

Abstract
Inhibitors of the ubiquitin-proteasome system (UPSIs) promote apoptosis of cancer cells and show encouraging anti-tumor activities in vivo. In this study, we evaluated the death activities of two different UPSIs: bortezomib and the isopeptidase inhibitor G5. To unveil whether these compounds elicit different types of death, we compared their effect both on apoptosis-proficient wild type mouse embryo fibroblasts and on cells defective for apoptosis (double-deficient Bax/Bak mouse embryo fibroblasts) (double knockout; DKO). We have discovered that (i) both inhibitors induce apoptosis in a Bax and Bak-dependent manner, (ii) both inhibitors elicit autophagy in WT and DKO cells, and (iii) only G5 can kill apoptosis-resistant DKO cells by activating a necrotic response. The induction of necrosis was confirmed by different experimental approaches, including time lapse analysis, HMGB1 release, and electron microscopy studies. Neither treatment with antinecrotic agents, such as antioxidants, poly(ADP-ribose) polymerase and JNK inhibitors, necrostatin, and the intracellular Ca2+ chelator, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid acetoxymethyl ester, nor overexpression of Bcl-2 and Bcl-xL prevented necrosis induced by G5. This necrotic death is characterized by the absence of protein oxidation and by the rapid cyclosporin A-independent dissipation of the mitochondrial membrane potential. Notably, a peculiar feature of the G5-induced necrosis is an early and dramatic reorganization of the actin cytoskeleton, coupled to an alteration of cell adhesion. The importance of cell adhesion impairment in the G5-induced necrotic death of DKO cells was confirmed by the antagonist effect of the extracellular matrix-adhesive components, collagen and fibronectin.
The ubiquitin-proteasome system (UPS)3 is the major route for the degradation of cellular proteins in eukaryotic cells (1). The UPS comprises a proteolytic machinery (the proteasome) and a complex network of enzymes characterized by the ability to interact with, add, or remove ubiquitin residues to target proteins (2, 3). Protein modification by ubiquitin controls many cellular functions, and it does not always determine the degradation of the substrate. Monoubiquitination, polymonoubiquitination, and even polyubiquitination, when not involving the Lys48 linkage, rather than promoting protein degradation, modulate protein activity in a signal-dependent manner (4).
Inhibitors of the ubiquitin-proteasome system (UPSIs) include a large cohort of compounds that can target not only the proteolytic chamber of the proteasome but also other components of the UPS, such as deubiquitinating enzymes (5). Compounds with the ability to modulate the ubiquitin turnover are emerging as new drug candidates for anti-tumor therapies (5, 6).
In multiple myeloma, both the FDA (United States Food and Drug Administration) and the EMEA (European Medicine Evaluation Agency) granted an approval for the use of the UPSI bortezomib in the treatment of relapsed multiple myeloma (7). Several clinical trials are exploring its use as a single agent or in combinatory therapies in different types of cancer (8).
UPSIs anti-tumor activities are mainly the result of their ability to promote apoptosis. Cell death after UPS inhibition occurs through multiple mechanisms, including induction of ER stress, activation of the death receptor pathway and of the JNK pathway, suppression of NF-κB, and induction of p53 and of the BH3-only proteins Noxa, Bim, and NBK/Bik (5, 6).
Resistance to drug treatment is a concern for many types of cancer. Often defects or dysregulation in elements of the apoptotic program are important determinants of the resistance to anti-cancer drugs (9). However, apoptosis is not the sole mechanism that causes cell death. Cell death can take place by at least three different routes historically divided by morphological criteria in apoptosis, necrosis, and autophagy (10). Although autophagy is considered a strategy of cell adaptation to adverse growth conditions, it may also result in cell death when induced massively and for prolonged periods of time (11).
In principle, the ability of a particular anti-cancer molecule to elicit different types of death in cancer cells might represent an advantage for its in vivo efficacy.
The multidomain proapoptotic Bcl-2 family members Bax and Bak are fundamental and redundant regulators of the intrinsic death pathway in response to a plethora of signals. Bax and Bak are ubiquitously expressed, and they represent the master controllers of the mitochondrial outer membrane permeabilization (12).
Murine embryonic fibroblasts obtained from mice double-deficient for Bax and Bak (double knock-out; DKO) are resistant to multiple apoptotic stimuli that provoke mitochondrial outer membrane permeabilization and caspase-9 activation (13). Although apoptosis is largely compromised in DKO cells, death can still occur through caspase-independent mechanisms. Cell death by necrosis or as a result of massive autophagy has been observed in DKO cells in response to specific signals or after prolonged exposure to apoptotic stresses (14). Hence, DKO cells could serve as a valuable tool in delineating multiple cell death pathways that can be engaged by cytotoxic agents, which may otherwise escape detection in cells with intact apoptotic machinery.
In this study, we have used DKO cells to compare the ability of two different UPSIs, bortezomib (7) and G5 (4H-thiopyran-4-one, tetrahydro-3,5-bis[(4-nitrophenyl)methylene]-1,1-dioxide), a recently identified isopeptidase inhibitor (15), to elicit different types of cell death. Previous studies have shown that these inhibitors can induce a similar apoptotic pathway, during which caspases can be activated independently from the apoptosome, as a result of the sustained levels of cytosolic Smac and the engagement of the death receptor pathway (5, 15).
Whether or not G5 and bortezomib can induce alternative death responses is presently unknown. Here we demonstrate that both drugs can elicit autophagy in WT and DKO cells and that only G5 can kill and activate caspases in DKO cells, through the engagement of the death receptor pathway. We have also demonstrated that death of DKO cells in response to G5 is caspase-independent and occurs via necrosis. G5 elicits a new necrotic pathway that is unrelated to the activation of PARP or of the mitogen-activated protein kinases and that cannot be inhibited by Bcl-2 or Bcl-xL. A peculiar feature of this necrotic death is its dependence on the adhesion to the extracellular matrix. In fact, cells grown on collagen or fibronectin are resistant to G5-necrotic death. In conclusion, our studies provide evidence that G5 and bortezomib, by targeting different elements of the UPS, can activate alternative types of cell death and may therefore represent nonredundant regimens of anti-cancer therapy.
MATERIALS AND METHODS
Reagents—ERK inhibitor U0126, JNK inhibitor SP600125, and p38 inhibitor SB203580 were purchased from Calbiochem. Caspase inhibitors Z-Val-Ala-dl-Asp(OMe)-fluoromethylketone and Z-Val-dl-Asp-fluoromethylketone were purchased from Bachem AG (Bubendorf, Switzerland). Mitotracker, MitoSOX red, and 5-(and-6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate were purchased from Molecular Probes, Inc. (Eugene, OR). Rotenone, cyclosporin A, 1,5-isoquinolinediol, 3,4-dihydro-5-[4-(1-piperidinyl)butoxyl]-1(24)-isoquinolinone, bafilomycin A1, methyl methanesulfonate, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid acetoxymethyl ester, 2,3-dimethoxy-1,4-naphthoquinone, phalloidin-TRITC, and 3-methyladenine were purchased from Sigma. TNF was acquired from Roche Applied Science. Velcade/Bortezomib was from Janssen-Cilag International N.V. (Beerse, Belgium), and necrostatin was from Alexis (Lausen, Switzerland).
Culture Conditions, Cell Death, and Retroviral Infection—SV40 T-antigen-transformed mouse embryo fibroblasts from Bax and Bak wild type (WT) mice or Bax and Bak DKO mice were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, penicillin (100 units/ml), glutamine (2 mm), and streptomycin (100 μg/ml) at 37 °C in a 5% CO2 atmosphere. In all trypan blue exclusion assays, 400 cells, from three independent samples, were counted for each data point. Data were represented as arithmetic means ± S.D. for at least three independent experiments.
