and Akt [72-77]. The increased concentration of Hsp90 in tumor cells inherently results in greater drug accumulation. A second mechanism, introduced by Conforma Therapeutics in 2003, proposes that the Hsp90 heteroprotein complex (2c, Fig. (2)) exhibits a higher affinity for N-terminal inhibitors than the inactive homodimer (2a, Fig. (2)). In cancer cells, Hsp90 exists predominantly in a heteroprotein complex due to the over abundance of mutated, denatured, and naturally expressed proteins. In contrast, the primary form of Hsp90 in normal cells is the homodimer, which explains why N-terminal inhibitors accumulate in the high-affinity Hsp90 complex found in tumor cells [2, 78, 79]. Finally, a 2006 report by Duvvuri and co-workers [80] suggests a physiochemical explanation for selectivity. Under normal physiological conditions, lysosomal pH is around 4–5. Hsp90 inhibitors, like many chemotherapeutic agents, often contain basic nitrogens, and in a process known as pH partitioning [81] become protonated as the ammonium salt within the lysosome, trapping them in the organelle and preventing interaction with Hsp90, which resides in the cytosol. Conversely, the lysosomal pH in cancerous cells is essentially neutral, favoring an unprotonated amine [82, 83] and suggesting that the equilibrium drug concentration between lysosome and cytosol favors cytosolic interaction with tumor-derived Hsp90 more than in normal cells (Fig. (3)).
Fig. (3).
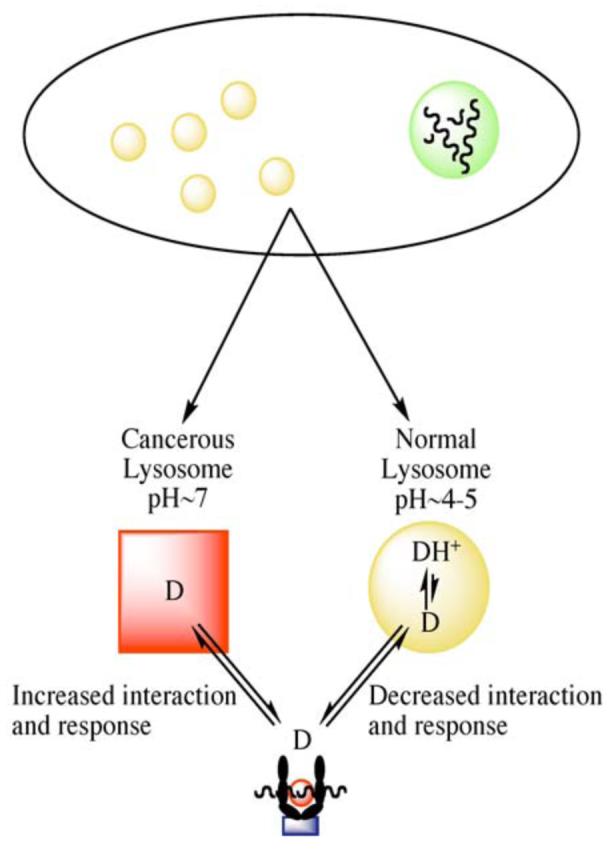
The effects of lysosomal pH on cellular drug distribution.
Neurodegenerative Applications
Neurodegenerative disorders such as Alzheimer's, Parkinson's, Huntington's, and spinal and bulbar muscular atrophy (SBMA), are in part characterized by the accumulation of misfolded protein aggregates. Under normal circumstances, this buildup can be prevented through resolubilization and rematuration of proteins by molecular chaperones. However, when suffering from these pathological conditions, aggregation exceeds the capacity of normal chaperone function, resulting in neuronal death [84].
Inhibition of Hsp90 stimulates the release of HSF-1, which in turn translocates to the nucleus, promoting transcription of HSPs [21, 25]. Increased levels of Hsp70 and Hsp90 have shown to be inversely proportional to β-amyloid and tau aggregation, and directly proportional to the binding of tau to microtubules, suggesting that inhibition of Hsp90 could serve as a neuroprotective approach for the treatment of Alzheimer's disease through dissolution of protein aggregates [22]. Recent studies by Shen and co-workers have affirmed this hypothesis by demonstrating the protective effects of GDA in vivo against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic toxicity associated with the formation of Lewy bodies (α-synuclein aggregation) in a Parkinson's disease model through inhibition of Hsp90 [19]. A similar study by Waza showed that 17-allylamino-17-demethoxygeldanamycin (17-AAG, Fig. (4)), a highly selective and more potent analogue of GDA, can diminish the effects of SBMA by inhibiting Hsp90, thus promoting degradation of mutant androgen receptor (AR) resulting from expansion of trinucleotide CAG repeats in the AR gene [20]. Ansar and co-workers have produced a library of non-toxic Hsp90 inhibitors that further highlight this concept through neuronal protection against Aβ-induced toxicity [85].
Fig. (4).
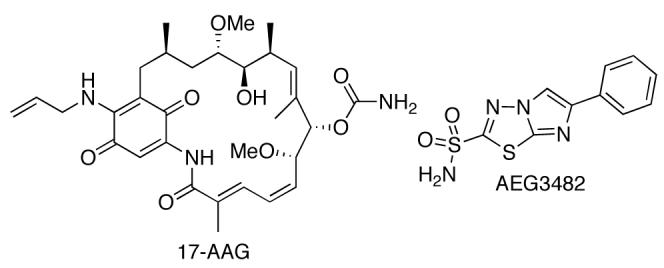
Structures of 17-AAG and AEG3482.
Finally, Huntington's, Parkinson's, and Alzheimer's disease attribute neuronal apoptosis to activation of the c-jun N-terminal kinase (JNK) signaling pathway. Hsp70 can prevent this cascade by binding to JNK, disrupting substrate interactions necessary for the initiation of apoptosis. In 2006, Gallo and co-workers demonstrated that the imidazothiadiazole sulfonamide AEG3482 (Fig. (4)) inhibits Hsp90, causing the release of HSF-1 and inducing Hsp transcription. This in turn provides neuroprotection through Hsp70 inhibition of the JNK signal transduction pathway [5]. Studies are underway to further refine the potential significance of modulating the Hsp90 protein folding machinery in a manner that can alleviate the accumulation of protein aggregates while providing a large therapeutic window and low cytotoxicity [85].
Natural Product Inhibitors of Hsp90
Clinical trials have shown that Hsp90 inhibitors are not only potent as anti-cancer agents, but are also well tolerated by patients. In fact, the toxicities and side effects discovered have not been directly linked to Hsp90 inhibition, but rather to hepatotoxicity, gastrointestinal irritation, and constitutional symptoms [86, 87]. It is not surprising, therefore, that medicinal chemists have become interested in discovering new scaffolds that exhibit Hsp90 modulatory activity for the treatment of cancer and neurodegenerative diseases. Given the inherent diversity and vast array of scaffolds that allow for protein interaction, natural products have become a key component in Hsp90 research [88].
A. Geldanamycin and Herbimycin
Geldanamycin and herbimycin (Fig. (5)) are naturally occurring benzoquinone ansamycin antibiotics that can be isolated through fermentation of Streptomyces hygroscopicus [89, 90]. The first total synthesis of herbimycin was reported by Nakata and co-workers in 1991 [91]. However, the total synthesis of GDA was not available until 2003, when Andrus and co-workers reported a procedure that afforded the natural product as a 1:10 mixture with (−)-o-quinogeldanamycin in low yield [92]. This result was due primarily to problematic oxidation of the trimethoxy precursor to the paraquinone. Andrus improved upon this methodology [93], and a subsequent 20 step total synthesis (2% overall yield) was achieved by Panek and Qin in 2008 [94].
Fig. (5).
