

Abstract
Okazaki fragments are initiated by short RNA/DNA primers, which are displaced into flap intermediates for processing. Flap endonuclease 1 (FEN1) and Dna2 are responsible for flap cleavage. Replication protein A (RPA)-bound flaps inhibit cleavage by FEN1 but stimulate Dna2, requiring that Dna2 cleaves prior to FEN1. Upon cleavage, Dna2 leaves a short flap, which is then cut by FEN1 forming a nick for ligation. Both enzymes require a flap with a free 5′-end for tracking to the cleavage sites. Previously, we demonstrated that FEN1 disengages the tracking mechanism of Dna2 to remove it from the flap. To determine why the disengagement mechanism evolved, we measured FEN1 dissociation of Dna2 on short RNA and DNA flaps, which occur during flap processing. Dna2 tracked onto these flaps but could not cleave, presenting a block to FEN1 entry. However, FEN1 disengaged these nonproductively bound Dna2 molecules, proceeding on to conduct proper cleavage. These results clarify the importance of disengagement. Additional results showed that flap substrate recognition and tracking by FEN1, as occur during fragment processing, are required for effective displacement of the flap-bound Dna2. Dna2 was recently shown to dissociate flap-bound RPA, independent of cleavage. Using a nuclease-defective Dna2 mutant, we reconstituted the sequential dissociation reactions in the proposed RPA/Dna2/FEN1 pathway showing that, even without cutting, Dna2 enables FEN1 to cleave RPA-coated flaps. In summary, RPA, Dna2, and FEN1 have evolved highly coordinated binding properties enabling one protein to succeed the next for proper and efficient Okazaki flap processing.
During eukaryotic DNA replication, synthesis of the leading strand occurs in continuous fashion in the direction of DNA unwinding. In contrast, the lagging strand is replicated in a discontinuous fashion via short Okazaki fragments. Each Okazaki fragment is between 100 and 150 nucleotides (nt)3 in length. In Saccharomyces cerevisiae, ∼100,000 fragments are created per replication cycle. These fragments are initiated by the DNA polymerase (pol) α-primase complex, which synthesizes 10–12 nt of RNA followed by 20–30 nt of DNA (1, 2). The pol α-primase complex is then displaced by the clamp loader, replication factor C. The toroidal sliding DNA clamp, proliferating cell nuclear antigen, and DNA pol δ are then loaded onto the DNA.
pol δ then synthesizes DNA until it encounters the downstream Okazaki fragment. The downstream RNA/DNA primer is then displaced into a flap intermediate by pol δ. The flap must then be removed and the fragments joined to avoid genome instability (3, 4). Removal of the primer is proposed to occur by at least two parallel-acting pathways in S. cerevisiae (2).
In one pathway, flap endonuclease 1 (FEN1) cleaves the flap intermediate to create a nicked product for ligation (5, 6). FEN1 is a structure-specific, single-stranded nuclease that recognizes and cleaves at the base of a flap structure on both DNA and RNA (7). The FEN1-only model suggests that strand displacement synthesis by pol δ produces short flaps, which are successively cleaved by FEN1 until the primer has been removed. DNA ligase I then joins the resultant nicked product.
Another model of primer removal involves both FEN1 and the nuclease/helicase Dna2 (8). Dna2 possesses both single-stranded DNA (ssDNA) nuclease and 5′ to 3′ ATP-dependent helicase activities (9, 10). It is functionally conserved from yeast to humans (11–14). Originally identified in a screen for DNA replication mutants, S. cerevisiae Dna2 has also been shown to play a role in telomere processing and DNA repair (15–21). Recently, it was identified as a major nuclease for resection of double strand breaks, in both S. cerevisiae and Xenopus laevis (22, 23).
In S. cerevisiae, Dna2 was shown to physically interact with FEN1 (24). Also, the overexpression of FEN1 rescued the temperature-sensitive phenotype of the dna2-1 nuclease-impaired mutant, and overexpression of Dna2 rescued the temperature-sensitive rad27Δ (FEN1-null) strain. Moreover, Dna2 interacts with the single-stranded binding protein, replication protein A (RPA), which is involved in both DNA replication and repair (25). RPA stimulates flap cleavage by Dna2, while repressing cleavage by FEN1. Based on these findings, Seo and co-workers proposed that pol δ displaces flaps that become long enough to be coated by RPA (8). Once RPA is bound, Dna2 cleavage is required, because FEN1 is inhibited. After cleavage by Dna2, the shortened flap is then free of RPA but must be further processed because Dna2, unlike FEN1, cannot cut at the base of the flap. Instead it leaves a short flap of ∼5 nt (8, 26), which is removed by FEN1 to create a nick for ligation.
The FEN1-only model is consistent with results obtained from the reconstitution of Okazaki fragment processing with S. cerevisiae proteins. These results showed that the coordination between pol δ and FEN1 is highly efficient, resulting in mostly mononucleotide cleavage products and the production of nicked replication intermediates for ligation (6). Later, however, we showed that although mostly short flaps were created during strand displacement by pol δ, a minor subset of longer flaps arose (27). This subset reached a length at which RPA could stably bind, suggesting a role for Dna2 in processing at least some flaps.
Relevant to this issue, Budd et al. (15) showed that the elimination of Pif1, a 5′ to 3′ helicase, rescued the lethality of the dna2Δ strain in S. cerevisiae. Cell growth was even more robust when both Pif1 and Pol32, the nonessential subunits of pol δ, were simultaneously deleted in the dna2Δ strain. Significantly, the pol δ mutant lacking Pol32 exhibits decreased strand displacement activity (28). Furthermore, we have recently shown that the addition of Pif1 in reconstituted Okazaki fragment processing augmented the subset of longer flaps that escaped FEN1 cleavage and were bound by RPA (29). These results suggest that Pif1 aids pol δ strand displacement in creating long flap substrates that require Dna2 nuclease function. Although the FEN1-only pathway is likely to be the dominant mechanism of flap removal, employment of both pathways appears to be critical to process and join all Okazaki fragments.