WT and DKO cells expressing CrmA, Bcl-2, and Bcl-xL were generated by retroviral infection. Briefly, crmA and bcl-xL were cloned in the retroviral vector pWZL-hygro, whereas bcl-2 was inserted into pBABE-puro retroviral vector. Next, retroviral vectors expressing these genes or empty vectors expressing only the resistance gene (HYGRO or PURO) were used to singularly transfect the ecotropic packaging cell line LinX-E (16). Transfection was performed by the calcium phosphate method. At 60 h post-transfection, viral supernatants were collected, filtered, supplemented with 8 μg/ml Polybrene, and combined with fresh medium in order to infect WT and DKO cells.
24-well multiwell plates coated with human fibronectin or rat collagen I were obtained from BD Biosciences. 6 h after seeding, cells were treated with the different compounds. Cell death was analyzed 12 h later.
The caspase activity was evaluated using the Apo-ONE caspase-3/7 homogeneous assay (Promega). The assay includes a profluorescent caspase-3/7 consensus substrate, rhodamine 110 bis-(N-Z-l-aspartyl-l-glutamyl-l-valyl-aspartic acid amide). Cells grown in 96-well plates were treated with the different insults and tested for caspase activity as recommended by the vendor.
Time Lapse Imaging and Electron Microscopy—Images of cells treated with different compounds were collected with a cooled CCD camera mounted on a time-lapse imaging system (Leica AF6000 LX), and they were analyzed using MetaMorph software (17). To observe actin dynamic in vivo DKO cells were transfected 24 h after plating by adding to the medium Opti-MEM containing Lipofectamine (Invitrogen) plus enhanced yellow fluorescent protein-actin (Clontech). 24 h later, cells were subjected to the time lapse analysis with a confocal microscope (Leica TCS-SP). Throughout the experiments, cells were grown under a 5% CO2 atmosphere at 37 °C.
Mitochondrial membrane potential was measured using Δψm-sensitive dye tetramethylrhodamine methyl ester, as previously described (17). Since individual cells depolarize mitochondria at different times from the addition of G5 or bortezomib, the beginning of depolarization (TMRM decrease) was set to 0 min.
The image analysis was performed using the MetaMorph 6.04 software. For TMRM analysis in single cells, fluorescent mitochondria (MT) were analyzed by drawing a region around them and by measuring the total brightness (integrated fluorescence intensity or IITMRM) of such a region. The background to be subtracted from the IITMRM (BkTMRM) was calculated, for each frame, by using a region of interest (ROI) positioned in a nonfluorescent region according to the following equation.
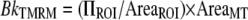 |
(Eq. 1) |
Finally, the background-corrected IITMRM values were presented as the percentage of the average of the five values detected before the IITMRM started decreasing. The IITMRM values have been taken for 10 min before and 52 min after the beginning of the Δψm decrease (set to 0 min).
For electron microscopy, cells were fixed with a mixture of 2% paraformaldehyde and 3% glutaraldehyde in 0.1 m phosphate buffer, pH 7.4, for 3 h and then postfixed in phosphate-buffered 1% osmium tetroxide for 1.5 h, dehydrated in graded ethanol series, and embedded in Epon 812. Thin sections were counterstained with uranyl acetate and lead citrate and examined in a Philips CM 12 electron microscope at 80 kV.
Western Blotting and Antibodies—Proteins obtained after an SDS denaturating lysis and sonication were transferred to a 0.2-μm pore size nitrocellulose membrane and incubated with the following antibodies: anti-caspase-2 and anti-tubulin (18); anti-caspase-3, anti-ERK1/ERK2, anti-SAPK/JNK, anti-p38 mitogen-activated protein kinase, anti-phospho-ERK1/ERK2, anti-phospho-p38 mitogen-activated protein kinase (Thr180/Tyr182), anti-phospho-SAPK/JNK (Thr183/Tyr185), and anti-PARP (Cell Signaling, Boston, MA); anti-HMGB1 (AbCam, Cambridge, UK); anti-HDAC4 (19); anti-LC3 (20); anti-Bcl-xL, anti-MEF2C, and anti-poly(ADP-ribose) (BD Biosciences Clontech); and anti-Bcl-2 and anti-vesicular stomatitis virus (Sigma). Blots were then rinsed three times with Blotto/Tween 20 and incubated with peroxidase-conjugated goat anti-rabbit or goat anti-mouse (Euroclone, Milano I) for 1 h at room temperature. Blots were then washed three times in Blotto/Tween 20, rinsed in phosphate-buffered saline, and developed with Super Signal West Pico, as recommended by the vendor (Pierce).
Detection of carbonyl groups was performed with the Oxy-Blot oxidized protein detection kit (Chemicon, UK) according to the manufacturer's protocol. Briefly, 5 μg of total extract proteins were incubated for 20 min at room temperature with 2,4-dinitrophenylhydrazine to form the carbonyl derivative dinitrophenylhydrazone before SDS-PAGE separation.
For subcellular fractionation, cells were washed twice in PBS, scraped, washed, and resuspended in 3 volumes of ice-cold extraction buffer (20 mm Hepes, pH 7.5, 1.5 mm MgCl2, 1 mm EDTA, 1 mm EGTA, 0.05% Nonidet P-40, 10 μg/ml cytochalasin B, 1 mm dithiothreitol, 1 mm phenylmethylsulfonyl fluoride, and 10 μg/ml each of chymostatin, leupeptin, antipain, and pepstatin). After 5 min of incubation on ice, lysates were centrifuged at 800 × g for 10 min. The postnuclear supernatant was then centrifuged at 8000 × g for 5 min to obtain the crude cytosolic fraction. The nuclear pellet was resuspended in extraction buffer to the same final volume as the cytosolic fraction.
Immunofluorescence Microscopy—Antibodies were applied to paraformaldehyde-fixed cytospin preparations or cells grown directly on coverslips. Briefly, after washes with PBS, 0.1 mol/liter glycine, pH 7.5, cells were permeabilized with 0.1% Triton X-100 in PBS for 5 min. Next, the coverslips were treated with the anti-HMGB1 antibodies diluted in PBS for 30 min in a moist chamber at 37 °C. They were then washed twice with PBS and incubated with 488-Alexa-conjugated secondary antibodies (Molecular Probes) for 30 min at 37 °C.
Phalloidin-TRITC (Sigma) was used to visualize actin filaments. Cells were examined by epifluorescence with a Leica DMI6000B microscope equipped with a CCD camera or with a Leica SP confocal microscope equipped with a 488 λ argon laser and a 543 λ helium neon. Cell images for deconvolution were taken using the Leica DMI6000B microscope at ×63 magnification, and z-stacks of ∼21 incremental slices (0.3-μm step size) were captured for each cell. The LAS AF Deconvolution software was used for image deconvolution and three-dimensional view reconstruction. Mitochondria were labeled in vivo for 1 h with 25 nmol/liter Mitotracker Red CMX Ros (Molecular Probes).
RESULTS
Cell Death and Caspase Activation in WT and DKO Cells Treated with G5 or Bortezomib—bax+/+bak+/+ (WT) and bax–/–bak–/– (DKO) cells were used as model systems in order to compare the killer activities of bortezomib and G5 and to study their involvement in multiple types of cell death (14).
Initially, we tested various concentrations of bortezomib and G5 in order to define the dose-response curves both in WT and DKO cells. Trypan blue staining was used to score cell death. Dose-dependent studies indicated that G5 induces 70% of death in WT cells when used at a 2.5 μm concentration (Fig. 1A). In DKO cells, treated with the same concentration of G5, death occurred in less than 20% of cells. However, doubling the concentration (5 μm) was enough to induce 50% of death in DKO cells. Cell death of DKO cells was further increased up to 80% in the presence of 10 μm G5 (Fig. 1A).
FIGURE 1.