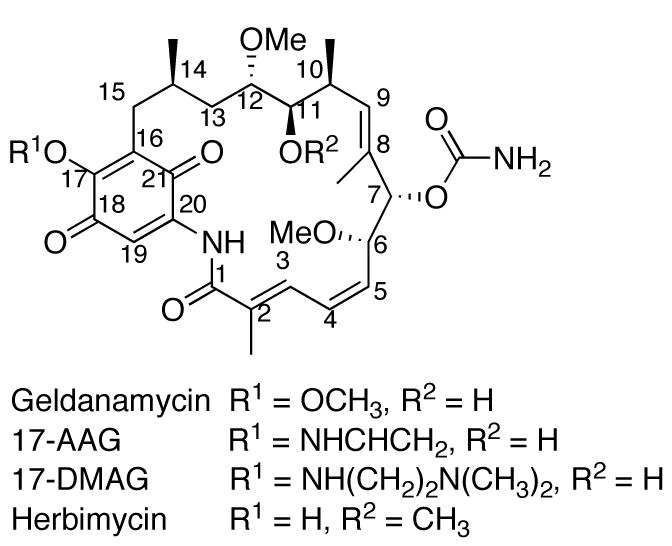
Structures of geldanamycin, structural analogues 17-AAG and 17-DMAG, and herbimycin.
The antitumor properties of GDA were first reported in 1986, and were initially attributed to its ability to inhibit v-Src phosphorylation in whole cells via Src tyrosine kinase [95, 96]. However a direct link between v-Src and GDA was never reported, as they were unable to directly inhibit the purified recombinant protein [97]. This suggested there might be a non-explicit interaction between the kinase and GDA. In 1994, Whiteshell and Neckers proved this relationship as a downstream effect of GDA's ability to specifically bind and antagonize Hsp90, a chaperone for v-Src [98, 99]. Using affinity purification, immobilized GDA affixed to agarose beads was incubated with reticulocyte lysate, resulting in identification of Hsp90. Further investigation proved that GDA specifically inhibited the Src-Hsp90 heteroprotein complex, facilitating degradation of the client protein. This observation was consistent with all prior work linking GDA to Src tyrosine kinase.
Initial reports by Roe and co-workers reported that GDA acted as a polypeptide mimic, interacting with Hsp90 at a highly conserved, 15 Å polypeptide substrate binding pocket involved in protein folding and maturation [48, 50]. However, Roe's co-crystallization of Hsp90 with GDA later revealed that it was actually binding to a previously unknown ATP binding pocket [100]. This seminal work opened the door for structure-activity relationship (SAR) studies that have led to the development of several analogues.
Although respectable IC50 values have been reported for GDA and herbimycin against various cancer cell lines, their poor solubility and hepatotoxicity in animals has prevented them from successfully completing clinical trials as anti-cancer agents [101]. SAR studies have shown that modifications to the carbamate group of GDA substantially decrease the potency of newly formed derivatives, as it serves to mimic the exocyclic amino and imino nitrogens of adenine. A similar loss in activity can be observed upon reduction of the 2–3 double bond, as the target–specific conformation of the macrocycle is compromised [88, 100]. Modification of the 17-methoxy substituent appears to be the most effective option, as it projects away from the ATP binding pocket and exhibits a minimal affect on Hsp90 affinity [50, 102]. Substituting an electron donating group for the 17-methoxy group decreases toxicity by stabilizing the quinone moiety and retarding formation of the semiquinone, which is capable of reacting with molecular oxygen, and producing superoxide radicals [103, 104].
The synthetic analogue 17-AAG (Fig. (5)), produced by Schulte and Neckers, displayed a 100-fold increase in differential selectivity at doses similar to GDA, as well as decreased hepatotoxicity [105]. Although 17-AAG proved to be more potent than GDA, solubility issues and its moderately persistent toxicity proved to be a factor in clinical development [106]. Additional work has produced 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (17-DMAG, Fig. (5)), which displays lower toxicity, higher potency, and improved bioavailability with respect to 17-AAG [106, 107]. 17-DMAG has entered phase I clinical trials and has demonstrated sensitization of therapeutically resistant cancer stem cells to chemotherapy [108].
Since the development of 17-AAG, several analogues of GDA have exhibited improved antitumor properties, as well as demonstrated neuroprotective activity. These include bioengineered compounds developed through site-directed mutagenesis of the polyketide synthase gene cluster [109], semi-synthesized analogues from biosynthetically generated metabolites [110], as well as several compounds arising from traditional synthetic techniques [111, 112]. The biologically modified synthetic approaches have offered alternative pathways to GDA analogues that were previously hindered by the lack of an efficient total synthesis. Traditional synthetic work, while effective, has been limited to alteration at the 17-position and the quinone moiety itself.
B. Radicicol and Pochonin
To date, radicicol (RDC, Fig. (6)) is the most potent natural product inhibitor of Hsp90, manifesting an IC50 value of 23 nM [100]. Its mechanism of action is similar to that of GDA, in that it binds to the N-terminal ATP-binding pocket of Hsp90. However, it does not manifest selectivity for the activated heteroprotein complex over the inactive form, as is the case for GDA. This can be attributed to the more rigid structure of RDC [100, 113]. RDC, isolated from Diheterospora chlamydosporia, has proven to have many downstream oncogenic effects through Hsp90 inhibition, including activity against 17-AAG resistant retinoblastoma cells [114]. However, no activity has been demonstrated in vivo, as RDC is rapidly converted to inactive metabolites due to the electrophilicity of the epoxide ring and α,β,γ,δ-unsaturated carbonyl [115].
Fig. (6).
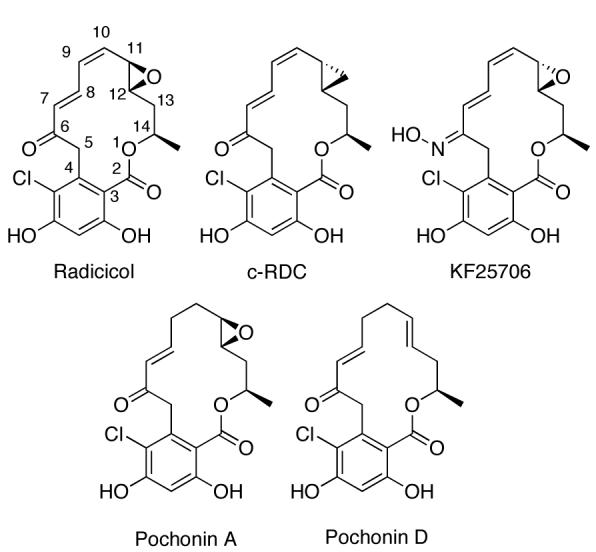
Structures of radicicol, c-RDC, KF25706 and Pochonin A & D.
Several analogues of RDC have been synthesized that minimize in vivo metabolism by decreasing its electrophilic nature [116-121]. These compounds display nanomolar activity in vivo. For example, synthesis of radicicol 6-oxime derivatives, such as KF25706 (Fig. (6)), has produced several compounds that demonstrate potent antiproliferative activity. Furthermore, the oxime stereochemistry has shown to be critical, as higher potency is observed with the E isomer [116, 117, 122]. Other studies have determined that Hsp90 inhibitory activity is dependent on the RDC scaffold being constrained to a bent conformation. This can be assisted by an sp2-hybridized C6 or a β-oriented oxygen close in proximity to C10 and C12 [123]. However, the best resource for synthetic SAR studies has been the total synthesis of RDC and analogues by Danishefsky and co-workers [124]. This route offers a remarkably straightforward synthesis, providing several opportunities for diversification. By replacing the electrophilic allylic epoxide with a cyclopropyl group (c-RDC, Fig (6)), these researchers were able to reintroduce activity comparable to GDA in vivo.
Isolation of the structurally similar pochonin family of natural products from Pochonia chlamydosporia has also shown promise in Hsp90 inhibition, particularly with pochonin A and pochonin D [125, 126]. Pochonin A and D have been shown to directly inhibit Hsp90 [126]. Pochonins A–F, while themselves displaying cytotoxicity in the micromolar range [125], provide an opportunity for conformational diversity that is not as easily achieved with radicicol [127]. As a result, several syntheses have now been completed [127, 128, 128].