A characteristic feature of both FEN1 and Dna2 is that they must enter a free flap 5′-end for substrate cleavage. If a double-stranded region or a streptavidin-biotin conjugate is used to block the 5′-end of the flap, then cleavage is inhibited (30, 31). Because tracking is required for cleavage, we previously tested whether a bound nuclease-defective mutant of Dna2, E675A, inhibited FEN1 cleavage (32). We were surprised to find that cleavage was not inhibited and discovered that FEN1 disengages the tracking mechanism of Dna2 to allow dissociation. FEN1 also dissociated Dna2 from RNA flaps, which Dna2 cannot cleave. Furthermore, we recently demonstrated the ability of Dna2 to dissociate flap-bound RPA (33). These findings suggest a sequential dissociation of RPA by Dna2 followed by the dissociation of Dna2 by FEN1.
Here we are investigating the significance of the FEN1 disengagement of Dna2 on relevant substrates of the proposed RPA/Dna2/FEN1 pathway. Dna2 binds but cannot cleave RNA (32, 34). Additionally, cleavage by Dna2 produces short ∼5-nt flaps, which Dna2 cannot cleave. In this study, we analyzed these substrates for Dna2 binding and FEN1 dissociation of flap-bound Dna2. We also probed the role of tracking and flap structure for disengagement of Dna2 by FEN1. Finally, we tested the proposed sequential dissociation reactions by reconstituting the RPA/Dna2/FEN1 pathway with the nuclease-defective Dna2 E675A.
EXPERIMENTAL PROCEDURES
Materials—Synthetic oligonucleotides, including ones with biotin modifications, were synthesized by Integrated DNA Technologies. Radioactive [α-32P]dCTP and [γ-32P]ATP were acquired from PerkinElmer Life Sciences. Both the polynucleotide kinase and the Klenow fragment of Escherichia coli DNA polymerase I, used for labeling, were purchased from Roche Applied Sciences. All other reagents were the best available commercial grade.
Oligonucleotides—Primers used in this study are listed in Table 1. 32P was incorporated at either the 5′- or 3′-end of the downstream primers for visualization as described (32). For 5′-end labeling [γ-32P]ATP was incorporated using polynucleotide kinase, and for 3′-end labeling [α-32P]dCTP was added by the Klenow fragment. Substrates were then PAGE-purified and resuspended in 1× TE. Radiolabeled primers were then annealed in a 1:2:4 ratio of downstream primer to template to upstream primer to create a flap substrate. Substrates containing RNA included Protector RNase (Roche Applied Science) during substrate purification and annealing to prevent degradation.
TABLE 1.
Oligonucleotides
| Primer | Length (nt) | Sequence | ||
|---|---|---|---|---|
| Downstreama,b,c(5′-3′) | ||||
| D1 | 23 | GCC GU C CAC CCG U CC ACC CGA CG | ||
| D2 | 28 | GCC GTC GTT TTA CAA CGA CGT GAC TGG G | ||
| D3 | 53 | TTC ACG CCT GTT AGT TAA TTC ACT GGC CGT CGT TTT ACA ACG ACG TGA CTG GG | ||
| D4 | 76 | GTA CCG AGC TCG AAT TCG CCC GTT TCA CGC CTG TTA GTT AAT TCA CTG GCC GTC GTT TTA CAA CGA CGT GAC TGG G | ||
| D5 | 76 | GTA CCG AGC TCG AAT TCG CCC GTT TCA CGC CTG TTA GTT AAT TCA CTG GCC GTC GTT TTA CAA CGA CGT GAC TGG G | ||
| D6 | 50 | GTA CCG AGC TCG AAT TCG CCC GTT TCA CGC CTG TTA GTT AAT TCA CTG GC | ||
| D7 | 46 | CAC TGG CCG TCG TTT TAC GGA CCC GTC CAC CCG ACG CCA CCT CCT G | ||
| Upstream (5′-3′) | ||||
| U1 | 26 | CGA CCG TGC CAG CCT AAA TTT CAA GA | ||
| U2 | 26 | CGC CAG GGT TTT CCC AGT CAC GAC CA | ||
| U3 | 26 | CGA CCG TGC CAG CCT AAA TTT CAA TA | ||
| Template (3′-5′) | ||||
| T1 | 44 | GCT GGC ACG GTC GGA TTT AAA GTT CGG TGG GCA GGT GGG CTG CG | ||
| T2 | 49 | GCG GTC CCA AAA GGG TCA GTG CTG GGC AAA ATG TTG CTG CAC TGA CCC G | ||
| T3 | 51 | GCT GGC ACG GTC GGA TTT AAA GTT AGG GCA GGT GGG CTG CGG TGG AGG ACG | ||
Nucleotides shown in boldface type are biotinylated.
RNA segment is shown in italic type.
Underlined nucleotide indicates the last annealed nucleotide.
Protein Purification—Wild-type and E675A Dna2 proteins from S. cerevisiae were overexpressed in baculovirus High Five cells and purified as described (35). Dna2 E675A was created using site-directed mutagenesis as described (35). S. cerevisiae FEN1 (36) and RPA (37) were overexpressed in E. coli and then purified as described.
Surface Plasmon Resonance—Association and dissociation of wild-type Dna2 with a single-stranded segment of DNA was analyzed using a Reichert SR7000 dual channel instrument (Reichert Inc., Depew, NY). A mixture of N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide/N-hydroxysuccinimide was used to activate the dithiol carboxyl surface of the sensor chip as described (38). Approximately 900 micro-refractive index units of Dna2 were then immobilized over one channel, whereas the other served as a control to detect nonspecific binding, refractive index changes, and instrument drift. Following Dna2 immobilization, the chip was inactivated by flowing 1 m ethanolamine, pH 8.5, over both chambers. The running buffer then consisted of 30 mm HEPES, pH 7.5, 0.1 mg/ml bovine serum albumin, 40 mm KCl, 2 mm CaCl2, 50 μm ATP, and 0.05% Tween 20. For association, ssDNA (D4) was run over the immobilized Dna2 at a flow rate of 50 μl/min for 3 min. For dissociation, the reaction buffer only was run over the chip for 3 min at the same flow rate of 50 μl/min. After each run the chip was regenerated for 2 min with 1 m NaCl in the running buffer to remove the bound DNA. Regeneration was verified by a return to the base line established prior to each run. The resulting data were then analyzed using Scrubber 2 software (Biologic Software Pty. Ltd.).