Cell death in WT and DKO cells in response to G5 or bortezomib treatments. A, WT and DKO cells were treated with the indicated concentrations of G5 for 20 h, and the appearance of cell death was scored by trypan blue staining (means ± S.D., n = 3). B, WT and DKO cells were treated with the indicated concentrations of bortezomib for 20 h, and the appearance of cell death was scored by trypan blue staining (means ± S.D., n = 3). C, DEVDase activity in WT and DKO cells treated with the indicated concentrations of G5 for 20 h (means ± S.D., n = 3). D, DEVDase activity in WT and DKO cells treated with the indicated concentrations of bortezomib for 20 h (means ± S.D., n = 3). E, processing of caspase-3 and HDAC4 in WT and DKO cells treated with the indicated concentrations of G5 for 20 h. Equal amounts of cell lysates were subjected to SDS-PAGE electrophoresis. Immunoblots were performed using the indicated antibodies. Tubulin was used as loading control. F, WT cells were treated for 20 h with the indicated concentrations of G5 or with 50μm of etoposide, in the presence or absence of 100μm of the two pancaspase inhibitors Z-VAD-fmk (zVAD) and Z-VD-fmk (zVD). Cell death was scored by trypan blue staining (means ± S.D., n = 3). G, DKO cells were treated for 20 h with the indicated concentrations of G5 in the presence or absence of 100μm of the two pancaspase inhibitors Z-VAD-fmk and Z-VD-fmk. Cell death was scored by trypan blue staining (means ± S.D., n = 3). H, processing of caspase-3 and HDAC4 in DKO cells treated for 20 h with the indicated concentrations of G5, in the presence or absence of a 100 μm concentration of the two pancaspase inhibitors Z-VAD-fmk and Z-VD-fmk. Equal amounts of cell lysates were subjected to SDS-PAGE. Immunoblots were performed using the indicated antibodies. Tubulin was used as loading control.
Bortezomib can induce 70% of death in WT cells when used at 25 nm concentration. However, in contrast to G5, dose escalation up to 10 μm of bortezomib failed to efficiently kill DKO cells (Fig. 1B).
In summary, a 4-fold increase in the concentration of G5 used to kill WT cells was sufficient to induce the same level of death in DKO cells. On the contrary, increasing up to 4000-fold the concentration of bortezomib used to kill WT cells was insufficient to trigger death in DKO cells.
Next, we analyzed caspase-3 and -7 activities in WT and DKO cells treated with escalating amounts of G5 or bortezomib. In WT cells, the DEVD activity peak was observed with 1.25 μm of G5. At higher concentrations of G5, the DEVD activity progressively decreased (Fig. 1C). In DKO cells, there was a modest increase of DEVD activity in response to incremental doses of G5. However, similarly to WT cells, this activity diminished when higher doses of G5 were employed. Bortezomib in contrast induced full DEVD activity also at higher doses in WT cells, whereas only a minimal activity was noted in DKO cells (Fig. 1D).
To confirm that G5 can activate caspases also in DKO cells, we performed immunoblot analysis of caspase-3 and HDAC4 processing in WT and DKO cells treated with G5. Fig. 1E shows that cleavage of caspase-3 and HDAC4 is evident in WT but also in DKO cells in the presence of 5 μm G5 and that this processing was reduced in the presence of higher doses of G5 (10 μm). Finally, we also confirmed that G5 induces cell death with undistinguishable kinetics in WT and DKO cells (Fig. S1).
Having noted that the isopeptidase inhibitor G5 induces cell death and some caspase activity in DKO cells, we next investigated whether caspases play a role during this cell death response. Cells were grown in the presence of two pancaspase inhibitors, Z-VAD-fmk and Z-VD-fmk, and treated with high concentrations of G5 (5 and 10 μm) or with etoposide, as control. In WT cells, death was only partially inhibited by the two inhibitors, thus indicating only a limited involvement of caspases (Fig. 1F). In DKO cells, Z-VAD-fmk and Z-VD-fmk were unable to influence the appearance of cell death (Fig. 1G), although they efficiently suppressed caspase-3 and HDAC4 processing (Fig. 1H). These results suggest that, although G5 may activate caspases in DKO cells, these cells die via a caspase-independent mechanism.
G5 Engages the Extrinsic Pathway to Trigger Caspase Activation in DKO Cells—Rare apoptotic signals can activate caspases in DKO cells. Among them, death receptor ligands, which act through the extrinsic pathway, activate caspases and induce cell death independently from Bax and Bak (13). Hence, it is possible that the processing and activation of caspases, observed in G5-treated DKO cells, takes place through the engagement of the extrinsic pathway.
To explore this hypothesis, we ectopically expressed, by retroviral infection, the natural caspase-8 inhibitor CrmA (21) in DKO cells. DKO cells expressing CrmA were resistant to TNFα-induced apoptosis (Fig. 2). Cell death, caspase activation, and PARP processing were all impaired in response to TNFα in DKO-CrmA cells compared with control, empty vector-infected cell line (DKO-Hygro) expressing only the resistance gene Hygro (Fig. 2, A–C).
FIGURE 2.

G5 induced caspase activation in DKO cells depends on caspase-8. A, DKO cells expressing vesicular stomatitis virus-tagged crmA or Hygro (hygromycin resistance) genes treated with increasing concentrations of TNF-α (1, 10, or 80 ng/ml) and cycloheximide (CHX; 1 mg/ml), with cycloheximide alone (1 mg/ml) or left untreated. 36 h later, cell death was scored by trypan blue staining (means ± S.D., n = 3). B, DEVDase activity in DKO cells expressing vesicular stomatitis virus-tagged crmA or Hygro genes treated as in A (means ± S.D., n = 3). C, processing of PARP in DKO cells expressing Hygro or crmA genes and treated for 36 h with TNF-α (10 ng/ml). Equal amounts of cell lysates were subjected to SDS-PAGE. Immunoblots were performed using the indicated antibodies. Tubulin was used as a loading control. D, DKO cells expressing crmA or Hygro genes were treated for 20 h with the indicated concentrations of G5. Cell death was scored by trypan blue staining (means ± S.D., n = 3). E, processing of HDAC4 and caspase-3 in DKO cells expressing Hygro or crmA genes and treated for 20 h with the indicated concentrations of G5. Equal amounts of cell lysates were subjected to SDS-PAGE. Immunoblots were performed using the indicated antibodies. Tubulin was used as loading control. The asterisk indicates a unspecific band.
When DKO-CrmA and DKO-Hygro cells were compared for the resistance to G5-induced cell killing, we scored identical percentages of death (Fig. 2D). In contrast, caspase-3 and HDAC4 cleavages in response to G5 were dramatically reduced in DKO-CrmA compared with DKO-Hygro cells (Fig. 2E). From these experiments, we can conclude that although the death receptor pathway plays a fundamental role in the G5-induced activation of caspases observed in DKO cells, caspase activation is dispensable for the death of these cells.
G5 and Bortezomib Induce Autophagy, Which Is Unrelated to the Appearance of Cell Death—DKO cells when exposed for prolonged times to etoposide can die by type II PCD or autophagic cell death (22). LC3 (light chain 3) is a specific marker to detect autophagosomes in mammalian cells (11, 20, 23). During autophagy, LC3 undergoes a conversion from the LC3-I isoform to the LC3-II isoform that is specific for autophagosomes.
To directly tackle the question of whether autophagy is elicited by G5 in DKO cells, we analyzed endogenous LC3 conversion by immunoblot. WT and DKO cells were incubated for 6 and 12 h (Fig. 3A) with G5 or with bortezomib, as a comparison (Fig. 3B). Tamoxifen was used as a positive control (20). Both UPSIs induced autophagy in mouse embryo fibroblast cells, although autophagy was more pronounced in WT with respect to DKO cells. Induction of autophagy was confirmed by electron microscopy. Fig. 3, C–E, illustrates the appearance of autophagosomes and multivesicular bodies in DKO cells treated with G5. Autophagosomes were similarly observed in WT cells treated with G5 (data not shown).