C. Chimeric Analogues of Geldanamycin and Radicicol
SAR studies have shown that GDA activity is dependent upon the structural integrity of the quinone ring as well as the stereochemistry of the carbamate. Similarly, RDC activity is dependent on the resorcinol ring, and to a lesser extent the epoxide [129]. In addition, the amide functionality in GDA appears to impart high differential selectively towards the Hsp90 heteroprotein complex [130].
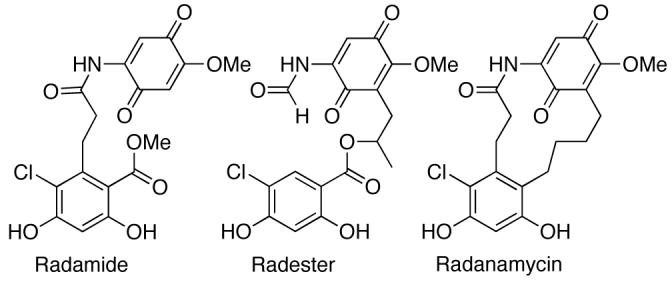
Seminal work by Shen and co-workers utilized this knowledge to compose a new class of chimeric analogues, combining the pro-inhibitory properties of GDA and RDC to form radamide, radester, and radanamycin [131-134]. Each chimera improved upon Hsp90 inhibitory activity with respect to the parent compounds (IC50 = 42 μM, 7.1μM, and 1.2 μM against MCF-7 breast cancer cells, respectively), and was synthesized in a minimal number of steps that allows for diversification of this potential drug class. Early studies by this group identified the hydroquinone species to be more active than the corresponding quinone, which was later confirmed with GDA by several other research laboratories [134].
D. Novobiocin, Coumermycin A1, and Clorobiocin
The coumermycin family of antibiotics, isolated from Streptomyces spheroids, has long been used clinically for antimicrobial purposes [135, 136]. Mechanistically, they bind the ATP binding pocket of DNA gyrase, another member of the GHKL superfamily [47], thus preventing ATP hydrolysis [137, 138]. Novobiocin in particular has been shown to display anti-cancer properties, and has been used in the clinic for many years [139]. Ground breaking work by Neckers and co-workers demonstrated that this activity could be ascribed to novobiocin's Hsp90 inhibitory activity. Using affinity chromatography, Neckers determined that novobiocin could competitively displace immobilized GDA bound to Hsp90, however GDA could not displace immobilized novobiocin when the reciprocal experiment was performed. Further studies revealed that novobiocin bound to a previously unrecognized C-terminal binding pocket, and induced degradation of Hsp90-dependent client proteins [45, 46]. These studies laid the groundwork for a vast library of novobiocin and coumermycin analogues that have since been prepared [85, 140-143].
SAR studies from our laboratory have revealed significant features that can control the activity manifested by these novobiocin analogues. Synthesis of A4 in 2005, along with DHN1 and DHN2 in 2006, highlighted key structural differences necessary for distinguishing between inhibition of DNA gyrase and Hsp90 [139, 140]. The 4-hydroxyl and 3′-carbamate of novobiocin are critical for DNA gyrase activity. Removing the 4-hydroxyl moiety and hydrolysis of the carbamate provided a 500-fold increase in selectivity towards Hsp90. A methyl group at the C8 position also moderately increased activity. As of 2006, A4 was not only the most potent novobiocin analogue to date, but interestingly displayed no growth inhibitory activity. This feature was exploited in its development as a neuroprotective agent in 2007 when Burlison and co-workers demonstrated that A4 could provide significant protection against Aβ-induced toxicity of neurons at non-cytotoxic concentrations [85]. Subsequent SAR studies concluded that the benzamide functionality of novobiocin was necessary for cytotoxicity [143]. It was also found that addition of a p-hydrogen bond acceptor and an m-aryl side chain were most effective at increasing anti-proliferative activity. Further derivatization resulting in heterocyclic analogues of the benzamide side chain revealed the most potent novobiocin analogue to date, KU-122. Installation of a 2-indole moiety in lieu of the native benzene ring resulted in a significant increase in anti-proliferative properties (IC50 = 0.37 μM in SKBr3 breast cancer cells, 0.17 μM in HCT-116 colon cancer cells). This variation in activity can be credited to the hydrogen bond donating capability and the rigid 2,3-olefin on the indole ring. These novobiocin analogues are unique in that rational modification of these compounds can provide molecules that selectively treat bacterial infections, cancer, or neurodegenerative diseases. Multiple projects are currently underway to further elucidate these properties and to create more potent Hsp90 inhibitors.
E. EGCG
Epigallocatechin-3-gallate (EGCG, Fig. (10)) is a naturally occurring polyphenol extract from the green tea, Camellia sinensis [144]. Although green tea has been marketed in Eastern medicine for years as an anticancer agent, it wasn't until 2003 that Palermo and co-workers ascribed this feature to inhibition of Aryl Hydrocarbon Receptor (AhR) activity [144]. Shortly thereafter, affinity studies concluded that EGCG did not bind directly to AhR, but instead antagonized Hsp90. Affinity purification studies concluded that EGCG, like novobiocin, binds to the C-terminus of Hsp90 [145, 146].
Figure 10.
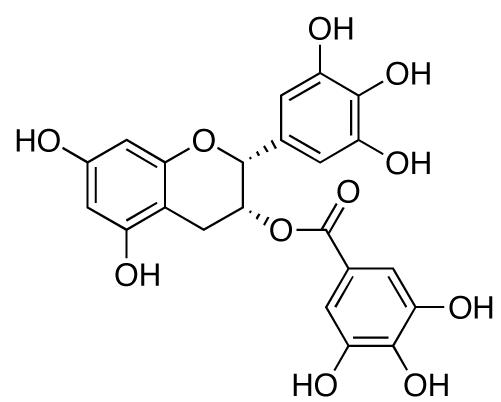
Structure of EGCG.
More recent studies have described EGCG's potential as a neuroprotective agent [147]. Although Weinreb and co-workers attribute this property to EGCG's ability to chelate iron in areas of the brain associated with Parkinson's and Alzheimer's disease, one cannot overlook the vastly growing library of Hsp90 inhibitors known to display neuroprotective qualities.
F. Taxol
Taxol's (Fig. (11)) biological activity as an anticancer agent has been attributed to its stabilization of microtubules and prevention of mitosis, and has been used clinically for over twenty years [148]. Through the activation of kinases and transcription factors, it has also been shown to elicit cell signaling in a manner indistinguishable from bacterial lipopolysaccharide (LPS) [149, 150].
Fig. (11).
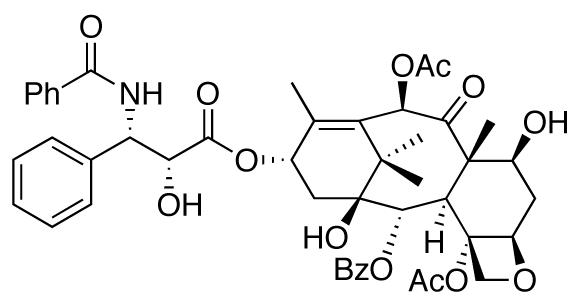
The structure of taxol.
Its isolation from the English yew tree, Taxus baccata L., by Monroe Wall, and his subsequent discovery of its anticancer properties, stands as one of the most significant findings in the history of natural product research [151, 152]. What is interesting, however, is that in recent years Rosen and co-workers have been able to show through affinity purification that taxol binds Hsp90, producing a stimulatory response [153-155]. This stimulatory response not only sensitizes malignant tumors to taxol, but could even prove useful in the future development of neuroprotective agents. The site to which taxol binds Hsp90 has not yet been elucidated.
G. Derrubone
Derrubone (Fig. (12)) is a prenylated isoflavone that was first isolated from the Indian tree, Derris robusta, in 1969 [156]. A total synthesis was reported three years later by Jain and Jain that consisted of 14 steps [157]. This feat was matched by Hossain and co-workers in 2006, and then improved upon by Hastings in 2007, reducing its preparation to 8 linear steps [158, 159]. Through utilization of the HTS luciferase assay discussed earlier [67], Hadden and co-workers identified derrubone as a C-terminal inhibitor of Hsp90 that yielded an IC50 value of 11.9 μM against MCF-7 breast cancer cells [160].