Gel Shift Assay—Reactions contained 5 fmol of radiolabeled substrate and various amounts of Dna2 and/or FEN1, as indicated. The reaction buffer contained 50 mm Tris-HCl, pH 8.0, 2 mm dithiothreitol, 30 mm NaCl, 0.1 mg/ml bovine serum albumin, 5% glycerol, and 50 μm ATP. Dna2 was pre-bound to the substrate for 5 min at room temperature prior to the addition of FEN1, which was then incubated with the reaction for 5 min at room temperature. When streptavidin was added, it was incubated with the substrate for 10 min prior to the addition of protein, unless otherwise indicated. Reactions were then loaded onto a 5% polyacrylamide gel and subjected to electrophoresis at 150 V for 30–40 min.
Nuclease Assays—Samples contained 5 fmol of radiolabeled substrate and various amounts of protein, as stated in the figure legends. The reaction buffer contained 50 mm Tris-HCl, pH 8.0, 2 mm dithiothreitol, 30 mm NaCl, 0.1 mg/ml bovine serum albumin, 5% glycerol, 50 μm ATP, and 2 mm MgCl2. In Fig. 1A, Dna2 was bound to the radiolabeled substrate for 5 min at room temperature. Unlabeled substrate (1 pmol) was then added at time 0. At each time point, MgCl2 was added, to a final concentration of 2 mm, to initiate the reaction. Reactions were then incubated at 37 °C for 10 min. In Fig. 5, RPA, Dna2 E675A, and FEN1 were mixed followed by the addition of the flap substrate. The reactions were then incubated at 37 °C for 10 min. After incubation, all reactions were then stopped by the addition of 2× termination dye, consisting of 90% formamide (v/v), 10 mm EDTA, 0.01% bromphenol blue, and 0.01% xylene cyanole. Reactions were then incubated at 95 °C for 5 min and loaded onto a 15% polyacrylamide gel, containing 7 m urea and subjected to electrophoresis at 80 watts for 1–1.5 h.
FIGURE 1.

Slow dissociation of Dna2 from DNA substrates. A, Dna2 (200 fmol) and 5 fmol of a radiolabeled 53-nt flap substrate (D4:U2:T2) were incubated followed by the addition of 200-fold excess unlabeled flap substrate (D4:U2:T2). MgCl2 was then added at the indicated time points. Dna2 cleavage was then measured by denaturing PAGE. Lane 1 is the substrate alone. Lane 2 is Dna2 with labeled and unlabeled substrate without MgCl2. In lane 12, the labeled and excess unlabeled substrates were mixed prior to the addition of Dna2. B, graphical analysis of A. Points were fit to a single (gray line) or double (black line) exponential decay curve using nonlinear least squares regression. Cleavage is defined as (cleaved/(cleaved + uncleaved)) × 100 and values were then normalized to cleavage at time zero. For the double exponential decay curve, the dissociation amplitude described by the first curve was 21% and the second 78%. C, surface plasmon resonance was used to measure the affinity between Dna2 and ssDNA (D4). Dna2 was immobilized, and increasing amounts of substrate (62.5, 125, 250, and 500 nm) were flowed over the chip. Measurements at each concentration were repeated twice. After 3 min, the flow of ssDNA was discontinued, and buffer alone was flowed over the chip to measure dissociation.
FIGURE 5.

Dna2 E675A overcomes RPA inhibition of FEN1. RPA (200 fmol), FEN1 (0.25 fmol), and Dna2 E675A (10, 20, 100, and 200 fmol) were mixed followed by the addition of a 21-nt flap substrate (D7:U3:T3) (lanes 8–11). Denaturing PAGE was then used to separate the products. Lane 1 is the substrate alone control. Lanes 2–4 are substrate with Dna2 E675A (200 fmol), substrate with FEN1 (0.25 fmol), and substrate with RPA (200 fmol), respectively. Lanes 5–7 are substrate with Dna2 E675A (200 fmol) and FEN1 (0.25 fmol), substrate with RPA (200 fmol) and FEN1 (0.25 fmol), and substrate with Dna2 E675A (200 fmol) and RPA (200 fmol), respectively. B, graphical analysis of A. Each bar of the graph represents the conditions shown in the corresponding lane in A. The bars are an average of four independent experiments, and error bars represent the S.D. The substrate is depicted above the gel in A, and the asterisk indicates the site of the 3′-32P radiolabel.
Gel Analysis—At least two independent experiments were performed for each figure, and representative gels are shown. After electrophoresis, the gels were placed on filter paper and dried on a gel dryer (Bio-Rad) with vacuum (Savant). Dried gels were then exposed to a phosphor screen, visualized by phosphorimaging (GE Healthcare), and analyzed using ImageQuantMac, version 1.2.
Calculation of Dissociation Rates—Data points in Fig. 1B are an average of five independent experiments and were fit using nonlinear least squares regression of either the single exponential decay Equation 1,
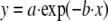 |
(Eq. 1) |
or the double exponential decay Equation 2,
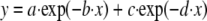 |
(Eq. 2) |
where y is the relative cleavage; a and c are the amplitudes of each dissociation curve, and b and d are the rates of dissociation for each curve. In Fig. 1C, data were fit using the Scrubber 2 software (Biologics Software Pty. Ltd.).
RESULTS
Dna2 Dissociates Slowly from a Flap Substrate—Previously, we observed that FEN1 disengages Dna2 from a flap substrate to gain access for cleavage (32). To understand the details of FEN1-promoted disengagement of Dna2, we used a DNA competition assay and surface plasmon resonance (SPR) to assess Dna2 dissociation (Fig. 1). We reasoned that if Dna2 dissociates slowly from flap substrates the disengagement reaction is likely to have evolved to facilitate rapid joining of Okazaki fragments. However, if spontaneous dissociation of Dna2 were rapid, the disengagement process has another purpose.
