

Abstract
Disposition of the second messenger nitric oxide (NO) in mammalian tissues occurs through multiple pathways including dioxygenation by erythrocyte hemoglobin and red muscle myoglobin. Metabolism by a putative NO dioxygenase activity in non-striated tissues has also been postulated, but the exact nature of this activity is unknown. In the present study, we tested the hypothesis that cytoglobin, a newly discovered hexacoordinated globin, participates in cell-mediated NO consumption. Stable expression of small hairpin RNA targeting cytoglobin in fibroblasts resulted in decreased NO consumption and intracellular nitrate production. These cells were more sensitive to NO-induced inhibition of cell respiration and proliferation, which could be restored by re-expression of human cytoglobin. We also demonstrated cytoglobin expression in adventitial fibroblasts as well as vascular smooth muscle cells from various species including human and found that cytoglobin was expressed in the adventitia and media of intact rat aorta. These results indicate that cytoglobin contributes to cell-mediated NO dioxygenation and represents an important NO sink in the vascular wall.
Nitric oxide (NO)2 plays a central role in the vasculature and regulates oxygen supply by relaxing smooth muscle and inhibiting mitochondrial respiration. NO is produced by nitric-oxide synthase and through mobilization of storage pools such as nitrite. Less is known on the mechanism of NO inactivation and removal in the vasculature, although it would be surprising that blood vessels rely on nonspecific chemical reactions to control its disposition.
Blood removes NO through reaction with excess oxyhemoglobin in erythrocytes, but diffusional barriers such as the red blood cell-free layer near the vessel wall allow autocrine NO to occur in significant amounts in the vascular wall (1, 2). In striated muscles, oxymyoglobin is an NO scavenger, and studies using transgenic mice lacking myoglobin have demonstrated a role for oxymyoglobin in attenuating NO-mediated cardiac dysfunction (3, 4). In contrast, NO inactivation in non-striated tissues is poorly understood, and examination of NO consumption in various cells suggests mechanisms that differ in NO saturation, oxygen and cyanide sensitivity, and product formation (5–8). Specific pathways include increased partition of NO in cell membranes (7) and consumption of NO by cytochrome c oxidase (9–11), NADPH oxidase (12), 15-lipoxygenase (13), prostaglandin-H synthase (14), and myeloperoxidase (15).
Many of the above mechanisms implicate metalloproteins as modulators of NO bioavailability among which the role of oxyglobins such as oxyhemoglobin and oxymyoglobin have been already highlighted (16). One of the primary mechanisms is the dioxygenation of NO to form nitrate and the ferric (Fe(III)) form of the protein. The recent discovery of two new mammalian globins, cytoglobin and neuroglobin, would suggest that the reactions of NO with globins are not limited to red blood cells and striated muscles but could extend to other cell types including those of blood vessels (17–19). In the case of cytoglobin, its expression is thought to be restricted to fibroblast lineages, but its presence in adventitial fibroblasts or other cell types in blood vessels is unknown (20). In addition, cytoglobin lacks an intrinsic reductase activity required for the reduction of the heme iron after NO oxidation, a situation that would rapidly deplete the NO-metabolizing pool of cytoglobin in cells unless the cellular environment may provide adequate reducing capacity (21).
Based on molecular manipulations to decrease and increase cytoglobin levels, we now show that cytoglobin is expressed and dioxygenates NO in fibroblasts. The effect of cytoglobin on cellular NO consumption, nitrate accumulation, cellular respiration, and NO-induced cytostasis reveals an activity associated with cytoglobin that represents an important physiological pathway for the metabolism of NO in tissues. Our observations that cytoglobin is expressed in aortic adventitial fibroblasts and vascular smooth muscle cells in vivo indicate that cytoglobin may be critical to the regulation of NO bioactivity in the vessel wall.
EXPERIMENTAL PROCEDURES
Reagents—DEA/NO, DETA/NO, PROLI/NO, and Sp/NO were all purchased from Cayman Chemical. A rabbit polyclonal antibody to CYGB using a peptide to the N terminus (Ac-CMEDPLEMERSPQLRK-amide) was generated (QCB). The mouse fibroblast cell line NIH-3T3 was obtained from the American Type Culture Collection (Manassas, VA). All other reagents were purchased from Sigma-Aldrich.
Stable Knockdown of Cytoglobin in Murine Fibroblasts—To derive the stably transfected cell line 4 μg of pSilencer-shCYGB 2 or pSilencer-shScrambled were transfected into one 1 × 105 NIH-3T3 cells using Lipofectamine 2000. At 4 h post-transfection the medium was changed to growth medium containing 3 μg/ml puromycin. After 72 h the cells were trypsinized and counted using a Coulter counter. Cells were then seeded at 1 × 103 cells in T-75 flasks containing the puromycin selection medium. The cells were allowed to form colonies and grow under selection for ∼2 weeks. Individual colonies were picked using sterile cotton swabs and seeded in 24-well plates containing selection medium. The cells were allowed to grow and expand under selection (3 μg/ml puromycin) and were evaluated for protein knockdown. N2 (shScrambled) are used as control cells, and C13 (shCYGB) have reduced levels of cytoglobin. To create the re-expression cell line, the C13 cells were transfected as described above with a plasmid containing full-length human cytoglobin (Open Biosystems, Huntsville, AL) cloned into pcDNA3.1 (Invitrogen). These cells were grown under both puromycin (3 μg/ml) and G418S (50 μg/ml) selection.