Fig. (12).
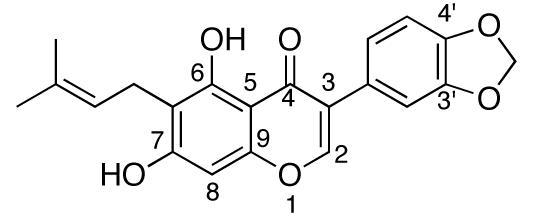
Structure of Derrubone.
SAR studies by Hastings and Hadden have identified key features of derrubone that allow optimal interactions with Hsp90, and several potent analogues have been synthesized [158]. First, the C3 aromatic ring substituent is essential for activity. Addition of an electron-withdrawing group at the C4′ position can further increase anti-proliferative activity, whereas substitution at C3′ results in complete loss of activity. Second, replacing the prenyl substituent with a more polar functionality results in decreased activity, whereas replacement with non-polar functionalities gives comparable activity to the prenyl group. A slight increase in activity was observed when the C6 substituent was translocated to the C8 position. Overall, this study produced analogues with IC50 values in the low micromolar range, and further development of the derrubone library is currently underway.
H. Gedunin and Celastrol
In recent years, gedunin (Fig. (13)), a tetranotriterpenoid isolated from the Indian neem tree Azadirachta Indica [161], and structurally related celastrol (Fig. (13)), a quinone methide triterpene from the Celastraceae family of plants, have become compounds of interest due to their anti-proliferative and neuroprotective properties [162-164]. Recently, studies identified these natural products as Hsp90 inhibitors [3, 165, 166]. Using a connectivity map, Lamb and co-workers were able to find high correlation scores between gedunin, celastrol, GDA, 17-AAG, and 17-DMAG, suggesting that the natural products exhibited their activity through Hsp90 modulation. A subsequent paper confirmed this hypothesis, however the mechanism of action was not fully revealed. In a fluorescence polarization assay, gedunin and celastrol failed to displace GDA, indicating the natural products were not binding competitively to the N-terminal ATP-binding site. Zhang and co-workers have reported that celastrol may disrupt Hsp90 function by blocking interactions between the molecular chaperone and the co-chaperone, Cdc37, preventing formation of the Hsp90 heteroprotein complex[162]. Based on structural similarities between gedunin and celastrol, it is likely that gedunin utilizes a similar mechanism of action towards Hsp90 inhibition.
Fig. (13).
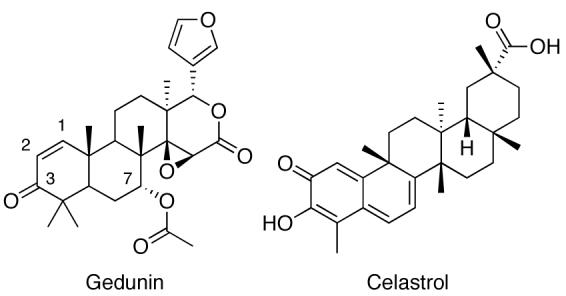
Structures of Gedunin and Celastrol.
In an attempt to elucidate structure–activity relationships between the molecular chaperone and natural products, multiple libraries have been synthesized [167]. Although the analogues made thus far have not proven more effective than gedunin in anti-proliferation assays, Brandt and co-workers1 have identified key structural features necessary for activity. Steric bulk applied to the C7 position has a pronounced effect on anti-proliferative activity, as inhibitory activity is diminished in response to size. Although it appears as though the electronic nature of the substituent is not imperative, the presence of a hydrogen bond acceptor can slightly improve anti-proliferative properties. C7 substituents also exhibit influence on the overall conformation of the molecule, and influence the binding of other substituents. The olefin of the α,β-unsaturated ketone is also essential for activity. One can assume this is due to the electrophilic nature of this moiety, however modifications to and reduction of the ketone itself have proven otherwise. Hydrogen bond accepting properties at the C3 substituent, as well as the rigidity of the 1,2-olefin are responsible for retention of activity. Studies are currently underway to further clarify gedunin's structure–activity relationship with Hsp90.
Conclusion
Natural products have long withstood the test of time for their contributions to medicinal chemistry. The development of new and interesting scaffolds, as well as small molecules that exhibit target selectivity, have been dependent on the isolation and modification of complex structures from Mother Nature. As Hsp90 continues to emerge as a target for the treatment of cancer, neurodegenerative diseases, and other disease states, the construction of viable inhibitors with drug-like properties becomes increasingly more important. The structures presented in this article have provided a summary of past achievements by natural product chemists and their recent impact on future applications.
Fig. (7).
Structure of the chimeric analogues of Geldanamycin and Radicicol.
Fig. (8).

The coumermycin family of antibiotics.
Fig. (9).

Structures of novobiocin analogues.
Acknowledgements
The authors gratefully acknowledge support of this work by the NIH (CA109265) and The University of Kansas Madison and Lila Self Fellowship program for their generous support of this project.
Footnotes
Brandt, G. E. L.; Schmidt, M. D.; Prisinzano, T. E.; Blagg, B. S. J. J. Med. Chem. 2008, in press.
Bibliography
- 1.Shimamura T, Shapiro GI. J. Thor. Oncol. 2008;3:S152–S159. doi: 10.1097/JTO.0b013e318174ea3a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bishop SC, Burlison JA, Blagg BSJ. Curr. Canc. Drug Targ. 2007;7:369–388. doi: 10.2174/156800907780809778. [DOI] [PubMed] [Google Scholar]
- 3.Powers MV, Workman P. FEBS Lett. 2007;581:3758–3769. doi: 10.1016/j.febslet.2007.05.040. [DOI] [PubMed] [Google Scholar]
- 4.Chaudhury S, Welch TR, Blagg BSJ. ChemMedChem. 2006;1:1331–1340. doi: 10.1002/cmdc.200600112. [DOI] [PubMed] [Google Scholar]
- 5.Gallo KA. J. Chem. Biol. 2006;13:115–116. doi: 10.1016/j.chembiol.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 6.Zhang H, Burrows F. J. Mol. Med. 2004;82:488–499. doi: 10.1007/s00109-004-0549-9. [DOI] [PubMed] [Google Scholar]
- 7.Sreedhar AS, Söti C, Csermely P. Biochim. Biophys. Acta. 2004;1697:233–242. doi: 10.1016/j.bbapap.2003.11.027. [DOI] [PubMed] [Google Scholar]
- 8.Pratt WB, Toft DO. Exp. Biol. Med. 2003;228:111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 9.Meyer P, Prodromou C, Hu B, Vaughan C, Roe SM, Panaretou B, Piper PW, Pearl LH. Molecular Cell. 2003;11:647–658. doi: 10.1016/s1097-2765(03)00065-0. [DOI] [PubMed] [Google Scholar]