To assess the rate of Dna2 dissociation from the flap, we incubated Dna2 with a radiolabeled 53-nt double flap substrate. After binding, an excess of unlabeled flap substrate was added to the reaction followed by the addition of MgCl2 at the indicated time points (Fig. 1A). Reactions were then incubated for 10 min to allow Dna2 cleavage. Because MgCl2 is required for Dna2 cleavage, the cleavage rate was proportional to the amount of Dna2 still bound to the labeled substrate at each time point. These results were compared with a control in which the labeled and excess unlabeled substrates were incubated prior to the addition of Dna2 (Fig. 1A, lane 12). A graph was then generated, and points were fit to an exponential decay curve to determine the dissociation rate (see “Experimental Procedures”) (Fig. 1B). Initially, we fit the curve to a single exponential decay equation, which showed a half-time of about 25 min (Fig. 1B, gray line). Based on the shape of the curve, we then utilized the double exponential decay equation and found a better fit (Fig. 1B, black line). This suggests two dissociation phases, an initial rapid dissociation followed by a much slower one. Based on the small amplitude (∼20% of the relative cleavage) and the short time frame (∼1 min), we believe that nonspecific binding or a weak binding mode accounts for the initial dissociation phase of Dna2. The second phase would account for the majority of Dna2 binding. Dna2 bound in this manner dissociates slowly from the DNA, with a half-time of about 40 min. These data show that binding of Dna2 to the substrate is quite stable.
To further assess the binding and dissociation rates of Dna2 to DNA, we performed SPR. Dna2 was immobilized onto a chip, and various amounts of ssDNA were allowed to flow over the chip while association was measured (Fig. 1C). This was followed by a dissociation phase with only buffer flowing over the chip. A second surface in which Dna2 was not immobilized served as a reference. When we attempted to fit the curves, they did not fit a simple 1:1 binding model, suggesting a complex interaction between Dna2 and the DNA. Although we were unable to simultaneously fit both the association and dissociation rates, we could independently fit the dissociation rate using the Scrubber 2 software. Because the curves appeared strikingly similar to those in Fig. 1B, we fit the data 30 s into the dissociation phase. By doing so, we were able to bypass the initial dissociation phase and fit a 1:1 binding model for the second dissociation phase. Again, these curves suggest a slow rate of dissociation, with a half-time of ∼50 min. Both the excess substrate and SPR dissociation measurements clearly indicate that the half-time for dissociation of Dna2 is in the range of ½ to 1 h. These findings are consistent with the conclusion that, because Dna2 binding to the flap is stable, FEN1 has evolved the ability to disengage Dna2 to efficiently gain access to the flap base for cleavage.
Dna2 Binds, but Does Not Cleave, Short RNA and DNA Flaps—Previously, we hypothesized that FEN1 evolved to remove Dna2 molecules that are unproductively bound to the flap. The need for disengagement is envisioned to arise at two stages of Okazaki fragment processing. Each Okazaki fragment is initiated by a short segment of RNA, 10–12 nt in length (1). The first stage requiring disengagement would occur during initial strand displacement by pol δ, when RNA flaps begin to emerge. Although FEN1 can readily cleave short RNA flap intermediates, RNA is not a substrate for the nuclease activity of Dna2 (34). A bound, inactive Dna2 molecule could block progressive FEN1 cleavage.
We previously showed that Dna2 bound but did not cleave a 30-nt RNA flap and that Dna2 was dissociated by FEN1 (32). Here we employed a substrate with 5 nt of RNA on the flap and an additional 8 nts of RNA in the annealed portion of the labeled primer. This substrate simulates the initial partial displacement of the RNA primer by pol δ. The substrate was used to test Dna2 cleavage and binding. Consistent with previous findings, Dna2 was unable to cleave the RNA flap (Fig. 2A). By way of a control, we measured robust Dna2 cleavage activity on a 30-nt DNA flap substrate. We then tested the ability of Dna2 to bind the 5-nt RNA flap. Dna2 was incubated with the substrate, and the reactions were then analyzed by gel shift (Fig. 2B). The labeled substrate band shifted upon the addition of Dna2 to indicate formation of a higher molecular weight complex.
FIGURE 2.

FEN1 disengagement of Dna2 from short RNA and DNA flaps to which Dna2 binds but cannot cleave. A, Dna2 (50, 100, 200, 500, and 1000 fmol) was incubated with 5 fmol of a 5-nt RNA flap substrate (D1:U1:T1) (squares), a 5-nt DNA flap substrate (D2:U2:T2) (circles), or a 30-nt DNA flap substrate (D3:U2:T2) (diamonds). Cleavage activity was then measured by denaturing PAGE. B, gel shift analysis was used to measure Dna2 (0.2, 0.5, 1 pmol) binding activity on the 5-nt RNA flap (lanes 2–4). Lane 1 is the substrate alone control. C, Dna2 (1 pmol) was pre-bound to the 5-nt RNA flap substrate followed by the addition of FEN1 (5, 10, 25, and 50 fmol) (lanes 3–6). Thesampleswerethenanalyzedbygelshift.Lanes1and7arethesubstratealone and substrate plus FEN1 (50 fmol), respectively. WT, wild type. D, as described in B, except a 5-nt DNA flap was used. E, as described in C, except a 5-nt DNA flap substrate was used. A, points are an average of three experiments, and the bars indicate the standard deviation. Percent cleavage is defined as (cleaved/(cleaved + uncleaved)) × 100. Percent Dna2 bound is defined as (Dna2 bound/(Dna2 bound + FEN1 bound + unbound substrate)) × 100. Percent FEN1 bound is defined as (FEN1 bound/(FEN1 bound + Dna2 bound + unbound substrate)) × 100. Substrates are depicted above gels with the RNA labeled in gray and DNA in black. The asterisk indicates the site of the 3′-32P radiolabel.
Next, we assessed FEN1 dissociation of Dna2 on the 5-nt RNA flap substrate (Fig. 2C). Dna2 was pre-bound to the flap. FEN1 was then added with the Dna2-bound substrate. The reactions were then analyzed by gel shift to separate the products and determine which protein remained bound to the substrate. Because Dna2 is three times the size of FEN1, the bound complexes of these proteins with the labeled substrate are easily distinguished (Fig. 2C, lanes 2 and 7). With increasing amounts of FEN1, the bands were shifted from a Dna2-bound substrate to a FEN1-bound substrate (Fig. 2C, lanes 3–6). This shift is indicative of the removal of Dna2 from the 5-nt RNA flap by FEN1. These results suggest that FEN1 disengagement of flap-bound Dna2 would promote FEN1 cleavage on initially displaced flaps consisting only of RNA.
The second stage requiring Dna2 disengagement would occur after Dna2 cleavage on RPA-coated flaps. When flaps become long enough to bind RPA, Dna2 is needed to shorten them so that RPA will no longer bind and block FEN1. The properties of the Dna2 nuclease function appear ideal for this task, in that it cleaves flaps to a terminal length of about five nucleotides. However, Dna2 may remain bound unless dissociated by FEN1.