FIGURE 3.

Autophagy in G5 and bortezomib treated cells. A, processing of LC3 in DKO and WT cells treated for 6 or 12 h with G5 (10 μm) or tamoxifen (12.5 μm). Equal amounts of cell lysates were subjected to SDS-PAGE. Immunoblots were performed using the indicated antibodies, and the LC3 conversion ratio was quantified by densitometric scanning of the Western blot shown. Tubulin was used as loading control. B, processing of LC3 in DKO and WT cells treated for 6 or 12 h with bortezomib (1 μm) or tamoxifen (12.5 μm). Equal amounts of cell lysates were subjected to SDS-PAGE. Immunoblots were performed using the indicated antibodies, and the LC3 conversion ratio was quantified by densitometric scanning of the Western blot shown. Tubulin was used as loading control. C, the electron micrograph shows the ultrastructure of multivesicular bodies (arrowhead) in DKO cells treated with 10 μm G5 for 12 h. D, electron micrograph at higher magnification showing multivesicular bodies in DKO cells treated with 10 μm G5 for 12 h. E, the electron micrograph at higher magnification showing detailed autophagosome structure in DKO cells treated with 10 μm G5 for 12 h. F, DKO cells were pretreated for 1 h with 3-methyladenine (3MA) (10 mm) or bafilomycin (0.1 μm) or left untreated (–). Next, G5 (10 μm), bortezomib (10 μm), or tamoxifen (12.5 μm) were added to the culture medium as indicated. 20 h later, cell death was scored by trypan blue staining (means ± S.D., n = 3).
The fact that both inhibitors induced autophagic markers in DKO seems to rule out a role for autophagy as a major death pathway differentially elicited by G5 in apoptosis-defective cells. To further corroborate this conclusion, we used two pharmacological inhibitors of autophagy, such as 3-methyladenine and bafilomycin (20). As illustrated in Fig. 3F, death of DKO cells in response to G5 was not influenced by the presence of the two inhibitors.
In conclusion, both cell lines activate autophagy in response to bortezomib and G5. However, autophagy is dispensable for the death of DKO cells in response to G5.
G5 Can Trigger Necrosis—Having excluded the involvement of caspases and autophagy in the death of DKO cells treated with G5, we investigated whether these cells die by type III cell death/necrosis (10, 24).
To determine whether cell death in response to G5 was occurring by necrosis, WT and DKO cells were incubated with annexin-V-FITC, which binds to phosphatidylserine exposed on the cell surface during apoptosis, and propidium iodide, which accumulates in late apoptotic or necrotic cells. The FACS analysis (see Fig. S2) revealed that in the presence of 10 μm G5, few cells were in early apoptosis (single positive for annexin V), whereas many cells were in late apoptosis/necrosis (double-positive for annexin V/PI).
To confirm the cytofluorometric analysis and to undoubtedly discern between late apoptosis and necrosis, we decided to adopt a new assay. We used time lapse epifluorescence microscopy in living cells to score the appearance, over time, of two different markers for each type of death (necrosis/apoptosis). Cell shrinkage together with membrane blebbing were considered as apoptotic markers; instead, the appearance of swelling (plasma membrane blistering) and the uptake of ethidium bromide were utilized as necrotic markers (25). Fig. 4 exemplifies a typical time lapse analysis of cells dying for apoptosis (Fig. 4A) or necrosis (Fig. 4B).
FIGURE 4.

Real time analysis of G5-induced cell death in WT cells. A, real time epifluorescence microscopy. Images were collected every 2 min after treatment of WT cells with 1.25μm G5. Representative images at the indicated times, differential interference contrast (upper panels) and ethidium bromide (lower panels), are shown. B, real time epifluorescence microscopy. Images were collected every 2 min after treatment of DKO cells with 10 μm G5. Representative images at the indicated times, differential interference contrast (upper panels) and ethidium bromide (lower panels), are shown.
WT cells were incubated with bortezomib, etoposide, or low concentrations of G5 (1.25 μm), all well established apoptotic stimuli. Intriguingly, with all the three insults, the first cells died 6–7 h after the addition of drugs. The apoptotic responses were asynchronous, spanning 6 h with etoposide or bortezomib and 12 h with G5. Membrane blebbing and cell shrinkage were the first two markers that appeared, thus confirming that the three drugs elicited apoptosis (Fig. 5). After a certain period of time, the two necrotic markers become visible (blister of the plasma membrane and ethidium bromide influx), which indicate the emerging of the secondary necrosis. In the case of G5, secondary necrosis was separated from apoptosis by 140 ± 54 min, whereas for bortezomib, the interval was 113 ± 30 min. In the case of etoposide, the lag time separating apoptosis from necrosis was even shorter, with 89 ± 44 min.
FIGURE 5.

Quantitative analysis of the time lapse studies. WT or DKO cells were treated with the indicated concentrations of bortezomib, G5, or etoposide and subjected to the time course analysis, as explained under “Materials and Methods.” The appearance of membrane blebbing, membrane blistering (cell swelling), and the uptake of ethidium bromide was scored in vivo. Each position along the x axis represents a single cell. ○, the appearance of membrane blebbing; ▵, membrane blister; ▪, ethidium bromide influx (EI).
When G5 was used at higher concentrations (5 or 10 μm), apoptosis was essentially undetectable in both WT and DKO cells, and membrane blistering and plasma membrane permeabilization (ethidium bromide uptake) become evident independently from apoptosis, suggesting that G5 at such concentrations directly activates a necrotic response. Interestingly, the time lapse analysis indicates that similarly to apoptosis and also necrosis in response to G5 is an asynchronous response, with some cells dying soon after the treatment (3–4 h after drug addition) and others 20 h later.
G5-induced Necrosis Is Characterized by HMGB1 Release—The time lapse analysis has indicated that G5 can activate a necrotic response. During necrotic cell death, the chromatin-associated protein HMGB1 is released in the extracellular environment, where it acts as a ligand for the monocyte/macrophage scavenger receptor RAGE (26–28). To further prove the induction of necrotic cell death by G5, DKO cells were evaluated for HMGB1 localization by immunofluorescence. In untreated cells, HMGB1 shows an exclusively nuclear localization; by contrast, in many G5-treated cells, HMGB1 was translocated into the cytosol (Fig. 6A). The quantitative analysis reported in Fig. 6B demonstrates that HMGB1 translocation into the cytosol can be observed only with a “pronecrotic” concentration of G5 (10 μm) in both WT and DKO cells but not when a “proapoptotic” concentration (1.25 μm) was used in WT cells. The translocation of HMGB1 into the cytosol was also investigated by subcellular fractionation. Fig. 6C confirms the accumulation of HMGB1 into the cytosol in response to pronecrotic concentrations of G5. Finally, we proved that HMGB1 was released in the extracellular medium of G5-treated DKO cells. The alkylating DNA damage agent methyl methanesulfonate was used as a positive control for HMGB1 release (Fig. 6D).
FIGURE 6.

G5-induced necrosis is characterized by HMGB1 release. A, DKO cells, grown on coverslips, were treated for 6 h with G5, as indicated. Immunofluorescences were performed, and epifluorescence microscopy followed by deconvolution analysis was used to visualize HMGB1 localization. Hoechst 33258 staining was applied to mark nuclei. B, quantitative analysis of the immunofluorescence studies exemplified in A. WT and DKO cells, grown on coverslips were treated for 6 h with G5 as indicated, and the cytosolic localization of HMGB1 was scored after immunofluorescence analysis. C, distribution of HMGB1 among subcellular fractions. WT and DKO cells were treated for 6 h with G5 as indicated. Crude nuclear and cytosolic fractions were obtained using detergent lysis, as described under “Materials and Methods.” Protein samples were prepared for Western blotting, and membranes were probed with the indicated antibodies. The transcription factor MEF2C and tubulin were used as controls for nuclear and cytosolic fractions. D, DKO cells were treated with methyl methanesulfonate (100 μg/ml) or G5 (10 μm), culture medium was collected 20 h later, and cells were lysed in SDS buffer. HMGB1 was detected by immunoblotting in both lysates and culture medium. Histones were visualized after Coomassie Blue staining. E, electron micrograph showing a WT cell in early necrosis after incubation with 10 μm G5. F, electron micrograph showing a DKO cell in early necrosis after incubation with 10 μm G5. G, electron micrograph showing a DKO cell in late necrosis after incubation with 10 μm of G5.