- 10.Chiosis G. Expert Opin. Ther. Targets. 2006;10:37–50. doi: 10.1517/14728222.10.1.37. [DOI] [PubMed] [Google Scholar]
- 11.Chiosis G, Vilenchik M, Kim J, Solit D. Drug Discovery Today. 2004;9:881–888. doi: 10.1016/S1359-6446(04)03245-3. [DOI] [PubMed] [Google Scholar]
- 12.Csermely P, Schnaider T, Soiti C, Prohaszka Z, Nardi G. Pharmacol. Ther. 1998;79:129–168. doi: 10.1016/s0163-7258(98)00013-8. [DOI] [PubMed] [Google Scholar]
- 13.Whiteshell L, Lindquist SL. Nat. Rev. Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 14.Adams J, Elliot PJ. Oncogene. 2000;19:6687–6692. doi: 10.1038/sj.onc.1204088. [DOI] [PubMed] [Google Scholar]
- 15.Yufu Y, Nishimura J, Nawata H. Leuk. Res. 1992;16:597–605. doi: 10.1016/0145-2126(92)90008-u. [DOI] [PubMed] [Google Scholar]
- 16.Franzen B, Linder S, Alaiya AA, Eriksson E, Fujioka K, Bergman AC. Electrophoresis. 1997;18:582–587. doi: 10.1002/elps.1150180341. [DOI] [PubMed] [Google Scholar]
- 17.Luparello C, Noel A, Pucci-Minafra I. DNA Cell Biol. 1997;16:1231–1236. doi: 10.1089/dna.1997.16.1231. [DOI] [PubMed] [Google Scholar]
- 18.Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. Cell. 1998;94:471–480. doi: 10.1016/s0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]
- 19.Shen HY, He JC, Wang Y, Huang QY, Chen JF. J. Biol. Chem. 2005;280:39962–39969. doi: 10.1074/jbc.M505524200. [DOI] [PubMed] [Google Scholar]
- 20.Waza M, Adachi H, Katsuno M, Minamiyama M, Sang C, Tanaka F, Inukai A, Doyu M, Sobue G. Nat. Med. 2005;11:1088–1095. doi: 10.1038/nm1298. [DOI] [PubMed] [Google Scholar]
- 21.Kim HR, Kang HS, Kim HD. IUBMB Life. 1999;48:429–433. doi: 10.1080/713803536. [DOI] [PubMed] [Google Scholar]
- 22.Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, Gouras GK. Proc. Natl. Acad. Sci. USA. 2003;100:721–726. doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dickey CA, Erikson J, Kamal A, Burrows F, Kasibhatla S, Eckman CB, Hutton M, Petrucelli L. Curr. Alzheimer Res. 2005;2:231–238. doi: 10.2174/1567205053585927. [DOI] [PubMed] [Google Scholar]
- 24.Hanahan D, Weinberg A. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 25.Workman P, de Billy E. Nature Med. 2007;13:1415–1417. doi: 10.1038/nm1207-1415. [DOI] [PubMed] [Google Scholar]
- 26.Pearl LH, Prodromou C, Workman P. Biochem. J. 2008;410:439–453. doi: 10.1042/BJ20071640. [DOI] [PubMed] [Google Scholar]
- 27.Söti C, Nagy E, Giricz Z, Vigh L, Csermely P, Ferdinandy P. Brit. J. Pharmacol. 2005;146:769–780. doi: 10.1038/sj.bjp.0706396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Welch WJ, Feramisco JR. J. Biol. Chem. 1982;257:14949–14959. [PubMed] [Google Scholar]
- 29.Lindquist S, Craig SE. Annu. Rev. Genet. 1988;22 doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- 30.Frydman J. Annu. Rev. Biochem. 2001;70:603–649. doi: 10.1146/annurev.biochem.70.1.603. [DOI] [PubMed] [Google Scholar]
- 31.Blagg BSJ, Kerr TD. Med. Res. Rev. 2006;26:310–338. doi: 10.1002/med.20052. [DOI] [PubMed] [Google Scholar]
- 32.Wright L, Barril X, Dymock B, Sheridan L, Surgenor A, Beswick M, Drysdale M, Collier A, Massey A, Davies N, Fink A, Fromont C, Aherne W, Boxall K, Sharp S, Workman P, Hubbard RE. Chem. Biol. 2004;11:775–785. doi: 10.1016/j.chembiol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 33.Toft DO. Trends Endocrinol. Metab. 1998;9:238–243. doi: 10.1016/s1043-2760(98)00060-5. [DOI] [PubMed] [Google Scholar]
- 34.Picard D. Hsp90 Interactors. www.picard.ch/downloads/Hsp90interactors.pdf (accessed Jul, 2008)
- 35.Prodromou C, Piper PW, Pearl LH. Proteins: Struct. Funct. Genet. 1996;25:517–522. doi: 10.1002/prot.13. [DOI] [PubMed] [Google Scholar]
- 36.Nemoto TN, Sato N, Iwanari H, Yamashita H, Takagi T. J. Biol. Chem. 1997;272:26179–26187. doi: 10.1074/jbc.272.42.26179. [DOI] [PubMed] [Google Scholar]
- 37.Young JC, Schneider C, Hartl FU. FEBS Lett. 1997;418:139–143. doi: 10.1016/s0014-5793(97)01363-x. [DOI] [PubMed] [Google Scholar]
- 38.Huai Q, Wang H, Liu Y, Kim HY, Toft D, Ke H. Structure. 2005;13:579–590. doi: 10.1016/j.str.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 39.Prodromou C, Panaretou B, Chohan S, Siligardi G, O'Brien R, Ladbury JE, Roe SM, Piper PW, Pearl LH. EMBO. 2000;19 doi: 10.1093/emboj/19.16.4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pearl LH, Prodromou C. Annu. Rev. Biochem. 2006;75:271–294. doi: 10.1146/annurev.biochem.75.103004.142738. [DOI] [PubMed] [Google Scholar]
- 41.Terasawa K, Minami M, Minami Y. J. Biochem. 2005;137:443–447. doi: 10.1093/jb/mvi056. [DOI] [PubMed] [Google Scholar]
- 42.Harris SF, Shiau AK, Agard DA. Structure. 2004;12:1087–1097. doi: 10.1016/j.str.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 43.Ali MU, Roe SM, Vaughan CK, Meyer P, Panaretou B, Piper PW, Prodromou C, Pearl LH. Nature. 2006;440:1013–1017. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Söti C, Radics L, Yahara I, Csermely P. Eur. J. Biochem. 1998;255:611–617. doi: 10.1046/j.1432-1327.1998.2550611.x. [DOI] [PubMed] [Google Scholar]
- 45.Marcu MG, Schulte TW, Neckers L. J. Natl. Canc. Inst. 2000;92:242–248. doi: 10.1093/jnci/92.3.242. [DOI] [PubMed] [Google Scholar]
- 46.Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM. J. Biol. Chem. 2000;275:37181–37186. doi: 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- 47.Dutta R, Inouye M. Trends Biochem. Sci. 2000;25:24–28. doi: 10.1016/s0968-0004(99)01503-0. [DOI] [PubMed] [Google Scholar]
- 48.Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, Pavletich NP. Cell. 1997;89:239–250. doi: 10.1016/s0092-8674(00)80203-2. [DOI] [PubMed] [Google Scholar]
- 49.Prodromou C, Roe SM, Piper PW, Pearl LH. Nat. Struct. Biol. 1997;4:477–482. doi: 10.1038/nsb0697-477. [DOI] [PubMed] [Google Scholar]
- 50.Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, Pearl LH. Cell. 1997;90:65–75. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- 51.Janin YL. J. Med. Chem. 2005;48:7503–7512. doi: 10.1021/jm050759r. [DOI] [PubMed] [Google Scholar]