RNA Extraction and cDNA Synthesis—RNA was extracted using TRIzol reagent and standard purification protocols. RNA was converted to cDNA using the Promega Reverse Transcription system with random hexamer primers and the supplied protocols. Prior to cDNA synthesis, mRNA was quantitated using a spectrophotometer so each reaction had the same total starting quantity (1 μg).
Quantitative PCR Analysis—Quantitative PCR was used to determine the presence of cytoglobin in NIH-3T3 mouse fibroblast cells. The primers were QPCR-Cygb-For (5′-CCA ACT GCG AGG ACG TGG-3′) and QPCR-Cygb-Rev (5′-ACT GGC TGA AGT ACT GCT TGG C-3′) (20). SYBR Green along with Taq polymerase was used (Abgene, Inc., Epsom, UK), and the amplification was performed in a regular three-step protocol: 94 °C for 15 s, 57.5 °C for 30 s, and 72 °C for 30 s, measuring fluorescence at the last step of each cycle. A melt curve was generated and observed to ensure one end product from the PCR. Data were normalized to β-actin levels and standard curves using a synthesized template.
Western Blot Analysis—Cells were lysed in radioimmune precipitation assay buffer, and equal amounts of proteins (∼50 μg) were loaded in each lane of 4–20% SDS-polyacrylamide gels (Bio-Rad) and electrophoresed at 75 V for 2.5 h. Proteins were transferred to nitrocellulose membranes, blocked in 5% nonfat milk/Tris-buffered saline for 1 h, and incubated with primary antibody overnight at 4 °C. Membranes were washed three times for 10 min with Tris-buffered saline with Tween 20, incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h, and washed again as described. Finally proteins were detected by enhanced chemiluminescence.
NO Consumption and Respiration Measurements—NO concentrations in solution were detected using an ISO-NO Mark II electrode (World Precision Instruments, Inc., Sarasota, FL). Experiments were conducted under sealed, stirred conditions where pH was adjusted to ∼7.4 prior to the start of experiment. NO consumption was measured with 2.5–3.75 × 106 cells/ml in a 2-ml total reaction volume of DPBS containing 5 mm glucose and in the presence of 100 units/ml superoxide dismutase. The NO donor was added to the chamber using a gas-tight Hamilton syringe. Heme sensitivity and oxygen dependence were determined through pretreatment of cell suspensions with 500 μm potassium cyanide (KCN) or glucose oxidase (4 units) for 15 min prior to injection of NO donors. All NO values obtained were normalized to an NaNO2 standard curve. Cell respiration and oxygen concentrations were measured using a Clark-type O2 electrode (YSI, Inc., Yellow Springs, OH). Oxygen consumption was measured with 5 × 106 cells/ml in a 4-ml total reaction volume of DPBS containing 5 mm glucose.
Nitrate and Nitrite Measurements—At the termination of NO
consumption experiments (∼30 min) the samples were centrifuged to pellet
the cells, and the medium was removed for analysis of
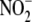 and
and
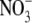 content. For intracellular
content. For intracellular
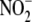 and
and
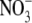 determination, 1 ml of cell
suspension was removed from the reaction vessel at the indicated times (1,
2.5, 5, 10, and 30 min) and centrifuged to pellet the cells. Cells were
resuspended in 1 ml of DPBS with 5 mm glucose and snap froze in a
dry ice, ethanol bath. Cells were subjected to three cycles of freeze-thaw.
Cell debris were pelleted, and the medium was removed for
determination, 1 ml of cell
suspension was removed from the reaction vessel at the indicated times (1,
2.5, 5, 10, and 30 min) and centrifuged to pellet the cells. Cells were
resuspended in 1 ml of DPBS with 5 mm glucose and snap froze in a
dry ice, ethanol bath. Cells were subjected to three cycles of freeze-thaw.
Cell debris were pelleted, and the medium was removed for
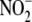 and
and
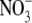 analysis. For DEA/NO experiments
analysis. For DEA/NO experiments
 and
and
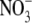 were determined using reductive
chemiluminescence with an NO Analyzer (NOA 280, GE Analytical Instruments,
Boulder, CO) as described previously
(22). Products obtained in
Sp/NO and DETA/NO experiments were analyzed by ion-pairing HPLC as described
previously (23).
were determined using reductive
chemiluminescence with an NO Analyzer (NOA 280, GE Analytical Instruments,
Boulder, CO) as described previously
(22). Products obtained in
Sp/NO and DETA/NO experiments were analyzed by ion-pairing HPLC as described
previously (23).