Next, the appearance of necrosis was investigated by electron microscopy. Both WT and DKO cells acquired necrotic characteristics upon treatment with 10 μm of G5. Fig. 6, E and F, represents early WT and DKO necrotic cells, respectively, whereas Fig. 6G represents a late DKO necrotic cell. Organelle swelling and intracellular vacuolization are well evident in the early necrotic cells, whereas nuclear disintegration without condensation was prominent in the late necrotic cell (Fig. 6G).
G5 Induces Rapid Mitochondrial Depolarization before Plasma Membrane Swelling—Mitochondrial membrane potential (ΔΨm) is often employed as an indicator of cellular viability, and its destruction has been implicated in many cell death processes (17). Therefore, we explored whether G5-induced necrosis is accompanied by ΔΨm dissipation. A time lapse analysis was performed to understand whether ΔΨm dissipation occurs before or after cell swelling. Fig. 7, A and B, illustrates that both in WT and DKO cells, ΔΨm dissipation is very rapid (within 5 min) and appears ∼5 min before plasma membrane blistering. We also analyzed for comparison ΔΨm in cells undergoing apoptosis (WT cells treated with bortezomib). Fig. 7C shows that ΔΨm dissipation during apoptosis is less pronounced and delayed when compared with the necrotic death (within 10 min).
FIGURE 7.

Mitochondrial depolarization induced by G5 in DKO cells. A, decrease of mitochondrial TMRM uptake in single WT cells as marker of ΔΨm. The ΔΨm was compared with the appearance of membrane blisters, a necrotic marker in cells treated with 5 μm G5. B, decrease of mitochondrial TMRM uptake in single DKO cells as marker of ΔΨm. The ΔΨm was compared with the appearance of membrane blisters, a necrotic marker in cells treated with 5 μm G5. C, decrease of mitochondrial TMRM uptake in single WT cells as a marker of ΔΨm. The ΔΨm was compared with the appearance of membrane blebbing, an apoptotic marker in cells treated with 25 nm bortezomib. Twenty typical WT or DKO cells from three independent experiments were analyzed. D, DKO cells were treated with G5 (10 μm) in the presence or absence of CsA (0.8 or 5 μm), and 12 h later, cell death was scored by trypan blue staining (means ± S.D., n = 3). E, WT and DKO cells were treated with rotenone (5 μm) or DMSO. 48 h later, cell death was scored by trypan blue staining (means ± S.D., n = 3).
Opening of the permeability transition pore (PTP) may result in ΔΨm dissipation and necrosis (29). In principle, G5 could induce mitochondrial stress, PTP opening, and mitochondrial depolarization.
CsA is commonly used inhibitor of the PTP (30). On this ground, DKO cells were incubated with CsA and treated with G5. Similar results were obtained when 5 or 10 μm concentrations of G5 were used (Fig. 7D). CsA was unable to prevent cell death in response to G5 treatment. We also verified that CsA did not inhibit G5-mediated Δψm dissipation (data not shown).
Having observed a dissipation of the mitochondrial membrane potential, we investigated whether the pronecrotic effect of G5 could be due to an inhibition of mitochondrial function and in particular of mitochondrial respiration. We used rotenone, a well known inhibitor of the mitochondrial respiration, as a model to evaluate the importance of mitochondrial respiration for the death of DKO cells (31, 32). In contrast to G5, rotenone was able to kill only WT and not DKO cells (Fig. 7F). This observation tends to exclude the possibility that G5 induces necrosis and death of DKO cells through the inhibition of the mitochondrial respiration.
Characterization of the Necrotic Pathway Activated by G5—We evaluated the involvement of previously described regulators of the necrotic death, such as PARP, JNK, receptor-interacting protein kinase, Bcl-2, Bcl-xL, intracellular Ca2+, and oxidative stress, in the necrotic death elicited by G5 (Figs. S3–S5). Our results indicate that not even one of these necrotic regulators plays a relevant role during G5-induced necrosis.
Interestingly, the time lapse analysis showed in Fig. 4B clearly points out that adding pronecrotic concentrations of G5 to the cells causes (i) suppression of plasma membrane protrusions, such as filopodia and lamellipodia, and (ii) reduction in adhesion/spreading, which possibly determines the cell round-up (Fig. 8A). These alterations are an early reaction to G5, detectable ∼5.31 ± 3.58 h before the ΔΨm collapse and the appearance of necrotic markers.
FIGURE 8.

Adhesion to the ECM counteracts G5-dependent necrosis. A, analysis of cell adhesion/spreading in cells treated with bortezomib or G5. Single images from the time lapse analysis of cells treated with G5 (10 μm) or bortezomib (10 μm) were subjected to morphological analysis. In G5-treated cells, the morphological analysis was performed before the appearance of necrosis (as marked by membrane blister and ethidium bromide influx staining) and within 20 h from the drug addition. Since bortezomib-treated cells do not show evident changes in cell shape, the morphological analysis was performed at 20 h from treatment. The cell spread area was quantified using MetaMorph. Results shown are pooled for 30 cells from three independent experiments. B, confocal pictures of DKO cells treated for 3 h with 10 μm G5 or bortezomib. Immunofluorescence analysis were performed to visualize HMGB1 subcellular localization. TRITC-phalloidin was used to decorate actin filaments. The arrows point to cells with evident alterations in cell morphology and adhesion but containing HMGB1 in the nucleus. The arrowheads point to cells with a cytoplasmic localization of HMGB1. C, quantitative analysis of stress fibers distribution in DKO cells treated as in B. 300 cells for each experiment were scored (means ± S.D., n = 3). D, time lapse images of a representative DKO cell treated with G5 (10 μm) and stained for enhanced yellow fluorescent protein-actin. The arrows indicate the disruption of actin filaments, and arrowheads show the uptake of ethidium bromide. The numbers indicate minutes from G5 addition. G and H, DKO cells expressing crmA or the relative control gene Hygro were grown on different ECM proteins, as indicated. 6 h after seeding, cells were incubated with G5 or 2,3-dimethoxy-1,4-naphthoquinone (DMNQ), as indicated. After 12 h, cell death was scored by trypan blue staining (means ± S.D., n = 3).
Hence, it is possible that G5 by inhibiting isopeptidases might interfere with the cellular processes involved in the regulation/maintenance of the cell shape/adhesion. To confirm the induction of early morphological changes by G5, we analyzed the organization of the actin cytoskeleton in cells that have been incubated with G5 for a short period of time (3 h) before necrosis occurred. Actin architecture was analyzed in bortezomib-treated or untreated cells for comparison. The subcellular localization of HMGB1 was used as a marker of cellular stress.
Fig. 8B demonstrates that, in response to G5 and before the appearance of the necrosis, the actin cytoskeleton and cell morphology are dramatically altered. Cells are rounded up, the adhesion surface is reduced, and stress fibers are barely detectable (see the quantitative analysis in Fig. 8C).
Bortezomib, when used at the same concentration and even for prolonged times (24 h) was never able to induce such dramatic alterations (Fig. 8B). However, although cell shape and adhesion were unaffected, some reorganization of the actin cytoskeleton, such as reduction of membrane ruffles and filopodia, were observed also in response to bortezomib.