We designed a substrate with a 5-nt DNA flap to simulate the terminal product of long flap cleavage by Dna2. We then tested this flap for Dna2 cleavage and binding. As expected, Dna2 cleavage did not occur (Fig. 2A). Dna2 binding was then measured (Fig. 2D). Gel shift analysis showed that the addition of Dna2 shifted the labeled substrate band. Finally, Dna2 was prebound to the 5-nt DNA flap followed by the addition of FEN1 to test for Dna2 removal (Fig. 2E). Again, the addition of FEN1 shifted the band distribution from a Dna2-bound substrate to a FEN1-bound substrate, indicative of Dna2 removal (Fig. 2E, lanes 3–6).
Notably, similar results were obtained for both the 5-nt RNA and DNA flaps. Dna2 unproductively bound both substrates, potentially blocking FEN1, but in both cases FEN1 removed Dna2 to enable progressive FEN1 action. In addition, FEN1 showed nearly the same amount of displacement and binding on both the DNA and RNA flaps, suggesting that the interaction properties of these proteins are similar on both DNA and RNA. These results support the conclusion that FEN1 has evolved the disengagement mechanism to ensure that it is the dominant nuclease at all times it shares a flap with Dna2.
FEN1 Tracking Mechanism Is Required to Dissociate Dna2—Both FEN1 and Dna2 must track from the 5′-end of the flap to display nuclease activity (30, 31). Our previous results indicate that FEN1 disengages the tracking mechanism of Dna2 to dissociate it from the flap (32). Here we tested whether FEN1 must be in its tracking mode, as expected during natural flap processing, to disengage Dna2.
To block FEN1 tracking, we employed a 53-nt flap with a biotin attached at the 5′-end (Fig. 3, lanes 1–7). Dna2 was preincubated with this substrate to allow tracking. The Dna2-bound substrate was then incubated with streptavidin, which blocked the FEN1 tracking mechanism. FEN1 was then added into the reaction. Interestingly, FEN1 was unable to remove the flap-bound Dna2 (Fig. 3, lanes 3–6), indicating that it must be tracking to dissociate the Dna2.
FIGURE 3.

FEN1 requires its tracking mechanism to disengage flap-bound Dna2. Gel shift analysis was used to test Dna2 dissociation by FEN1 when tracking was blocked. Dna2 (500 fmol) was bound to a 53-nt flap substrate with a biotin attached at either the 5′ flap end (D4:U2:T2) or in the middle of the flap (D5:U2:T2). Streptavidin (Strept) was then added to the reaction for conjugation with the biotin. Following conjugation, FEN1 (5, 10, 20, and 50 fmol) was added (lanes 3–6 and 10–13). Lanes 1 and 8 are streptavidin-bound substrate alone. Lanes 2 and 9 are streptavidin-bound substrate plus Dna2 (500 fmol). Lanes 7 and 14 are streptavidin-bound substrate plus FEN1 (50 fmol). Percent Dna2 bound is defined as (Dna2 bound/(Dna2 bound + FEN1 bound + unbound substrate)) × 100. Percent FEN1 bound is defined as (FEN1 bound/(FEN1 bound + Dna2 bound + unbound substrate)) × 100. Substrates are depicted above gels, and the asterisk indicates the site of the 3′-32P radiolabel. WT, wild type. B indicates the site of biotin modification.
We then questioned whether Dna2 would be disengaged if the FEN1 began tracking but was not allowed to track all the way to the position of the flap-bound Dna2. In this scenario, the FEN1 would be in the tracking mode and could still potentially contact the Dna2 by a looping process. However, it would not likely form the exact contacts that it could make if it tracked in the natural manner. To achieve this situation, we employed a substrate with a biotin attached to a nucleotide in the middle of the flap (Fig. 3, lanes 8–14). This substrate was previously used to test the Dna2 tracking mechanism (30). The Dna2 cleavage pattern was unaltered indicating the biotin modification did not interfere with Dna2 tracking. By placing the biotin in the middle of the flap, the addition of streptavidin would permit FEN1 tracking but prevent full travel along the flap. Dna2 was bound to the flap followed by the addition of streptavidin. FEN1 was then added, and gel shift analysis was performed (Fig. 3, lanes 10–13). As with the 5′-end blocked flap, a block to the middle of the flap prevented FEN1 removal of flap-bound Dna2. Based on these findings, we propose that natural tracking by FEN1 is a requirement for the removal of Dna2 from the flap.
FEN1 Is Unable to Remove Dna2 from ssDNA—FEN1 is a structure-specific enzyme that recognizes the 5′ flap junction between ssDNA and double-stranded DNA for cleavage and needs a 1-nt 3′ tail for maximal cleavage efficiency (36). We reasoned that Dna2 removal by FEN1 might also require a genuine flap structure. Accordingly, a linear ssDNA segment was used to measure Dna2 dissociation by FEN1 (Fig. 4). A biotin was attached to the 3′-end of the DNA, which, when conjugated with streptavidin, would prevent Dna2 or FEN1 from tracking off the 3′-end of the single strand. Experiments were done with and without streptavidin to compare its effect.
FIGURE 4.

FEN1 cannot dissociate Dna2 on an ssDNA segment. A 50-nt ssDNA segment with a biotin attached at the 3′-end (D6) was used to test FEN1 disengagement of Dna2. In lanes 8–14, streptavidin (Strept) was preincubated with the substrate. Dna2 (500 fmol) was bound to the ssDNA segment followed by the addition of FEN1 (5, 10, 25, and 50 fmol) (lanes 3–6 and 10–13). Gel shift was then used to separate the products. Lanes 1, 2, and 6 are substrate alone, substrate with Dna2 (500 fmol), and substrate with FEN1 (50 fmol), respectively. Lanes 8, 9, and 14 are the same as lanes 1–3, respectively, except with streptavidin. Percent Dna2 bound is defined as (Dna2 bound/(Dna2 bound + unbound substrate)) × 100. Substrates are depicted above gels, and the asterisk indicates the site of the 5′-32P radiolabel. WT, wild type. B indicates the site of biotin modification.