Immunohistochemistry—Aortas from rats were harvested and embedded in optimum temperature compound and frozen by submersion in liquid nitrogen. Tissue sections (7 μm) were cut using a Leica CM3050 cryostat, and the sections were mounted onto glass slides and stored at -80 °C. The aorta sections were fixed with precooled acetone for 10 min at 4 °C and rinsed with 1× phosphate-buffered saline. The sections were then incubated in a phosphate-buffered saline washing buffer containing 0.1% Triton X-100 for 10 min. The sections were incubated for 30 min in blocking buffer (1× phosphate-buffered saline, 5% goat serum, 0.5% fish gelatin, 0.1% Triton X-100). The sections were then treated overnight with primary antibody at 4 °C. An anti-cytoglobin primary antibody was diluted 1:10 in blocking buffer. Anti-cytoglobin and -myosin heavy chain antibodies were combined and diluted 1:10 and 1:25, respectively, in blocking buffer for co-staining experiments. The sections were rinsed with washing buffer and then incubated in secondary antibody for 1 h at room temperature. For cytoglobin staining, a 488 nm anti-rabbit secondary antibody (Molecular Probes) was diluted 1:300 in blocking buffer. For cytoglobin/myosin heavy chain co-staining, a 488 nm anti-rabbit secondary was combined with a 594 nm anti-mouse secondary, and both were diluted 1:300 in blocking buffer. Finally the sections were mounted with a mounting medium containing 4′,6-diamidino-2-phenylindole. The sections were later imaged using confocal microscopy.
Statistics—For groups of three or more, the data were analyzed by analysis of variance with post hoc Tukey analysis using Graph Pad Prism software. Comparisons restricted to two groups were analyzed using the Student's t test. A probability value of less than 0.05 was considered to represent a statistically significant difference.
RESULTS
Characterization of NO Consumption by Murine Fibroblasts—Fig. 1A illustrates experiments in which the generation of NO in solution was determined upon addition of the diazeniumdiolate DEA/NO (1 μm final concentration in DPBS, pH = 7.4). The NO signal in the absence of cells increased at an initial rate of 5.4 ± 0.1 nm·s-1 (n = 4), corresponding to a half-life for DEA/NO of ∼2.8 min at 37 °C. The concentration of NO reached a maximum of 718 ± 21 nm and then decreased because of NO autoxidation. In accordance, oxygen depletion raised NO concentration to ∼1.4 μm and did not change significantly over the 1000-s time frame of the experiment. Addition of 3.75 × 106 cells/ml fibroblasts to the medium accelerated NO removal as evidenced by a decrease in the apparent rate of NO formation (2.7 ± 0.2 nm·s-1; n = 4) and a leftward shift with a maximum NO concentration ∼37% of that attained with NO alone (Fig. 1A). Oxygen removal in the presence of cells raised the apparent amount of NO detected demonstrating the oxygen sensitivity of fibroblast-mediated NO consumption (Fig. 1A).
FIGURE 1.

NO consumption by mouse fibroblasts is oxygen-dependent, inhibited by
KCN, and produces intracellular nitrate. The concentration of NO in
solution was measured using an ISO-NOP NO electrode with 3.75 ×
106 cells/ml and 1 μm DEA/NO
(t½ ∼3 min) in DPBS (pH = 7.4). Cells were
depleted of oxygen (A) as described under “Experimental
Procedures” or treated with 500 μm KCN (B) prior
to injection of the NO donor to evaluate the effects of oxygen removal and
inhibition of heme proteins on NO consumption. The tracings represent mean
± S.E. of four independent experiments. C, time course of
intracellular nitrate (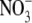 ) and nitrite
(
) and nitrite
(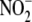 ) production in response to
addition of 1 μm DEA/NO to 3.75 × 106 cells/ml
mouse fibroblasts in DPBS. Nitrate/nitrite content was evaluated using
reductive chemiluminescence of cellular lysates. The effect of KCN on nitrate
(
) production in response to
addition of 1 μm DEA/NO to 3.75 × 106 cells/ml
mouse fibroblasts in DPBS. Nitrate/nitrite content was evaluated using
reductive chemiluminescence of cellular lysates. The effect of KCN on nitrate
(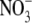 )/nitrite
(
)/nitrite
(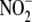 ) intracellular accumulation at 2.5
min (D) and in the medium at 30 min post-NO donor addition
(E) is shown. Values represent the mean ± S.E., n =
4. * indicates p < 0.01 as compared with cells; #
indicates p < 0.01 as compared with no cells. w/o,
without.
) intracellular accumulation at 2.5
min (D) and in the medium at 30 min post-NO donor addition
(E) is shown. Values represent the mean ± S.E., n =
4. * indicates p < 0.01 as compared with cells; #
indicates p < 0.01 as compared with no cells. w/o,
without.
Addition of the heme poison KCN (500 μm; Fig. 1B) decreased NO consumption by fibroblasts as indicated by an increase in the apparent rate of NO release to a value that was not statistically different from the rate obtained in the absence of cells (5.2 ± 0.4 nm·s-1; n = 4; p < 0.05 as compared with DEA/NO alone). The maximum NO concentration attained in solution was increased from ∼37 to ∼69% of control (Fig. 1B). Although pharmacological application of cyanide could be indicative of multiple mechanisms, a role for the mitochondrial respiratory chain could be excluded because treatments of the cells with the mitochondrial inhibitors rotenone, myxothiazol, or antimycin A were ineffective in changing NO concentrations (Fig. 2).
FIGURE 2.

NO consumption is independent of mitochondrial respiration. The effects of various mitochondrial inhibitors on NO consumption were measured by preincubation of cells (5 × 106 cells/ml) in suspension with the inhibitor prior to addition of DEA/NO 2 μm. The y axis depicts the maximum NO concentration measured in solution. NO consumption was inhibited by addition of the heme poison KCN but not by other mitochondrial inhibitors (mean ± S.E., n = 3). * indicates p < 0.05 as compared with control cells.