- 52.Hu Y, Mivechi NF. J. Biol. Chem. 2003;278:17299–17306. doi: 10.1074/jbc.M300788200. [DOI] [PubMed] [Google Scholar]
- 53.Rutherford SL, Lindquist S. Nature. 1998;396:336–342. doi: 10.1038/24550. [DOI] [PubMed] [Google Scholar]
- 54.Carrello A, Ingley E, Minchin RF, Tsai A, Ratajczak T. J. Biol. Chem. 1999;274:2682–2689. doi: 10.1074/jbc.274.5.2682. [DOI] [PubMed] [Google Scholar]
- 55.Knight CA. Science. 2002;296:2348–2349. doi: 10.1126/science.1073846. [DOI] [PubMed] [Google Scholar]
- 56.Kosano H, Stensgard B, Charlesworth MC, McMahon N, Toft D. J. Biol. Chem. 1998;273:32973–32979. doi: 10.1074/jbc.273.49.32973. [DOI] [PubMed] [Google Scholar]
- 57.Prodromou C, Siligardi G, O'Brien R, Woolfson DN, Regan L, Panaretou B, Ladbury JE, Piper PW, Pearl LH. EMBO. 1999;18:754–762. doi: 10.1093/emboj/18.3.754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Forsythe HL, Jarvis JL, Turner JW, Elmore LW, Holt SE. J. Biol. Chem. 2001;276:15571–15574. doi: 10.1074/jbc.C100055200. [DOI] [PubMed] [Google Scholar]
- 59.Chen S, Sullivan WP, Toft DO, Smith DF. Cell Stress Chaperones. 1998;3:118–129. doi: 10.1379/1466-1268(1998)003<0118:diopat>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ratajczak T, Carrello A. J. Biol. Chem. 1996;271:2961–2965. doi: 10.1074/jbc.271.6.2961. [DOI] [PubMed] [Google Scholar]
- 61.Obermann WMJ, Sondermann H, Russo AA, Pavletich N, Hartl FU. J. Cell Biol. 1998;143:901–910. doi: 10.1083/jcb.143.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Panaretou B, Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, Pearl LH. EMBO. 1998;17:4829–4836. doi: 10.1093/emboj/17.16.4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chadli A, Bouhouche I, Sullivan WP, Stensgard B, McMahon N, Catelli M, Toft DO. Proc. Natl. Acad. Sci. USA. 2000;97:12524–12529. doi: 10.1073/pnas.220430297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu LX, Xu JH, Zhang KZ, Lin Q, Huang XW, Wen CX, Chen YZ. Leukemia. 2008;22:1402–1409. doi: 10.1038/leu.2008.89. [DOI] [PubMed] [Google Scholar]
- 65.Eleuteri AM, Cuccioloni M, Bellesi J, Lupidi G, Fioretti E, Angeletti M. Proteins: Struc. Funct. Genet. 2002;48:169–177. doi: 10.1002/prot.10101. [DOI] [PubMed] [Google Scholar]
- 66.Yonehara M, Minami Y, Kawata Y, Nagai J, Yahara I. J. Biol. Chem. 1996;271:2641–2645. doi: 10.1074/jbc.271.5.2641. [DOI] [PubMed] [Google Scholar]
- 67.Galam L, Hadden MK, Ma Z, Ye Q, Yun B, Blagg BSJ, Matts RL. Bioorg. Med. Chem. 2007;15:1939–1946. doi: 10.1016/j.bmc.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hondermarck H, Tastet C, Yazidi-Belkoura IE, Toillon R, Le Bourhis X. J. Prot. Res. 2008;7:1403–1411. doi: 10.1021/pr700870c. [DOI] [PubMed] [Google Scholar]
- 69.Biamonte MA, Shi J, Hurst D, Hong K, Boehm MF, Kasibhatla SR. J. Org. Chem. 2005;70:717–720. doi: 10.1021/jo048522a. [DOI] [PubMed] [Google Scholar]
- 70.Martin CJ, Gaisser S, Challis IR, Carletti I, Wilkinson B, Gregory M, Prodromou C, Roe SM, Pearl LH, Boyd SM, Zhang MQ. J. Med. Chem. 2008;51:2853–2857. doi: 10.1021/jm701558c. [DOI] [PubMed] [Google Scholar]
- 71.Cortajarena AL, Yi F, Regan L. ACS Chem. Biol. 2008;3:161–166. doi: 10.1021/cb700260z. [DOI] [PubMed] [Google Scholar]
- 72.Chiosis G, Huezo H, Rosen N, Mimnaugh E, Whiteshell L, Neckers L. Mol. Cancer Ther. 2003;2:123–129. [PubMed] [Google Scholar]
- 73.Xu W, Neckers L. Clin. Cancer Res. 2007;13:1625–1629. doi: 10.1158/1078-0432.CCR-06-2966. [DOI] [PubMed] [Google Scholar]
- 74.Jolly C, Morimoto RI. J. Natl. Cancer Inst. 2000;92:1564–1572. doi: 10.1093/jnci/92.19.1564. [DOI] [PubMed] [Google Scholar]
- 75.Neckers L. Trends Mol. Med. 2002;8:S55–S61. doi: 10.1016/s1471-4914(02)02316-x. [DOI] [PubMed] [Google Scholar]
- 76.Chiosis G, Timaul MN, Lucas B, Munster PN, Zheng FF, Sepp-Lorenzino L, Rosen N. Chem. Biol. 2001;8:289–299. doi: 10.1016/s1074-5521(01)00015-1. [DOI] [PubMed] [Google Scholar]
- 77.Schnur RC, Corman ML, Gallaschun RJ, Cooper BA, Dee MF, Doty JL, Muzzi ML, DiOrio CI, Barbacci EG, Miller PE, Pollack VA, Savage DM, Sloan DE, Pustilnik LR, Moyer JD, Moyer MP. J. Med. Chem. 1995;38:3813–3820. doi: 10.1021/jm00019a011. [DOI] [PubMed] [Google Scholar]
- 78.Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows F. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 79.Le Brazidec J, Kamal A, Busch D, Thao L, Zhang L, Timony G, Grecko R, Trent K, Lough R, Salazar T, Khan S, Burrows F, Boehm MF. J. Med. Chem. 2004;47:3865–3873. doi: 10.1021/jm0306125. [DOI] [PubMed] [Google Scholar]
- 80.Duvvuri M, Konkar S, Hong KH, Blagg BSJ, Krise J. ACS Chem. Biol. 2006;1:309–315. doi: 10.1021/cb6001202. [DOI] [PubMed] [Google Scholar]
- 81.de Duve C, de Barsy T, Poole B, Trouet A, Tulkens P, Van Hoof F. Biochem. Pharmacol. 1974;23:2495–2531. doi: 10.1016/0006-2952(74)90174-9. [DOI] [PubMed] [Google Scholar]
- 82.Altan N, Chen Y, Schindler M, Simon SM. J. Exp. Med. 1998;187:1583–1598. doi: 10.1084/jem.187.10.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kokkonen N, Rivinoja A, Kauppila A, Suokas M, Kellokumpu I, Kellokumpu S. J. Biol. Chem. 2004;279:39982–39988. doi: 10.1074/jbc.M406698200. [DOI] [PubMed] [Google Scholar]
- 84.Muchowski PJ, Wacker JL. Nature Rev. Neuro. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- 85.Ansar S, Burlison JA, Hadden MK, Yu XM, Desino KE, Bean J, Neckers L, Audus KL, Michaelis ML, Blagg BSJ. Bioorg. Med. Chem. Lett. 2007;17:1984–1990. doi: 10.1016/j.bmcl.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 86.Sausville EA. Curr. Cancer Drug Targets. 2003;3:377–383. doi: 10.2174/1568009033481831. [DOI] [PubMed] [Google Scholar]
- 87.Banerji U. Proc. Am. Assoc. Cancer Res. 2003;44:677. [Google Scholar]
- 88.Driggers EM, Hale SP, Lee J, Terrett NK. Nature Rev. Drug Disc. 2008;7:608–624. doi: 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]
- 89.DeBoer C, Meulman PA, Wnuk RJ, Peterson DH. J. Antibiot. (Tokyo) 1970;23:442–447. doi: 10.7164/antibiotics.23.442. [DOI] [PubMed] [Google Scholar]
- 90.Omura S, Iwai Y, Takahashi Y, Sadakane N, Nakagawa A. J. Antibiot. 1979;32:255–261. doi: 10.7164/antibiotics.32.255. [DOI] [PubMed] [Google Scholar]