Dna2 was preincubated with the ssDNA substrate, and increasing concentrations of FEN1 were then added into the reaction. Significantly, the addition of FEN1 did not affect the amount of Dna2 bound to the ssDNA (Fig. 4, compare lanes 3–6 with lanes 10–13). In fact, FEN1 alone was unable to bind the ssDNA substrate (Fig. 4, lanes 7 and 14). The inability of FEN1 to bind the ssDNA suggests that FEN1 must structurally identify the flap base for binding. This result implies that a collection of specific structural features of the DNA are important for Dna2 removal by FEN1.
Nuclease-defective Dna2 E675A Overcomes RPA Inhibition of FEN1—Is the ability of FEN1 to displace Dna2 significant in the context of the RPA/Dna2/FEN1 Okazaki fragment processing pathway? As described previously, flap-bound RPA inhibits FEN1 cleavage (8). We recently discovered that Dna2 displaces flap-bound RPA, independent of cleavage (33). In addition, FEN1 dissociation of Dna2 enables FEN1 cleavage (32). This offered us an opportunity to address the role of FEN1 dissociation of Dna2 in the actual RPA/Dna2/FEN1 pathway, in which RPA is blocking FEN1 cleavage. We envision that after a flap grows long and binds RPA, Dna2 would displace the RPA, cleave the flap, but then remain bound. FEN1 would then dissociate the Dna2, cleaving the flap to create a nick for ligation. Because Dna2 does not require nuclease activity to remove RPA, we asked whether Dna2 could permit FEN1 activity on an RPA-coated flap even without cutting it. To address this question, we reconstituted the RPA/Dna2/FEN1 pathway with the nuclease-defective Dna2 E675A.
RPA, Dna2 E675A, and FEN1 were incubated with a 21-nt flap substrate followed by denaturing PAGE analysis of the labeled primer (Fig. 5). Comparing the amount of substrate cleaved by FEN1 with or without RPA-bound, we measured an ∼3-fold inhibition of FEN1 cleavage activity in the presence of RPA (compare Fig. 5, A and B, lanes 3 and 6). Moreover, we observed that Dna2 E675A, as expected, does not inhibit FEN1 cleavage (Fig. 5, A and B, lane 5). In fact, Dna2 E675A slightly stimulated FEN1 nuclease activity. RPA, FEN1, and increasing amounts of Dna2 E675A were then incubated with the 21-nt flap substrate (Fig. 5, A and B, lanes 8–11). At the highest Dna2 E675A concentration, FEN1 cleavage was restored to near the level achieved without RPA (compare Fig. 5, A and B, lanes 3 and 11). Based on these results, we conclude that Dna2 E675A actively displaced the RPA from the flap followed by FEN1 removal of the now flap-bound Dna2 E675A. FEN1 was then free to cleave the flap devoid of either RPA or Dna2.
DISCUSSION
We previously demonstrated that FEN1 disengages Dna2 from the flap (32). Our current studies questioned why FEN1 evolved to remove flap-bound Dna2 in the context of Okazaki fragment processing. We conclude that Dna2 binding to short RNA and DNA flaps would potentially block FEN1 entry and cleavage. However, FEN1 displaces these flap-bound Dna2 molecules.
Dna2 does not cleave RNA, but it binds RNA flap substrates. In fact, it binds with a similar affinity to RNA as DNA and can track from the 5′-end of an RNA segment to cleave the DNA portion of an RNA-DNA flap substrate (34). Such properties would enable Dna2 cleavage of the long flap intermediates anticipated to occur naturally as pol δ displaces an RNA-primed Okazaki fragment. However, previous results indicate that FEN1 alone processes most flaps (6, 27). The ability of FEN1 to cleave an RNA flap indicates that it is designed to act constantly on the RNA, and then DNA, as the flap is generated. In support of this conclusion, biochemical reconstitution studies suggest that coordination between pol δ and FEN1 produces short cleavage products of 1–8 nt, with the majority of products being mononucleotides. This coordination is highly efficient and allows for the rapid processing of flap intermediates. We envision that a binding competition between Dna2 and FEN1 can arise as the RNA flap is beginning to be displaced. Dna2 binding ahead of FEN1 on RNA flaps would prove unproductive until the RNA and more than about five nucleotides of DNA have been displaced. The ability of FEN1 to remove Dna2 from short RNA flaps allows FEN1 to act as the sole nuclease, unless long flaps arise.
Once flaps escape FEN1 cleavage and become long, they are bound by RPA eliciting the need for the RPA/Dna2/FEN1 pathway. We showed that Dna2 dissociates RPA to access the flap for cleavage (33). In addition, RPA strand melting capacity stimulates Dna2 cleavage by removing DNA secondary structure. Although RPA binding inhibits FEN1 cleavage, RPA would also prevent structured flap formation, which would inhibit efficient cleavage by either Dna2 or FEN1. By this reasoning, RPA binding likely prepares long structured flaps for Dna2 cleavage. Dna2 tracks down the flap removing RPA and successively cleaves until the flap reaches ∼5 nt in length. At this length, Dna2 cannot cleave, necessitating FEN1 cleavage to make a product for ligation. Again, we found that FEN1 can disengage Dna2 that has reached this static state. In fact, the unproductively bound Dna2 may even act to recruit FEN1 to the shortened RPA-free flap for final processing.
Dna2 binding kinetics were consistent with a slow rate of dissociation from the DNA after initial binding (Fig. 1). Slow dissociation may be enhanced by the tracking mechanism of Dna2. When tracking, the protein behaves as a bead on a string, or as if it is encircling the flap. The Dna2 may also not readily slide back off of the 5′-end of the flap. This resistance to 5′ motion may be accentuated when the Dna2 helicase is acting to continuously drive Dna2 toward the flap base. Our demonstration that Dna2 can bind Okazaki fragment intermediates nonproductively, together with evidence of slow natural dissociation, highlights the reasons why FEN1 has evolved the ability to disengage Dna2.
Using the nuclease-defective Dna2 mutant, we showed that the RPA/Dna2/FEN1 pathway could be reconstituted in the absence of Dna2 cleavage activity (Fig. 5). Results of this experiment reveal the elegant coordination of functions that can be displayed by Okazaki fragment maturation proteins. We were impressed to see that the successive binding functions of Dna2 and FEN1 were sufficient to clear a long flap of RPA and allow FEN1 cleavage. Although this experiment allowed us to visualize more closely the sequential steps to proper flap removal, it seemingly questions the role of the nuclease activity of Dna2. Genetic evidence emphasizes the importance of Dna2 nuclease activity in DNA replication (39). The nuclease activity is essential in S. cerevisiae, and the temperature-sensitive mutant, dna2-1, which has reduced nuclease function, showed defects associated with DNA replication at the restrictive temperature. These include highly fragmented DNA, deficiency in DNA, but not RNA, synthesis, and undivided nuclei.