To further define the nature of the NO consumptive activity, we determined
the concentrations of nitrite (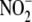 ) and
nitrate (
) and
nitrate (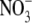 ) formed upon decomposition
of DEA/NO. We found that most of the NO could be recovered in fibroblasts as
nitrate with little to no change in nitrite
(Fig. 1C). The rise in
nitrate was transient and inhibited by the addition of KCN
(Fig. 1, C, D, and
E). Thus, NO was mainly oxidized to nitrate by the
fibroblasts and released such that all of it could be accounted for in the
medium at the 30-min time point (Fig.
1E). Treatment of the cells with KCN resulted in a switch
to nitrite as the primary metabolite of which accumulation was evident only in
the medium. Similarly most of the NO could be accounted for by nitrite in the
absence of cells consistent in this case with NO autoxidation
(Fig. 1E). In control
experiments, we found that nitrite (10 μm) did not oxidize to
nitrate in the presence of the same cell concentration (data not shown)
eliminating the possibility that NO consumption was a two-step process
generating first nitrite and then nitrate. Thus, the KCN-inhibitable NO
consumption in fibroblasts was consistent with an NO dioxygenase activity,
which generates nitrate.
) formed upon decomposition
of DEA/NO. We found that most of the NO could be recovered in fibroblasts as
nitrate with little to no change in nitrite
(Fig. 1C). The rise in
nitrate was transient and inhibited by the addition of KCN
(Fig. 1, C, D, and
E). Thus, NO was mainly oxidized to nitrate by the
fibroblasts and released such that all of it could be accounted for in the
medium at the 30-min time point (Fig.
1E). Treatment of the cells with KCN resulted in a switch
to nitrite as the primary metabolite of which accumulation was evident only in
the medium. Similarly most of the NO could be accounted for by nitrite in the
absence of cells consistent in this case with NO autoxidation
(Fig. 1E). In control
experiments, we found that nitrite (10 μm) did not oxidize to
nitrate in the presence of the same cell concentration (data not shown)
eliminating the possibility that NO consumption was a two-step process
generating first nitrite and then nitrate. Thus, the KCN-inhibitable NO
consumption in fibroblasts was consistent with an NO dioxygenase activity,
which generates nitrate.
Cytoglobin Dioxygenates NO in Fibroblasts—Recent studies indicate that cytoglobin is mainly expressed in fibroblast lineages in various organs and tissues (20, 24).
To test the hypothesis that cytoglobin contributes to NO dioxygenation in fibroblasts, we manipulated protein levels of cytoglobin in murine fibroblasts through stable expression of shRNA and forced expression of human cytoglobin. Fibroblasts stably expressing shRNA targeting mouse cytoglobin displayed a ∼50 and 80% decrease in cytoglobin content at the mRNA and protein levels, respectively (Fig. 3, A and B). The anti-cytoglobin antibody detected cytoglobin in protein extracts with bands at ∼25 and 50 kDa as reported by others (25). Forced expression of human cytoglobin in cells expressing shRNA specifically targeting mouse cytoglobin showed an increase in cytoglobin protein content with an additional band at ∼20 kDa (Fig. 3B).
FIGURE 3.

Cytoglobin consumes NO to produce nitrate. Quantitative PCR (QPCR) (A) and Western blot (WB) (B) of cytoglobin in mouse fibroblasts stably expressing plasmids containing a short hairpin scrambled sequence (shScrambled), shRNA targeting mouse cytoglobin (shCYGB), and full-length human cytoglobin (human CYGB). C, the concentration of NO in solution was measured using an ISO-NOP NO electrode with 2.5 × 106 cells/ml and 1 μm DEA/NO + 100 units/ml superoxide dismutase in DPBS (pH = 7.4). The arrow indicates addition of NO donor. D, nitrite and nitrate accumulation in the medium was assayed using reductive chemiluminescence 30 min after NO donor addition. Values represent the mean ± S.E., n = 4. * indicates p < 0.05 as compared with shScrambled, and # indicates p < 0.05 as compared with human CYGB. IB, immunoblot; hCYGB, human CYGB.
We next examined NO consumption using the NO donor drug DEA/NO (1 μm; Fig. 3C). NO concentrations rose at a rate of ∼2.7 nm·s-1 in the presence of control cells (shScrambled), 49% slower than the initial rate of NO production in the absence of cells indicative of cell-mediated NO consumption. As NO production decreased over time with decayed DEA/NO, NO concentration reached a maximum, which was only 30% of the maximal NO concentration attained in the absence of cells (Table 1). Nitric oxide concentration then decreased at a rate that could not be fitted to a second order process and that was too rapid to be NO autoxidation.
TABLE 1.