Importantly, in many G5-treated cells showing altered actin organization and morphology, HMGB1 was still detectable into the nucleus. By contrast, in the few cells with a cytoplasmic localization of HMGB1 (Fig. 8B, arrowheads) cell shape alterations were less evident. These observations suggest that G5 but not bortezomib triggers early changes in actin organization, cell shape, and adhesion and that these changes are uncoupled to the accumulation of HMGB1 into the cytosol.
To further characterize the G5-induced changes in cell adhesion and actin organization, we expressed enhanced yellow fluorescent protein-tagged actin protein in DKO cells. This fusion protein becomes incorporated in the actin fibers forming different structures within the cells; thus, their dynamics can be monitored in vivo. Fig. 8D shows a typical DKO cell expressing enhanced yellow fluorescent protein-actin. Actin cables enriched at the cell periphery can be observed. After treatment with 10 μm G5, we sequentially observed the disappearance of actin cables, the reduction of cell adhesion, and finally the emergence of the necrotic phenotype, attested by the loss of the plasma membrane integrity (ethidium uptake).
If alterations in actin cytoskeleton and cell adhesion, as induced by G5, are critical events in the appearance of the necrotic phenotype, by strengthening cell-ECM interactions, it would be possible to counteract the necrotic response.
To evaluate this hypothesis, we compared the cell death induced by G5 in cells cultured on the ECM components fibronectin and collagen with respect to cells plated on uncoated Petri dishes. To exclude the secondary induction of apoptosis, these studies were performed in DKO cells expressing CrmA or hygromycin as control.
Fig. 8, E and F, illustrates that both ECM components can counteract the necrotic response of G5. Importantly, this effect was specific for G5. In fact, fibronectin and collagen were unable to antagonize the necrotic response induced by the oxidation-inducing agent 2,3-dimethoxy-1,4-naphthoquinone. We also evaluated the effect of cell-ECM interactions on the death induced by bortezomib. Since DKO cells are resistant to bortezomib, scoring 10–20% of trypan blue-positive cells after 20 h of treatment, the analysis on cell death was extended also at 24 h. Fig. 8, G and H, demonstrates that adhesion to fibronectin or collagen was unable to modulate the death of DKO cells in response to bortezomib.
DISCUSSION
During tumor progression, neoplastic cells accumulate mutations in the common components of the apoptotic program (9), which render them particularly refractory to chemotherapeutic treatments and more prone to spread into the organism. In principle, a further possibility to eradicate neoplastic cells that escaped apoptosis is to endorse alternative death responses, such as autophagy and necrosis (10, 11). Hence, it is important to identify and characterize compounds capable of triggering multiple death responses. UPS inhibitors are emerging as interesting compounds for anti-cancer therapy. They induce apoptosis through the engagement of several pathways that finally cause mitochondrial outer membrane permeabilization (6–8). Here we have investigated the ability of bortezomib and G5, two different UPS inhibitors, to activate, in addition to apoptosis, autophagy and necrosis.
Apoptosis—Our data clearly demonstrate that bortezomib and low doses of G5 induce apoptosis. This apoptotic response is strictly dependent on the activity of Bax and Bak. In addition, we have also found that G5 can activate caspases in DKO cells. This activation requires caspase-8 activity and possibly the engagement of the death receptor pathway (15). Interestingly, NPI-0052, a recently identified molecule able to inhibit all three proteasome activities, in contrast to bortezomib, relies more on the FADD-caspase-8-mediated cell death signaling pathway (33). Hence, it is possible that blocking isopeptidases and ubiquitin disassembling or inhibiting all of the catalytic activities of the proteasome generates similar stress signals, which promote caspase-8 activation. However caspase-8, as activated by G5, is unable to efficiently sustain apoptosis, since with time, the necrotic response dominates and determines the ultimate fate of death. The appearance of necrosis explains the paradox of a reduced caspase activity when cells are incubated with higher doses of G5.
Autophagy—Bortezomib and G5 can also elicit autophagy both in WT and DKO cells. Our studies suggest that the autophagic response cannot be responsible for the death of DKO cells. On the contrary, it is possible that autophagy, when elicited by UPSIs, represents an effort to escape cell death. In agreement with our observations, recent data reported that toxicity dependent on proteasome impairment can be alleviated by autophagy (34). Cells can compensate for the block of protein turnover by increasing autophagy. Hence, these data imply that the co-treatment with inhibitors of autophagy could strengthen the antiproliferative effect of the UPSIs, including bortezomib. This hypothesis is supported by recent more general observations, proving that autophagy inhibition enhances the pro-death properties of a certain number of anti-cancer compounds, including alkylating agents (11, 35) and histone deacetylase inhibitors (36).
Necrosis—In our studies, we have observed that only G5 can trigger necrosis. A large body of evidence, such as time lapse analysis, HMGB1 release, and electron microscopy images, confirmed the appearance of necrosis. The necrotic response activated by G5 is responsible for the death of Bax/Bak mutant cells (DKO).
Necrosis has often been referred to as an unscheduled, passive form of cell death (10, 24). An important characteristic of the necrotic cell death is the rupture of the plasma membrane and the consequent spillage of cellular components. This release triggers an inflammatory response and immune amplification of the damage signal. Although recent studies have pointed out the existence of genes that regulate the necrotic death, this phenomenon is mostly obscure, and further studies are required to decipher whether or not a form of “programmed necrosis” exists.
In normal and leukemia cells, Drp-1 (dynamin-related protein-1) seems to play a relevant role when necrosis is activated by ligation of CD47 (37). Nonselective pore opening of P2X7R, a purinergic receptor regulated by extracellular ATP, is responsible for the necrotic death of thymocytes (38). PARP is a critical element of the necrotic death in response to DNA-alkylating agents caused by NAD consumption (27). Genetic studies have revealed the molecular elements governing the necrotic signaling activated by PARP. RIP1 (receptor-interacting protein 1) and TRAF2 (tumor necrosis factor receptor-associated factor 2) are important elements of this pathway. RIP1 is also involved in necrotic death induced, under particular circumstances, by death receptors Fas and TNF-α (39–41). A downstream effector of this pathway is JNK. In response to PARP activation and metabolic stress, JNK is important for triggering mitochondrial dysfunction and necrotic cell death (42).
In this work, we have tested different inhibitors of well known elements of the necrotic responses, such as JNK, PARP, RIP1, Ca2+, mitochondrial PTP, and reactive oxygen species. None of them was able to inhibit the necrotic death induced by G5. Clearly, it is possible that more than a single necrotic pathway could be activated by G5; hence, different combinations of inhibitors should be tested in the future.
In particular, the failure of CsA to block the necrotic response induced by G5 was unexpected. G5-induced necrosis is characterized by the rapid and dramatic loss of ΔΨm, which precedes the appearance of necrotic markers. In several conditions, mitochondrial depolarization is caused by the opening of the mitochondrial PTP, a high conductance channel that can be desensitized by CsA through its binding to CyP-D, a matrix peptidyl-prolyl cis-trans isomerase (30, 43). However, studies in CyP-D null mice have led to the conclusion that the PTP only regulates some forms of necrotic cell death (44, 45). It should be stressed that experiments in CyP-D null mice only allow conclusions to be made about the role of CyP-D, not of the PTP. Indeed, all studies agree that the PTP can open even in the absence of CyP-D, provided that a sufficient Ca2+ load is delivered (44–47).
This consideration also applies to studies using CsA, including our own. Indeed, our results show that G5 belongs to the class of compounds/stresses that induce necrosis and ΔΨm loss in a CsA-insensitive manner (46, 48–50). Since CsA is not a blocker of the PTP, we feel that further studies will be necessary to clarify whether G5 induces mitochondrial depolarization through a pore-dependent or -independent mechanism.