An answer to this puzzle is suggested by the results of an attempted repeat of the reconstitution of the RPA/Dna2/FEN1 pathway shown in Fig. 5, but with a 53-nt RPA-coated flap substrate. With this substrate, Dna2 E675A was unable to stimulate FEN1 cleavage (data not shown). We interpret this result to mean that Dna2 dissociated the flap-bound RPA and then stalled at the base, unable to cleave. RPA then rebound behind the Dna2 to inhibit FEN1 cleavage. The 21-nt flap likely represents a length at which Dna2 removes RPA without allowing stable RPA rebinding. Reconstitutions of Okazaki fragment processing suggest that a fraction of flaps grows into the 40–50-nt size range (27, 29). If so, the need to process such flaps is anticipated to require the nuclease activity of Dna2. Even on shorter flaps, it is likely that the nuclease function of Dna2 accelerates the rate of flap removal in a way that makes its function essential, as suggested by the nearly but not totally complete rescue of FEN1 cleavage in Fig. 5.
FEN1 tracking, a prerequisite for cleavage, was also necessary for the removal of Dna2 (Fig. 3). Apparently, when the flap is long, FEN1 must recognize the 5′-end, bind, and track down the flap until it encounters Dna2. The interaction between FEN1 and Dna2 then results in disengagement of Dna2 from the flap. In contrast, when the flap is short the situation could differ. On a 5-nt flap, the natural terminal product of Dna2 cleavage, Dna2 may occlude the entire flap. If so, FEN1 could not track. Yet, it still dissociates Dna2 from the flap. How might FEN1 tracking be required on long but not short flaps? Dna2, like FEN1, is a tracking enzyme (30). For cleavage, it must load onto the 5′-end and track down the flap. Interestingly, although tracking is required for cleavage activity, both FEN1 and Dna2 can bind the flap independent of tracking (32, 40), but the ability to bind the flap was not sufficient for FEN1 to promote the dissociation of Dna2 (Fig. 3). Like FEN1, it is envisioned that the flap is threaded through Dna2. The site where the 5′-end of the flap exits Dna2 is likely near the required area for proper protein-protein contacts with FEN1. Likewise, the region on FEN1 critical for protein contacts with Dna2 would be located near the site of flap entry on the smaller nuclease. Consistent with this interpretation, FEN1 could not loop around a streptavidin block in the middle of a long flap to remove the flap-bound Dna2 (Fig. 3). Instead the streptavidin block prevented FEN1 from removing Dna2. Natural tracking by FEN1 would allow direct contact of the appropriate protein surfaces to induce the disengagement of Dna2 and release it from the flap. In the case of a short flap, the surfaces of interaction would be unobstructed by the flap, permitting FEN1 to properly interact with Dna2. In addition, the flap likely facilitates proper protein contacts by bringing the proteins into close proximity. The actual orientations of FEN1, Dna2, and DNA during these processes await high resolution structural analysis.
Finally, we showed that FEN1 did not disengage Dna2 on a single-stranded segment of DNA (Fig. 4). Recognition of a genuine flap substrate is required for efficient cleavage by FEN1 (36). FEN1 does not cleave linear ssDNA segments at all, and here we show that FEN1 is unable even to bind such DNA. Binding was not achieved even with a streptavidin block at the 3′-end of the ssDNA, suggesting at least two possibilities. Structural features of the flap must be recognized prior to binding and tracking. Alternatively, the flap base stabilizes FEN1 binding after tracking.
Previously, we envisioned that FEN1 begins tracking by first recognizing the 5′-end of the flap, followed by threading of the flap through the protein until it reached the base. Upon encountering the base, FEN1 would then identify the structure features required to activate cleavage of the substrate. Based on this model, FEN1 should still track on the ssDNA segment, with the streptavidin block preventing FEN1 from tracking off the 3′-end. Because the substrate does not possess the flap base structure that activates FEN1 for cleavage, FEN1 would be stopped by the streptavidin block until it tracks back off the 5′-end for dissociation (31). In addition, FEN1 should still remove flap-bound Dna2. Instead, FEN1 could not bind the ssDNA or remove Dna2. Interestingly, Dna2, also a tracking enzyme, was unable to dissociate RPA on a segment of ssDNA (33). Together these findings support the idea that structural recognition of the flap base is required prior to tracking.
In conclusion, our results have defined the significance of FEN1 disengagement of Dna2 during Okazaki fragment processing. Unproductive binding by Dna2 creates a potential block to FEN1 tracking and cleavage during multiple steps of flap processing. FEN1 has evolved to disengage Dna2 from the flap to permit rapid progression toward the final product and proper fragment joining. In addition, our results highlight the coordinated protein action of the RPA/Dna2/FEN1 pathway in which each protein is specifically designed to succeed the next for efficient flap processing.
Acknowledgments
We thank Drs. Sara Binz and Marc Wold for the purified RPA protein. We thank Dr. Tom Ryan and team at Reichert Inc. and the Sullivan laboratory at the University of Rochester for training and assistance with the SPR equipment. We also thank the Bambara and Campbell laboratories for beneficial discussion and review of the manuscript.
This work was supported by National Institutes of Health Grant GM024441 (to R. A. B.), with additional support from Grant GM087666 (to J. L. C.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: nt, nucleotide; pol, polymerase; FEN1, flap endonuclease 1; RPA, replication protein A; ss, single-stranded; SPR, surface plasmon resonance.