Apparent rate constants and maxima of NO accumulation upon DEA/NO (2 μm) decomposition in the presence of cells with different levels of cytoglobin hCYGB, human CYGB.
| Initial rate | Maximum [NO] | Percentage of control (buffer)a | Rate of NO decomposition | |
|---|---|---|---|---|
| nm·s–1 | nm | ×10–3 s–1 | ||
| Buffer alone | 5.4 ± 0.1 | 717 ± 21 | ||
| shScrambled | 2.7 ± 0.1 | 167 ± 14 | 33.2 ± 3.0 | 3.2 ± 0.1 |
| shCYGB | 4.0 ± 0.1b | 309 ± 18c | 56.7 ± 4.7c | 2.6 ± 0.2c |
| shCYGB + hCYGB | 1.1 ± 0.1b,d | 102 ± 12c,d | 18.6 ± 2.2b,d | 2.4 ± 0.1c |
Corrected for maximum NO in buffer alone at 120 s
Indicates p < 0.001 as compared with shScrambled
Indicates p < 0.05 as compared with shScrambled
Indicates p < 0.001 as compared with shCYGB
We observed an ∼50% increase in the initial rate of NO release and maximum concentration of NO recovered when cytoglobin was silenced (Fig. 3C). This was accompanied with a decrease in nitrate production and reciprocal increase in nitrite (Fig. 3D). Re-expression of human cytoglobin in the knockdown cells rescued cell-dependent NO consumption as demonstrated by a return of the initial rate of NO generation, NO maximum, and nitrate and nitrite concentrations at or below levels of control cells (Fig. 3 and Table 1). Interestingly the decay rate of the remaining NO was decreased upon cytoglobin silencing, indicating a requirement for cytoglobin (Table 1). Expression of human cytoglobin failed to rescue this effect suggesting a tight coupling in NO decomposition that could not be complemented by expression of the human protein into the mouse system.
To determine whether the cytoglobin-mediated dioxygenation of NO in fibroblasts could occur over longer periods of time, we used the NO donor Sp/NO (t½ ∼37 min at 37 °C compared with ∼2.8 min for DEA/NO). A 50 μm concentration was selected so that the initial rate of NO generation was similar to the rate observed in the preceding experiments with 1 μm DEA/NO. However, NO generation was sustained long enough for steady state NO concentration to be achieved in the absence of cells (Fig. 4A). The expectation was that if the NO dioxygenase activity became saturated we would obtain a secondary rise in the NO concentration. In contrast, we found that under these conditions NO dioxygenation was biphasic, sustained, and cytoglobin-dependent. The presence of shScrambled cells (3.75 × 106 cells/ml) increased NO removal such that the initial rate of NO appearance was first decreased to ∼2 nm·s-1 and then further decreased to reach a rate of 1 nm·s-1 past 120 s (Fig. 4A and Table 2). The secondary increase in the rate of NO decomposition was unlikely due to a change in oxygen concentration because the rate of oxygen consumption in the presence of the cell density used only varied by 20% over the 1000 s of the experiment (data not shown).
FIGURE 4.

Cytoglobin mediates sustained NO consumption. A, the concentration of NO in solution was measured using an ISO-NOP NO electrode with 3.75 × 106 cells/ml as described in Fig. 1 except that DEA/NO was replaced with 50 μm Sp/NO (t½ ∼37 min). The arrow indicates addition of donor. B, nitrate accumulation in the medium at 2 and 10 min postaddition of NO donor was assay using ion-pairing HPLC. AU, arbitrary units; hCYGB, human CYGB.
TABLE 2.
Apparent rate constants of NO accumulation upon Sp/NO (50 μm) decomposition in the presence of cells with different levels of cytoglobin hCYGB, human CYGB.
| Initial rate (±S.E.) | Second rate (±S.E.) | |
|---|---|---|
| nm·s–1 | nm·s–1 | |
| Buffer alone | 4.9 ± 0.3 | |
| shScrambled | 2.1 ± 0.1a,b | 1.0 ± 0.1 |
| shCYGB | 2.8 ± 0.1a,c | 1.4 ± 0.1c |
| shCYGB + hCYGB | 2.2 ± 0.2a,b | 1.3 ± 0.1c |
Indicates p < 0.001 as compared with buffer
Indicates p < 0.05 as compared with shCYGB
Indicates p < 0.05 as compared with shScrambled
Cytoglobin silencing (shCYGB) resulted in a leftward shift in the NO curvature and an increase in the rates of NO appearance indicative of a decrease in NO consumption (Fig. 4A and Table 2). Re-expression of human cytoglobin completely rescued the initial phase of consumption but did not rescue the second phase. There was a striking correlation between the levels of cytoglobin and the amount of nitrate accumulated during the first phase of NO consumption (2-min time point) but also later when the rate of NO consumption was further increased (10 min; Fig. 4B).
Cytoglobin Regulates NO Bioactivity—With the demonstration that cytoglobin participates in NO dioxygenation in fibroblasts, we next determined whether cytoglobin regulated NO-mediated inhibition of cell respiration. Forced expression of cytoglobin increased the basal rate of oxygen consumption as compared with the control (shScrambled) and knockdown cells (shCYGB) as measured with 5 × 106 cells/ml in a sealed system (Fig. 5). Bolus addition of 2 μm PROLI/NO (half-life 1.8 s) at ∼70 μm oxygen concentration to control cells (shScrambled) resulted in a shallow plateau of ∼120 s followed by a return to initial rate of oxygen consumption, indicating a reversible inhibition of cell respiration by NO (Fig. 6). In cells with reduced levels of cytoglobin (shCYGB) we observed a prolonged plateau, ∼220 s in length, that was reversed by re-expression of human cytoglobin. These results demonstrated that levels of cytoglobin expressed in fibroblasts are sufficient to regulate NO-mediated inhibition of cell respiration.