Why is G5, in contrast to bortezomib, able to trigger a necrotic response? Altering proteins turnover, promoting the accumulation of polyubiquitinated or misfolded proteins is not enough to trigger necrosis. G5 is a nonselective isopeptidase inhibitor, thus potentially able to inhibit not only deubiquitinating enzymes but also enzymes that remove a ubiquitin-like molecule, for instance SUMO (51). It is evident that cellular functions under isopeptidase control (DNA repair, membrane trafficking) are broader than those modulated by the proteasome (3, 4). Hence, it is possible that the cellular stress induced by isopeptidases inhibition is less governable by the cells, and at the end it causes necrosis.
As a matter of fact, we have noted that G5 can elicit profound alterations in the actin cytoskeleton organization and in cell adhesion. The partial but consistent anti-necrotic effect of the ECM-adhesive components collagen and fibronectin clearly suggests that these alterations are important for the pro-death effect of G5. Notably, this antinecrotic effect of collagen and fibronectin seems to be specific for G5, since, for the reactive oxygen species producer 2,3-dimethoxy-1,4-naphthoquinone-induced necrotic response, adhesion to ECMs was unable to have the antinecrotic effect.
The influence of the ECM on the execution of the apoptotic program through the actions of adhesion receptors is well established, whereas the relationships between necrosis and ECM are less characterized. Even so, mitochondria-dependent necrosis seems to have a central role in mediating myocyte and myofiber loss in skeletal muscle as a consequence of disease states associated with dysfunctions in cell-ECM interactions (52).
Can we exclude off-target effects of G5 responsible for the necrotic death? Since G5 a broad isopeptidase inhibitor, it is difficult to answer this question. Nevertheless, we can exclude at least one putative off-target of G5: the glutathione. In fact, it has been hypothesized that some of the effects of compounds similar to G5 could be caused by a depletion of glutathione (53). This is not the case in our experimental system. Indeed, treatment of DKO cells with l-buthionine-(SR)-sulfoximine, a selective inhibitor of γ-glutamylcysteine synthetase, which depletes glutathione (54), was unable to induce morphological changes and cell death (data not shown). The inability of both bortezomib and glutathione depletion to trigger alterations in adhesion to ECM and necrosis supports the notion that G5 exerts its effect via inhibition of the isopeptidases.
In conclusion, we have shown that cells can activate multiple death pathways in response to UPSIs and that G5 can promote a peculiar necrotic response. It is evident that further investigations and the generation of more specific isopeptidase inhibitors will be necessary to establish the role of specific DUB or ubiquitin-like proteases in the induction of necrosis. Interestingly, since cancer cells usually show reduced adhesion to the ECM, they could be more prone to the pronecrotic effect of G5. Clearly, a therapy that induces a necrotic cell death will initiate an immune response to tumor cells. Whether the inflammation associated with necrosis is desirable or leads to further tumor growth is still a matter of debate (11).
Supplementary Material
Acknowledgments
We thank Michela Faleschini, Elisa Trevisan, and Emanuela Aleo (Dipartimento di Scienze e Tecnologie Biomediche, Università di Udine) for helping in some experiments.
This work was supported by grants from the Associazione Italiana Ricerca sul Cancro, Regione Friuli-Venezia Giulia (Progetto AITT), and Ministero della Salute (to C. B. and R. M.) and Ministero dell'Università e Ricerca (to C. B.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5.
Footnotes
The abbreviations and trivial name used are: UPS, ubiquitin-proteasome system; UPSI, ubiquitin-proteasome system inhibitor; CsA, cyclosporin A; DKO, double knock-out; PARP, poly(ADP-ribose) polymerase; JNK, c-Jun N-terminal kinase; ERK, extracellular signal-regulated kinase; Z, benzyloxycarbonyl; TRITC, tetramethylrhodamine isothiocyanate; TNF, tumor necrosis factor; WT, wild type; SAPK, stress-activated protein kinase; PBS, phosphate-buffered saline; fmk, fluoromethyl ketone; PTP, permeability transition pore; ECM, extracellular matrix; G5, 4H-thiopyran-4-one, tetrahydro-3,5-bis[(4-nitrophenyl)methylene]-1,1-dioxide.
References
- 1.Ciechanover, A. (2005) Cell Death Differ. 12 1178–1190 [DOI] [PubMed] [Google Scholar]
- 2.Hershko, A., and Ciechanover, A. (1998) Annu. Rev. Biochem. 67 425–479 [DOI] [PubMed] [Google Scholar]
- 3.Amerik, A. Y., and Hochstrasser, M. (2004) Biochim. Biophys. Acta 1695 189–207 [DOI] [PubMed] [Google Scholar]
- 4.Sun, L., and Chen, Z. J. (2004) Curr. Opin. Cell Biol. 16 119–126 [DOI] [PubMed] [Google Scholar]
- 5.Brancolini, C. (2008) Curr. Mol. Pharm. 1 24–37 [DOI] [PubMed] [Google Scholar]
- 6.Demarchi, F., and Brancolini, C. (2005) Drug Resist. Updates 8 359–368 [DOI] [PubMed] [Google Scholar]
- 7.Rajkumar, S. V., Richardson, P. G., Hideshima, T., and Anderson, K. C. (2005) J. Clin. Oncol. 23 630–639 [DOI] [PubMed] [Google Scholar]
- 8.Milano, A., Iaffaioli, R. V., and Caponigro, F. (2007) Eur. J. Cancer 43 1125–1133 [DOI] [PubMed] [Google Scholar]
- 9.Rodriguez-Nieto, S., and Zhivotovsky, B. (2006) Curr. Pharm. Des. 12 4411–4425 [DOI] [PubMed] [Google Scholar]
- 10.Golstein, P., and Kroemer, G. (2007) Trends Biochem. Sci. 32 37–43 [DOI] [PubMed] [Google Scholar]
- 11.Amaravadi, R. K., and Thompson C. B. (2007) Clin. Cancer Res. 13 7271–7279 [DOI] [PubMed] [Google Scholar]
- 12.Lindsten, T., Ross, A. J., King, A., Zong, W. X., Rathmell, J. C., Shiels, H. A., Ulrich, E., Waymire, K. G., Mahar, P., Frauwirth, K., Chen, Y., Wei, M., Eng, V. M., Adelman, D. M., Simon, M. C., Ma, A., Golden, J. A., Evan, G., Korsmeyer, S. J., MacGregor, G. R., and Thompson, C. B. (2000) Mol. Cell. 6 1389–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruiz-Vela, A., Opferman, J. T., Cheng, E. H., and Korsmeyer, S. J. (2005) EMBO Rep. 6 379–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindsten, T., and Thompson, C. B. (2006) Cell Death Differ. 13 1272–1276 [DOI] [PubMed] [Google Scholar]
- 15.Aleo, E., Henderson, C. J., Fontanini, A., Solazzo, B., and Brancolini, C. (2006) Cancer Res. 66 9235–9244 [DOI] [PubMed] [Google Scholar]
- 16.Henderson, C., Mizzau, M., Paroni, G., Maestro, R., Schneider, C., and Brancolini, C. (2003) J. Biol. Chem. 278 12579–12589 [DOI] [PubMed] [Google Scholar]