- 91.Nakata M, Osumi T, Ueno A, Kimura T, Tamai T, Tatsuta K. Tetrahedron Lett. 1991;32:6015–6018. [Google Scholar]
- 92.Andrus MB, Meredith EL, Simmons BL, Soma Sekhar BBV, Hicken EJ. Org. Lett. 2002;4:3549–3552. doi: 10.1021/ol0267432. [DOI] [PubMed] [Google Scholar]
- 93.Andrus MB, Hicken EJ, Meredith EL, Simmons BL, Cannon JF. Org. Lett. 2003;5:3859–3862. doi: 10.1021/ol035400g. [DOI] [PubMed] [Google Scholar]
- 94.Qin H, Panek JS. Org. Lett. 2008;10:2477–2479. doi: 10.1021/ol800749w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Uehara Y, Hori M, Takeuchi T, Umezawa H. Mol. Cell. Biol. 1986;6:2198–2206. doi: 10.1128/mcb.6.6.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jove R, Hanafusa H. Ann. Rev. Cell Biol. 1987;3:31–56. doi: 10.1146/annurev.cb.03.110187.000335. [DOI] [PubMed] [Google Scholar]
- 97.Whiteshell L, Shifrin SD, Schwab G, Neckers LM. Cancer Res. 1992;52:1721–1728. [PubMed] [Google Scholar]
- 98.Whiteshell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Proc. Natl. Acad. Sci. USA. 1994;91:8323–8328. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Neckers L, Schulte TW, Mimnaugh EG. Invest. New Drugs. 1999;17:361–373. doi: 10.1023/a:1006382320697. [DOI] [PubMed] [Google Scholar]
- 100.Roe SM, Prodromou C, O'Brien R, Ladbury JE, Piper PW, Pearl LH. J. Med. Chem. 1999;42:260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- 101.Supko JG, Hickman RL, Grever MR, Malspeis L. Cancer Chemother. Pharmacol. 1995;36:305–315. doi: 10.1007/BF00689048. [DOI] [PubMed] [Google Scholar]
- 102.An WG, Schnur RC, Neckers L, Blagosklonny MV. Cancer Chemother. Pharmacol. 1997;40:60–64. doi: 10.1007/s002800050626. [DOI] [PubMed] [Google Scholar]
- 103.Dikalov S, Rumyantseva GV, Piskunov AV, Weiner LM. Biochemistry. 1992;31:8947–8953. doi: 10.1021/bi00152a034. [DOI] [PubMed] [Google Scholar]
- 104.Dikalov S, Landmesser U, Harrison DG. J. Biol. Chem. 2002;277:25480–25485. doi: 10.1074/jbc.M203271200. [DOI] [PubMed] [Google Scholar]
- 105.Schulte TW, Neckers LM. Cancer Chemother. Pharmacol. 1998;42:273–279. doi: 10.1007/s002800050817. [DOI] [PubMed] [Google Scholar]
- 106.Jez JM, Chen C, Rastelli G, Stroud RM, Santi DV. Chem. Biol. 2003;10:361–368. doi: 10.1016/s1074-5521(03)00075-9. [DOI] [PubMed] [Google Scholar]
- 107.Egorin MJ, Lagattuta TF, Hamburger DR, Covey JM, White KD, Musser SM, Eiseman JL. Cancer Chemother. Pharmacol. 2002;49:7–19. doi: 10.1007/s00280-001-0380-8. [DOI] [PubMed] [Google Scholar]
- 108.Wright MH, Calcagno AM, Salcido CD, Carlson MD, Ambudkar SV, Varticovski L. Breast Cancer Res. 2008;10:R10. doi: 10.1186/bcr1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Patel K, Piagentini M, Rascher A, Tian Z, Buchanan GO, Regentin R, Hu Z, Hutchinson CR, McDaniel R. Chem. Biol. 2004;11:1625–1633. doi: 10.1016/j.chembiol.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 110.Lee K, Ryu JS, Jin Y, Kim W, Kaur N, Chung SJ, Jeon Y, Park J, Bang JS, Lee HS, Kim TY, Lee JJ, Hong Y. Org. Biomol. Chem. 2008;6:340–348. doi: 10.1039/b713407j. [DOI] [PubMed] [Google Scholar]
- 111.Tadtong S, Meksurlyen D, Tanasupawat S, Isobe M, Suwanborirux K. Bioorg. Med. Chem. Lett. 2007;17:2939–2943. doi: 10.1016/j.bmcl.2006.12.041. [DOI] [PubMed] [Google Scholar]
- 112.Maroney AC, Marugan JJ, Mezzasalma TM, Barnakov AN, Garrabrant TA, Weaner LE, Jones WJ, Barnakova LA, Koblish HK, Todd MJ, Masucci JA, Deckman IC, Galemmo RA, Jr., Johnson DL. Biochemistry. 2006;45:5678–5686. doi: 10.1021/bi0524969. [DOI] [PubMed] [Google Scholar]
- 113.Cutler HG, Arrendale RF, Springer JP, Cole PD, Roberts RG, Hanlin RT. Agric. Biol. Chem. 1987;51:3331–3338. [Google Scholar]
- 114.Yamamoto K, Garbaccio RM, Stachel SJ, Solit DB, Chiosis G, Rosen N, Danishefsky SJ. Angew. Chem. Int. Ed. 2003;42:371–376. doi: 10.1002/anie.200390329. [DOI] [PubMed] [Google Scholar]
- 115.Geng X, Yang Z, Danishefsky SJ. Synlett. 2004;8:1325–1333. [Google Scholar]
- 116.Soga S, Neckers LM, Schulte TW, Shiotsu Y, Akasaka K, Narumi H, Agatsuma T, Ikuina Y, Murakata C, Tamaoki T, Akinaga S. Cancer Res. 1999;59:2931–2938. [PubMed] [Google Scholar]
- 117.Agatsuma T, Ogawa H, Akasaka K, Asai A, Yamashita Y, Mizukami T, Akinaga S, Saitoh Y. Bioorg. Med. Chem. 2002;10:3445–3454. doi: 10.1016/s0968-0896(02)00260-2. [DOI] [PubMed] [Google Scholar]
- 118.Ikuina Y, Amishiro N, Miyata M, Narumi H, Ogawa H, Akiyama T, Shiotsu Y, Akinaga S, Murakata C. J. Med. Chem. 2003;46:2534–2541. doi: 10.1021/jm030110r. [DOI] [PubMed] [Google Scholar]
- 119.Kitamura Y, Nara S, Nakagawa A, Nakatsu R, Nakashima T, Soga S, Kajita S, Shiotsu Y, Kanda Y. Japan Patent WO2005063222 2005
- 120.Drysdale MJ, Dymock BW, Barril-Alonso X. England Patent WO2005034950 2005
- 121.Dymock BW, Drysdale MJ, Fromont C, Jordan A. England Patent WO2005021552 2005
- 122.Soga S, Sharma SV, Shiotsu Y, Shimizu M, Tahara H, Yamaguchi K, Ikuina Y, Murakata C, Tamaoki T, Kurebayashi J, Schulte TW, Neckers LM, Akinaga S. Cancer Chemother. Pharmacol. 2001;48:435–445. doi: 10.1007/s002800100373. [DOI] [PubMed] [Google Scholar]
- 123.Turbyville TJ, Wijeratne EMK, Liu MX, Burns AM, Seliga CJ, Luevano LA, David CL, Faeth SH, Whiteshell L, Gunatilaka AAL. J. Nat. Prod. 2006;69:178–184. doi: 10.1021/np058095b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yang Z, Geng X, Solit D, Pratilas CA, Rosen N, Danishefsky SJ. J. Am. Chem. Soc. 2004;126:7881–7889. doi: 10.1021/ja0484348. [DOI] [PubMed] [Google Scholar]
- 125.Hellwig V, Mayer-Bartschmid A, Müller H, Greif G, Kleymann G, Zitzmann W, Tichy H, Stadler M. J. Nat. Prod. 2003;66:829–837. doi: 10.1021/np020556v. [DOI] [PubMed] [Google Scholar]
- 126.Moulin E, Zoete V, Barluenga S, Karplus M, Winssinger N. J. Am. Chem. Soc. 2005;127:6999–7004. doi: 10.1021/ja043101w. [DOI] [PubMed] [Google Scholar]
- 127.Barluenga S, Moulin E, Lopez P, Winssinger N. Chem. Eur. J. 2005;11:4935–4952. doi: 10.1002/chem.200500160. [DOI] [PubMed] [Google Scholar]
- 128.Moulin E, Barluenga S, Winssinger N. Org. Lett. 2005;7:5637–5639. doi: 10.1021/ol052263+. [DOI] [PubMed] [Google Scholar]