FIGURE 5.

Cytoglobin overexpression increases basal oxygen consumption. 5 × 106 cells/ml of the indicated cell type were added to a sealed, stirred chamber maintained at 37 °C. Oxygen concentration in solution was determined using a Clark oxygen electrode. Values represent the mean ± S.E., n = 4. * indicates p < 0.05 as compared with shScrambled, and # indicates p < 0.05 as compared with shCYGB.
FIGURE 6.

Cytoglobin regulates NO-mediated inhibition of cell respiration. A, representative tracings of O2 detectable in solution upon addition of 2 μm PROLI/NO (t½ 1.8 s) to 5 × 106 cells/ml of the indicated cell type. The arrow indicates addition of donor to both cell lines. The bar indicates 100 s. B, quantitation of the time it took each cell type to return to the rate of oxygen consumption prior to addition of NO. Values represent the mean ± S.E., n = 4. * indicates p < 0.01 as compared with shScrambled, and # indicates p < 0.01 as compared with shCYGB. hCYGB, human CYGB; shScrml, shScrambled.
The cytostatic effect of NO has been demonstrated for many different cell types and tissues (26), and we reasoned that changing levels of cytoglobin should impact NO-induced cytostasis if cytoglobin-mediated NO consumption was sustained. Fig. 7 demonstrates that cytoglobin inhibits the cytostatic effect of NO. The three cell lines shSCrambled, shCYGB, and shCYGB with human cytoglobin were seeded at uniform density and allowed to grow under normal growth conditions in the presence or absence of the NO donor DETA/NO (t½ ∼20 h at 37 °C; 50 μm) for 48 h. Fibroblasts with shScrambled showed a ∼45% inhibition of proliferation as normalized to shScrambled cells without NO. Reduced levels of cytoglobin (shCYGB) resulted in an increase in inhibition of cell proliferation to ∼70%. This was decreased to ∼55% in knockdown cells when cytoglobin levels were restored (Fig. 7). There was no significant change in viability between cell types in either control or NO-treated cells as measured by trypan blue exclusion (data not shown).
FIGURE 7.

Nitric oxide-induced cytostasis is inhibited by cytoglobin. Cells were seeded at uniform concentration (1.4 × 104 cells/well) in 12-well dishes containing normal growth medium. The cells were grown in the presence or absence of 50 μm DETA/NO (t½ 20 h) for 48 h, trypsinized, and counted using a Coulter counter. Samples were compared with the same cell type with no NO added (control). Cell viability was analyzed through trypan blue exclusion. Values represent the mean ± S.E., n = 4. * indicates p < 0.01 as compared with shScrambled, and # indicates p < 0.01 as compared with shCYGB.
Cytoglobin Is Expressed in Both Adventitial Fibroblasts and Smooth Muscle Cells in the Aorta—Results illustrated in Fig. 8A demonstrate cytoglobin expression in adventitial fibroblasts as well as medial smooth muscle cells in the intact rat aorta. These results were supported by co-localization with myosin heavy chain, an intracellular smooth muscle marker. Analysis of undispersed adventitial fibroblasts and medial smooth muscle cells indicated significant amounts of cytoglobin mRNA (Fig. 8B), and we detected cytoglobin message in cultured vascular smooth muscle cells of mouse, rat, and human origin. Western blot analysis of lysates obtained from cultured rat aortic adventitial fibroblasts and vascular smooth muscle cells showed typical bands for cytoglobin at 20, 25, and 50 kDa (Fig. 8C).
FIGURE 8.

Cytoglobin is expressed in vascular fibroblasts and smooth muscle cells. A, cellular localization of cytoglobin in rat aorta. Rat aortas were harvested and co-stained with cytoglobin and myosin heavy chain antibodies. Immunohistochemical analysis shows positive staining in the medial (noted M) smooth muscle layer as well as the adventitial (noted A) fibroblast layer. Lumen to noted L. Myosin heavy chain is used to show cytoglobin co-localization with an intracellular smooth muscle protein. B, copy number of cytoglobin mRNA detected in various tissues of the blood vessel normalized to total RNA (mean ± S.E., n = 3). C, Western blot analysis of cytoglobin expression in cultured rat aortic vascular smooth muscle cells (VSM) and cultured rat adventitial fibroblasts. IB, immunoblot.
DISCUSSION
The function of cytoglobin is unknown with recent studies suggesting that it acts as a tumor suppressor and protects against free radical-induced tissue injury (27, 28). Biochemical studies have focused mainly on the mechanism by which diatomic ligands including NO bind and react with cytoglobin (21, 29, 30). Oxygenated ferrous (Fe2+) cytoglobin deoxygenates NO in vitro to form nitrate and ferric (Fe3+) cytoglobin, a reaction that is common to many members of the globin family (21). However, the relevance of this reaction to NO metabolism and function is doubtful unless sustainable cytoglobin-mediated NO dioxygenation is demonstrated in mammalian cells. In the present study, we showed that cytoglobin dioxygenates NO to nitrate and regulates cell respiration and proliferation in fibroblasts. We also showed that cytoglobin is expressed in the vascular wall in both adventitial fibroblasts and medial smooth muscle cells.