- 17.Henderson, C. J., Aleo, E., Fontanini, A., Maestro, R., Paroni, G., and Brancolini, C. (2005) Cell Death Differ. 12 1240–1254 [DOI] [PubMed] [Google Scholar]
- 18.Paroni, G., Henderson, C., Schneider, C., and Brancolini, C. (2001) J. Biol. Chem. 276 21907–21915 [DOI] [PubMed] [Google Scholar]
- 19.Paroni, G., Mizzau, M., Henderson, C. J., Del Sal, G., Schneider, C., and Brancolini, C. (2004) Mol. Biol. Cell. 15 2804–2818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Demarchi, F., Bertoli, C., Copetti, T., Tanida, I., Brancolini, C., Eskelinen, E. L., and Schneider, C. (2006) J. Cell Biol. 175 595–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou, Q., Snipas, S., Orth, K., Muzio, M., Dixit, V. M., and Salvesen, G. S. (1997) J. Biol. Chem. 272 7797–7800 [DOI] [PubMed] [Google Scholar]
- 22.Shimizu, S., Kanaseki, T., Mizushima, N., Mizuta, T., Arakawa-Kobayashi, S., Thompson, C. B., and Tsujimoto, Y. (2004) Nat. Cell Biol. 6 1221–1228 [DOI] [PubMed] [Google Scholar]
- 23.Kabeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda, T., Kominami, E., Ohsumi, Y., and Yoshimori, T. (2000) EMBO J. 19 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zong, W. X., and Thompson, C. B. (2006) Genes Dev. 20 1–15 [DOI] [PubMed] [Google Scholar]
- 25.Sardão, V. A., Oliveira, P. J., Holy, J., Oliveira, C. R., and Wallace, K. B. (2007) BMC Cell Biol. 8 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scaffidi, P., Misteli, T., and Bianchi, M. E. (2002) Nature 418 191–195 [DOI] [PubMed] [Google Scholar]
- 27.Zong, W. X., Ditsworth, D., Bauer, D. E., Wang, Z. Q., and Thompson, C. B. (2004) Genes Dev. 18 1272–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ditsworth, D., Zong, W. X., and Thompson, C. B. (2007) J. Biol. Chem. 282 17845–17855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsujimoto, Y., and Shimizu, S. (2007) Apoptosis 12 835–840 [DOI] [PubMed] [Google Scholar]
- 30.Bernardi, P., Krauskopf, A., Basso, E., Petronilli, V., Blachly-Dyson, E., Di Lisa, F., and Forte, M. A. (2006) FEBS J. 273 2077–2099 [DOI] [PubMed] [Google Scholar]
- 31.Smaili, S. S., Hsu, Y. T., Sanders, K. M., Russell, J. T., and Youle, R. J. (2001) Cell Death Differ. 8 909–920 [DOI] [PubMed] [Google Scholar]
- 32.Marella, M., Seo, B. B., Matsuno-Yagi, A., and Yagi, T. (2007) J. Biol. Chem. 282 24146–24156 [DOI] [PubMed] [Google Scholar]
- 33.Chauhan, D., Catley, L., Li, G., Podar, K., Hideshima, T., Velankar, M., Mitsiades, C., Mitsiades, N., Yasui, H., Letai, A., Ovaa, H., Berkers, C., Nicholson, B., Chao, T. H., Neuteboom, S. T., Richardson, P., Palladino, M. A., and Anderson, K. C. (2005) Cancer Cell. 5 407–419 [DOI] [PubMed] [Google Scholar]
- 34.Pandey, U. B., Nie, Z., Batlevi, Y., McCray, B. A., Ritson, G. P., Nedelsky, N. B., Schwartz, S. L., DiProspero, N. A., Knight, M. A., Schuldiner, O., Padmanabhan, R., Hild, M., Berry, D. L., Garza, D., Hubbert, C. C., Yao, T. P., Baehrecke, E. H., and Taylor, J. P. (2007) Nature 447 859–863 [DOI] [PubMed] [Google Scholar]
- 35.Amaravadi, R. K., Yu, D., Lum, J. J., Bui, T., Christophorou, M. A., Evan, G. I., Thomas-Tikhonenko, A., and Thompson, C. B. (2007) J. Clin. Invest. 117 326–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carew, J. S., Nawrocki, S. T., Kahue, C. N., Zhang, H., Yang, C., Chung, L., Houghton, J. A., Huang, P., Giles, F. J., and Cleveland, J. L. (2007) Blood 110 313–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bras, M., Yuste, V. J., Roué, G., Barbier, S., Rancho, P., Virely, C., Rubio, M., Baudet, S., Esquerda, J. E., Merle-Béral, H., Sarfati, M., and Susin, S. A. (2007) Mol. Cell. Biol. 27 7073–7088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Auger, R., Motta, I., Benihoud, K., Ojcius, D. M., and Kanellopoulos, J. M. (2005) J. Biol. Chem. 280 28142–28151 [DOI] [PubMed] [Google Scholar]
- 39.Chan, F. K., Shisler, J., Bixby, J. G., Felices, M., Zheng, L., Appel, M., Orenstein, J., Moss, B., and Lenardo, M. J. (2003) J. Biol. Chem. 278 51613–51621 [DOI] [PubMed] [Google Scholar]
- 40.Holler, N., Zaru, R., Micheau, O., Thome, M., Attinger, A., Valitutti, S., Bodmer, J. L., Schneider, P., Seed, B., and Tschopp, J. (2000) Nat Immunol. 1 489–495 [DOI] [PubMed] [Google Scholar]
- 41.Temkin, V., Huang, Q., Liu, H., Osada, H., and Pope, R. M. (2006) Mol. Cell. Biol. 26 2215–2225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu, Y., Huang, S., Liu, Z. G., and Han, J. (2006) J. Biol. Chem. 281 8788–8795 [DOI] [PubMed] [Google Scholar]
- 43.Halestrap, A. P., and Davidson, A. M. (1990) Biochem. J. 268 153–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baines, C. P., Kaiser, R. A., Purcell, N. H., Blair, N. S., Osinska, H., Hambleton, M. A., Brunskill, E. W., Sayen, M. R., Gottlieb, R. A., Dorn, G. W., Robbins, J., and Molkentin, J. D. (2005) Nature 434 658–662 [DOI] [PubMed] [Google Scholar]
- 45.Nakagawa, T., Shimizu, S., Watanabe, T., Yamaguchi, O., Otsu, K., Yamagata, H., Inohara, H., Kubo, T., and Tsujimoto, Y. (2005) Nature 434 652–658 [DOI] [PubMed] [Google Scholar]
- 46.Basso, E., Fante, L., Fowlkes, J., Petronilli, V., Forte, M. A., and Bernardi, P. (2005) J. Biol. Chem. 280 18558–18561 [DOI] [PubMed] [Google Scholar]
- 47.Schinzel, A. C., Takeuchi, O., Huang, Z., Fisher, J. K., Zhou, Z., Rubens, J., Hetz, C., Danial, N. N., Moskowitz, M. A., and Korsmeyer, S. J. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 12005–12010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marques-Santos, L. F., Coqueiro, V. M., and Rumjanek, V. M. (2006) Cell Biol. Int. 30 197–204 [DOI] [PubMed] [Google Scholar]
- 49.Brustovetsky, N., and Dubinsky, J. M. (2000) J. Neurosci. 20 103–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Malkevitch, N. V., Dedukhova, V. I., Simonian, R. A., Skulachev, V. P., and Starkov, A. A. (1997) FEBS Lett. 412 173–178 [DOI] [PubMed] [Google Scholar]
- 51.Nicholson, B., Leach, C. A., Goldenberg, S. J., Francis, D. M., Kodrasov, M. P., Tian, X., Sanks, J., Sterner, D. E., Bernal, A., Mattern, M. R., Wilkinson, K. D., and Butt, T. R. (2008) Protein Sci. 17 1035–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Millay, D. P., Sargent, M. A., Osinska, H., Baines, C. P., Barton, E. R., Vuagniaux, G., Sweeney, H. L., Robbins, J., and Molkentin, J. D. (2008) Nat. Med. 14 442–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li, Z., Melandri, F., Berdo, I., Jansen, M., Hunter, L., Wright, S., Valbrun, D., and Figueiredo-Pereira, M. E. (2004) Biochem. Biophys. Res. Commun. 19 1171–1180 [DOI] [PubMed] [Google Scholar]
- 54.Griffith, O. W., and Meister, A. (1979) J. Biol. Chem. 254 7558–7560 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.