References
- 1.Rossi, M. L., Purohit, V., Brandt, P. D., and Bambara, R. A. (2006) Chem. Rev. 106 453–473 [DOI] [PubMed] [Google Scholar]
- 2.Burgers, P. M. (2009) J. Biol. Chem. 284 4041–4045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gordenin, D. A., Kunkel, T. A., and Resnick, M. A. (1997) Nat. Genet. 16 116–118 [DOI] [PubMed] [Google Scholar]
- 4.Kunkel, T. A., Resnick, M. A., and Gordenin, D. A. (1997) Cell 88 155–158 [DOI] [PubMed] [Google Scholar]
- 5.Ayyagari, R., Gomes, X. V., Gordenin, D. A., and Burgers, P. M. (2003) J. Biol. Chem. 278 1618–1625 [DOI] [PubMed] [Google Scholar]
- 6.Garg, P., Stith, C. M., Sabouri, N., Johansson, E., and Burgers, P. M. (2004) Genes Dev. 18 2764–2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu, Y., Kao, H. I., and Bambara, R. A. (2004) Annu. Rev. Biochem. 73 589–615 [DOI] [PubMed] [Google Scholar]
- 8.Bae, S. H., Bae, K. H., Kim, J. A., and Seo, Y. S. (2001) Nature 412 456–461 [DOI] [PubMed] [Google Scholar]
- 9.Bae, S. H., Choi, E., Lee, K. H., Park, J. S., Lee, S. H., and Seo, Y. S. (1998) J. Biol. Chem. 273 26880–26890 [DOI] [PubMed] [Google Scholar]
- 10.Budd, M. E., and Campbell, J. L. (1995) Proc. Natl. Acad. Sci. U. S. A. 92 7642–7646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu, Q., Choe, W., and Campbell, J. L. (2000) J. Biol. Chem. 275 1615–1624 [DOI] [PubMed] [Google Scholar]
- 12.Kang, H. Y., Choi, E., Bae, S. H., Lee, K. H., Gim, B. S., Kim, H. D., Park, C., MacNeill, S. A., and Seo, Y. S. (2000) Genetics 155 1055–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim, J. H., Kim, H. D., Ryu, G. H., Kim, D. H., Hurwitz, J., and Seo, Y. S. (2006) Nucleic Acids Res. 34 1854–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Masuda-Sasa, T., Imamura, O., and Campbell, J. L. (2006) Nucleic Acids Res. 34 1865–1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Budd, M. E., Reis, C. C., Smith, S., Myung, K., and Campbell, J. L. (2006) Mol. Cell. Biol. 26 2490–2500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choe, W., Budd, M., Imamura, O., Hoopes, L., and Campbell, J. L. (2002) Mol. Cell. Biol. 22 4202–4217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masuda-Sasa, T., Polaczek, P., Peng, X. P., Chen, L., and Campbell, J. L. (2008) J. Biol. Chem. 283 24359–24373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Budd, M. E., Tong, A. H., Polaczek, P., Peng, X., Boone, C., and Campbell, J. L. (2005) PLoS Genet 1 e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weitao, T., Budd, M., Hoopes, L. L., and Campbell, J. L. (2003) J. Biol. Chem. 278 22513–22522 [DOI] [PubMed] [Google Scholar]
- 20.Vernon, M., Lobachev, K., and Petes, T. D. (2008) Genetics 179 237–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tomita, K., Kibe, T., Kang, H. Y., Seo, Y. S., Uritani, M., Ushimaru, T., and Ueno, M. (2004) Mol. Cell. Biol. 24 9557–9567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liao, S., Toczylowski, T., and Yan, H. (2008) Nucleic Acids Res. 36 6091–6100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu, Z., Chung, W. H., Shim, E. Y., Lee, S. E., and Ira, G. (2008) Cell 134 981–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Budd, M. E., and Campbell, J. L. (1997) Mol. Cell. Biol. 17 2136–2142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wold, M. S. (1997) Annu. Rev. Biochem. 66 61–92 [DOI] [PubMed] [Google Scholar]
- 26.Kao, H. I., Veeraraghavan, J., Polaczek, P., Campbell, J. L., and Bambara, R. A. (2004) J. Biol. Chem. 279 15014–15024 [DOI] [PubMed] [Google Scholar]
- 27.Rossi, M. L., and Bambara, R. A. (2006) J. Biol. Chem. 281 26051–26061 [DOI] [PubMed] [Google Scholar]
- 28.Burgers, P. M., and Gerik, K. J. (1998) J. Biol. Chem. 273 19756–19762 [DOI] [PubMed] [Google Scholar]
- 29.Rossi, M. L., Pike, J. E., Wang, W., Burgers, P. M., Campbell, J. L., and Bambara, R. A. (2008) J. Biol. Chem. 283 27483–27493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kao, H. I., Campbell, J. L., and Bambara, R. A. (2004) J. Biol. Chem. 279 50840–50849 [DOI] [PubMed] [Google Scholar]
- 31.Murante, R. S., Rust, L., and Bambara, R. A. (1995) J. Biol. Chem. 270 30377–30383 [DOI] [PubMed] [Google Scholar]
- 32.Stewart, J. A., Campbell, J. L., and Bambara, R. A. (2006) J. Biol. Chem. 281 38565–38572 [DOI] [PubMed] [Google Scholar]
- 33.Stewart, J. A., Miller, A. S., Campbell, J. L., and Bambara, R. A. (2008) J. Biol. Chem. 283 31356–31365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bae, S. H., and Seo, Y. S. (2000) J. Biol. Chem. 275 38022–38031 [DOI] [PubMed] [Google Scholar]
- 35.Budd, M. E., Choe, W., and Campbell, J. L. (2000) J. Biol. Chem. 275 16518–16529 [DOI] [PubMed] [Google Scholar]
- 36.Kao, H. I., Henricksen, L. A., Liu, Y., and Bambara, R. A. (2002) J. Biol. Chem. 277 14379–14389 [DOI] [PubMed] [Google Scholar]
- 37.Henricksen, L. A., and Wold, M. S. (1994) J. Biol. Chem. 269 24203–24208 [PubMed] [Google Scholar]
- 38.Subramanian, A., Irudayaraj, J., and Ryan, T. (2006) Biosens. Bioelectron. 21 998–1006 [DOI] [PubMed] [Google Scholar]
- 39.Budd, M. E., Choe, W. C., and Campbell, J. L. (1995) J. Biol. Chem. 270 26766–26769 [DOI] [PubMed] [Google Scholar]
- 40.Hohl, M., Dunand-Sauthier, I., Staresincic, L., Jaquier-Gubler, P., Thorel, F., Modesti, M., Clarkson, S. G., and Scharer, O. D. (2007) Nucleic Acids Res. 35 3053–3063 [DOI] [PMC free article] [PubMed] [Google Scholar]