Production of nitrate in various mammalian cells and tissues has been previously reported to be sensitive to cyanide inhibition indicating a role for a heme-containing protein in NO metabolism, although the exact identity of this protein has never been reported (5, 6, 31). The experimental evidence obtained with four different NO donors in the present study showed a direct correlation between cytoglobin levels in fibroblasts and intracellular nitrate production indicating that cytoglobin contributes to NO dioxygenation in this cell type.
Past studies have indicated differences in product formation. Lancaster and co-workers (7) have shown that the primary product formed in hepatocytes is nitrite and not nitrate. The results obtained in the present study are consistent with those obtained by Gardner et al. (5), Schmidt and Mayer (6), and Garthwaite and co-workers (8), who all have found nitrate to be the primary product in various cell types, cell lysates, and tissue extracts. Overall this would suggest different pathways for NO consumption. It has been shown for example that 15-lipoxygenase, prostaglandin-H synthase, and myeloperoxidase all consume NO to yield nitrite, and it is possible that these pathways may prevail under specific conditions (13–15).
Although NO consumption was primarily oxygen-dependent, we observed a slow but significant NO consumption in the absence of oxygen (Fig. 1). Clarkson et al. (32) observed NO consumption by Chinese hamster ovary cells at low (less than 1 μm) oxygen concentration that was inhibited by cyanide. Because this could be recapitulated with isolated mitochondria, this anaerobic NO consumption was indicative of reductive NO metabolism mediated by the mitochondria. Our results regarding NO consumption in the absence of oxygen were consistent with this process.
In the presence of oxygen, NO kinetics in fibroblasts were biphasic with both phases being cytoglobin-dependent (Figs. 1 and 3). Although it is impossible to derive a mechanism for this observation at the present time, the first saturable component might be due to stoichiometric reaction of NO with cytoglobin to form nitrate and ferric cytoglobin. The second component might be determined by a rate-limiting step in which ferric cytoglobin is reduced back to the ferrous form to allow dioxygenase activity to continue in agreement with previous studies (21). Interestingly the rate of NO consumption increased past the first component in the presence of the NO donor Sp/NO but not with more rapidly decaying DEA/NO. This would suggest an important role for NO concentrations in controlling the increase in rate of NO dioxygenation after the first phase. It is also noteworthy that in the presence of Sp/NO the formation of nitrate continued past the first phase and was completely inhibited upon silencing of cytoglobin. Although expression of human cytoglobin restored nitrate production to some extent, this could not be matched with a statistically significant recovery in NO consumption suggesting important differences between human and mouse cytoglobin.
The reaction of NO with intraerythrocytic oxyhemoglobin is classically viewed as the major route of NO disposition in the vasculature (33). However, the discovery that diffusional barriers may limit scavenging of NO in the vascular wall opens the possibility that other mechanisms may control NO levels. Our observation that aortic adventitial fibroblasts and medial smooth muscle cells express cytoglobin is important because it provides an alternative mechanism for NO decomposition in the vascular wall. We have noted, at least at the mRNA level, differences (although not significant) between undispersed and cultured vascular cells (Fig. 8). Cultured vascular aortic smooth cells acquire an undifferentiated proliferating phenotype that is most representative of the phenotypes observed during vascular injury (34). This is in contrast to the differentiated contractile phenotype of cells from intact vessels. This would suggest differences in cytoglobin levels associated with different vascular phenotypes or potentially different vascular beds such as the aorta and microvasculature that might explain different sensitivity to NO.
The concentrations of NO generated from the NO donors used in this study mimic those of NO concentrations expected during inflammatory conditions when several sources of NO such as inducible nitric-oxide synthase are up-regulated (35, 36). Previous studies have shown an inhibition of tissue respiration upon cytokine stimulation of isolated vessels, an effect that is reversed upon nitric-oxide synthase inhibition (37). Similarly recent studies have shown that inflammatory levels of NO may serve to regulate specific signaling pathways including the tumor suppressor p53 and the mitogen-activated protein kinases to impact cell function including cell proliferation (26, 35). It is therefore possible to postulate that cytoglobin may play a role in buffering NO levels below cytostatic or cytotoxic levels and thus regulates tissue concentrations of NO attained during vascular inflammation.
As noted above, we specifically addressed the role of cytoglobin in regulating NO functions in the presence of concentrations that most notably affect cell respiration and proliferation. At the same time, the question arises as to whether cytoglobin may affect cGMP production through inhibition of NO activation of soluble guanylate cyclase. This could not be assessed in our mouse fibroblast cell lines because we could not detect any increase in cGMP production upon stimulation with different concentrations of NO donors (data not shown). This was most likely due to the lack of significant amounts of soluble guanylate cyclase in this cell type. Past studies have shown very little to no overlap between the NO concentrations that activate soluble guanylate cyclase and those required for inhibition of cell respiration (38, 39). Thus, the question of whether cytoglobin affects NO-mediated cGMP production awaits future studies addressing this specific issue.
Although we appreciate that the biochemical conditions upon which cytoglobin may scavenge NO in tissues need to be better defined, we propose that cytoglobin is a key element in determining the magnitude of the NO response in the vasculature. In addition, the present study would indicate that any alternate function of cytoglobin may be regulated through reaction with NO.