- 129.Schulte TW, Akinaga S, Soga S, Sullivan W, Stensgard B, Toft D, Neckers LM. Cell Stress Chaperones. 1998;3:100–108. doi: 10.1379/1466-1268(1998)003<0100:arbttn>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Logan IR, Gaughan L, McCracken SRC, Sapountzi V, Leung HY, Robson CN. Mol. Cell Biol. 2006;26:6502–6510. doi: 10.1128/MCB.00147-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Clevenger RC, Blagg BSJ. Org. Lett. 2004;2004:4459–4462. doi: 10.1021/ol048266o. [DOI] [PubMed] [Google Scholar]
- 132.Shen G, Blagg BSJ. Org. Lett. 2005;7 doi: 10.1021/ol050580a. [DOI] [PubMed] [Google Scholar]
- 133.Wang M, Shen G, Blagg BSJ. Bioorg. Med. Chem. Lett. 2006;16:2459–2462. doi: 10.1016/j.bmcl.2006.01.086. [DOI] [PubMed] [Google Scholar]
- 134.Hadden MK, Lubbers DJ, Blagg BSJ. Curr. Top. Med. Chem. 2006;6:1173–1182. doi: 10.2174/156802606777812031. [DOI] [PubMed] [Google Scholar]
- 135.Hooper DC, Wolfson JS, McHugh GL, Winters MB, Swartz MN. Antimicrob. Agents Chemother. 1982;22:662–671. doi: 10.1128/aac.22.4.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tanitame A, Oyamada Y, Ofuji K, Fujimoto M, Suzuki K, Ueda T, Terauchi H, Kawasaki M, Nagai K, Wachi M, Yamagishi J. Bioorg. Med. Chem. 2004;12:5515–5524. doi: 10.1016/j.bmc.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 137.Laurin P, Ferroud D, Schio L, Klich M, Dupuis-Hamelin C, Mauvais P, Lassaigne P, Bonnefoy A, Musicki B. Bioorg. Med. Chem. Lett. 1999;9:2875–2880. doi: 10.1016/s0960-894x(99)00492-8. [DOI] [PubMed] [Google Scholar]
- 138.Ali JA, Jackson AP, Howells AJ, Maxwell A. Biochemistry. 1993;32:2717–2724. doi: 10.1021/bi00061a033. [DOI] [PubMed] [Google Scholar]
- 139.Burlison JA, Neckers L, Smith AB, Maxwell A, Blagg BSJ. J. Am. Chem. Soc. 2006;128:15529–15536. doi: 10.1021/ja065793p. [DOI] [PubMed] [Google Scholar]
- 140.Yu XM, Shen G, Neckers L, Blake H, Holzbeierlein J, Cronk B, Blagg BSJ. J. Am. Chem. Soc. 2005;127:12778–12779. doi: 10.1021/ja0535864. [DOI] [PubMed] [Google Scholar]
- 141.Burlison JA, Blagg BSJ. Org. Lett. 2006;8:4855–4858. doi: 10.1021/ol061918j. [DOI] [PubMed] [Google Scholar]
- 142.Huang Y, Blagg BSJ. J. Org. Chem. 2007;72:3609–3613. doi: 10.1021/jo062083t. [DOI] [PubMed] [Google Scholar]
- 143.Burlison JA, Avila C, Vielhauer G, Lubbers DJ, Holzbeierlein J, Blagg BSJ. J. Org. Chem. 2008;73:2130–2137. doi: 10.1021/jo702191a. [DOI] [PubMed] [Google Scholar]
- 144.Palermo CM, Hernando JIM, Dertinger SD, Kende AS, Gasiewicz TA. Chem. Res. Toxicol. 2003;16:865–872. doi: 10.1021/tx025672c. [DOI] [PubMed] [Google Scholar]
- 145.Palermo CM, Westlake CA, Gasiewicz TA. Biochemistry. 2005;44:5041–5052. doi: 10.1021/bi047433p. [DOI] [PubMed] [Google Scholar]
- 146.Beltz LA, Bayer DK, Moss AL, Simet IM. Anti-Cancer Agents in Med. Chem. 2006;6:389–406. doi: 10.2174/187152006778226468. [DOI] [PubMed] [Google Scholar]
- 147.Weinreb O, Amit T, Youdim MBH. Free Rad. Biol. Med. 2007;43:546–556. doi: 10.1016/j.freeradbiomed.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 148.Schiff PB, Horwitz SB. Proc. Natl. Acad. Sci. USA. 1980;77:1561–1565. doi: 10.1073/pnas.77.3.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ding AH, Porteu F, Sanchez E, Nathan CF. Science. 1990;248:370–372. doi: 10.1126/science.1970196. [DOI] [PubMed] [Google Scholar]
- 150.Byrd CA, Bornmann W, Erdjument-Bromage H, Tempst P, Pavletich N, Rosen N, Nathan CF, Ding A. Proc. Natl. Acad. Sci. USA. 1999;96:5645–5650. doi: 10.1073/pnas.96.10.5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wani MC, Taylor HL, Wall ME, Coggon P, McPhail AT. J. Am. Chem. Soc. 1971;93:2325–2327. doi: 10.1021/ja00738a045. [DOI] [PubMed] [Google Scholar]
- 152.Kingston DGI. Pharmac. Ther. 1991;52:1–34. doi: 10.1016/0163-7258(91)90085-z. [DOI] [PubMed] [Google Scholar]
- 153.Basso AD, Solit D, Chiosis G, Giri B, Tsichlis P, Rosen N. J. Biol. Chem. 2002;277:39858–39866. doi: 10.1074/jbc.M206322200. [DOI] [PubMed] [Google Scholar]
- 154.Solit DB, Basso AD, Olshen AB, Scher HI, Rosen N. Cancer. Res. 2003;63:2139–2144. [PubMed] [Google Scholar]
- 155.Sawai A, Chandarlapaty S, Greulich H, Gonen M, Ye Q, Arteaga CL, Sellers W, Rosen N, Solit D. Cancer Res. 2008;68:589–596. doi: 10.1158/0008-5472.CAN-07-1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.East AJ, Ollis WD, Wheeler RE. J. Chem. Soc. 1969;C:365–373. [Google Scholar]
- 157.Jain AC, Jain SM. Tetrahedron. 1972;28:5063–5067. [Google Scholar]
- 158.Hastings JM, Hadden MK, Blagg BSJ. J. Org. Chem. 2008;73:369–373. doi: 10.1021/jo702366g. [DOI] [PubMed] [Google Scholar]
- 159.Hossain MM, Kawamura Y, Yamashita K, Tsukayama M. Tetrahedron. 2006;62:8625–8635. [Google Scholar]
- 160.Hadden MK, Galam L, Gestwicki JE, Matts RL, Blagg BSJ. J. Nat. Prod. 2007;70:2014–2018. doi: 10.1021/np070190s. [DOI] [PubMed] [Google Scholar]
- 161.Khalid SA, Duddeck H, Gonzalez-Sierra M. J. Nat. Prod. 1989;52:922–927. doi: 10.1021/np50065a002. [DOI] [PubMed] [Google Scholar]
- 162.Zhang T, Hamza A, Cao X, Wang B, Yu S, Zhan C, Sun D. Mol. Canc. Ther. 2008;7:162–170. doi: 10.1158/1535-7163.MCT-07-0484. [DOI] [PubMed] [Google Scholar]
- 163.Westerheide SD, Bosman JD, Mbadugha BNA, Kawahara TLA, Matsumoto G, Kim S, Gu W, Devlin JP, Silverman RB, Morimoto RI. J. Biol. Chem. 2004;279:56053–56060. doi: 10.1074/jbc.M409267200. [DOI] [PubMed] [Google Scholar]
- 164.Uddin SJ, Nahar L, Shilpi JA, Shoeb M, Borkowski T, Gibbons S, Middleton M, Byres M, Sarker S. Phytother. Res. 2007;21:757–761. doi: 10.1002/ptr.2159. [DOI] [PubMed] [Google Scholar]
- 165.Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A, Nieto M, Du J, Stegmaier K, Raj SM, Maloney KN, Clardy J, Hahn WC, Chiosis G, Golub TR. Cancer Cell. 2006;10:321–330. doi: 10.1016/j.ccr.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 166.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet J, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, Wei R, Carr SA, Lander ES, Golub TR. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 167.Vinson-Hieronymous H, Golub TR, Lamb J, Stegmaier K. United States Patent WO2007117466 2007