Acknowledgments
We thank Dr. Margarida Barroso and Nicole McGrath for help with the confocal microscopy. We also acknowledge the excellent technical contribution of Catherine Vincent.
This work was supported, in whole or in part, by National Institutes of Health Grant CA-89366 from the NCI (to D. J.). This work was also supported by Philip Morris USA, Inc. and Philip Morris International (to D. J.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: NO, nitric oxide; CYGB, cytoglobin; DEA/NO, diethylammonium-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate; DETA/NO, (Z)-1-[N-(2-aminoethyl)-N-(2-ammoniethyl)amino]diazen-1-ium-1,2-diolate; KCN, potassium cyanide; PROLI/NO, E-1-(hydroxyl-NNO-azoxy)-l-proline; shRNA, small hairpin RNA; Sp/NO, N-4-1-3-aminopropyl-2-hydroxy-2-nitrosohydrazinobutyl-1,3-propane; DPBS, Dulbecco's phosphate-buffered saline; HPLC, high pressure liquid chromatography.
References
- 1.Lancaster, J., Jr. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 8137-8141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liao, J. C., Hein, T. W., Vaughn, M. W., Huang, K.-T., and Kuo, L. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 8757-8761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Godecke, A., Molojavyi, A., Heger, J., Flogel, U., Ding, Z., Jacoby, C., and Schrader, J. (2003) J. Biol. Chem. 278 21761. [DOI] [PubMed] [Google Scholar]
- 4.Flogel, U., Merx, M. W., Godecke, A., Decking, U. K., and Schrader, J. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 735-740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gardner, P. R., Martin, L. A., Hall, D., and Gardner, A. M. (2001) Free Radic. Biol. Med. 31 191-204 [DOI] [PubMed] [Google Scholar]
- 6.Schmidt, K., and Mayer, B. (2004) FEBS Lett. 577 199-204 [DOI] [PubMed] [Google Scholar]
- 7.Thomas, D. D., Liu, X., Kantrow, S. P., and Lancaster, J. R., Jr. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 355-360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griffiths, C., Yamini, B., Hall, C., and Garthwaite, J. (2002) Biochem. J. 362 459-464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao, X. J., Sampath, V., and Caughey, W. S. (1995) Biochem. Biophys. Res. Commun. 212 1054-1060 [DOI] [PubMed] [Google Scholar]
- 10.Torres, J., Sharpe, M. A., Rosquist, A., Cooper, C. E., and Wilson, M. T. (2000) FEBS Lett. 475 263-266 [DOI] [PubMed] [Google Scholar]
- 11.Palacios-Callender, M., Hollis, V., Mitchison, M., Frakich, N., Unitt, D., and Moncada, S. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 18508-18513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rajagopalan, S., Kurz, S., Munzel, T., Tarpey, M., Freeman, B. A., Griendling, K. K., and Harrison, D. G. (1996) J. Clin. Investig. 97 1916-1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Donnell, V. B., Taylor, K. B., Parthasarathy, S., Kuhn, H., Koesling, D., Friebe, A., Bloodsworth, A., Darley-Usmar, V. M., and Freeman, B. A. (1999) J. Biol. Chem. 274 20083-20091 [DOI] [PubMed] [Google Scholar]
- 14.Coffey, M. J., Natarajan, R., Chumley, P. H., Coles, B., Thimmalapura, P., Nowell, M., Kühn, H., Lewis, M. J., Freeman, B. A., and O'Donnell, V. B. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 8006-8011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eiserich, J. P., Baldus, S., Brennan, M. L., Ma, W., Zhang, C., Tousson, A., Castro, L., Lusis, A. J., Nauseef, W. M., White, C. R., and Freeman, B. A. (2002) Science 296 2391-2394 [DOI] [PubMed] [Google Scholar]
- 16.Gardner, P. R., Gardner, A. M., Brashear, W. T., Suzuki, T., Hvitved, A. N., Setchell, K. D., and Olson, J. S. (2006) J. Inorg. Biochem. 100 542-550 [DOI] [PubMed] [Google Scholar]
- 17.Kawada, N., Kristensen, D. B., Asahina, K., Nakatani, K., Minamiyama, Y., Seki, S., and Yoshizato, K. (2001) J. Biol. Chem. 276 25318-25323 [DOI] [PubMed] [Google Scholar]
- 18.Trent, J. T., III, and Hargrove, M. S. (2002) J. Biol. Chem. 277 19538-19545 [DOI] [PubMed] [Google Scholar]
- 19.Burmester, T., Ebner, B., Weich, B., and Hankeln, T. (2002) Mol. Biol. Evol. 19 416-421 [DOI] [PubMed] [Google Scholar]
- 20.Schmidt, M., Gerlach, F., Avivi, A., Laufs, T., Wystub, S., Simpson, J. C., Nevo, E., Saaler-Reinhardt, S., Reuss, S., Hankeln, T., and Burmester, T. (2004) J. Biol. Chem. 279 8063-8069 [DOI] [PubMed] [Google Scholar]
- 21.Smagghe, B. J., Trent, J. T., III, and Hargrove, M. S. (2008) PLoS ONE 3 e2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feelisch, M., Rassaf, T., Mnaimneh, S., Singh, N., Bryan, N. S., Jourd'heuil, D., and Kelm, M. (2002) FASEB J. 16 1775-1785 [DOI] [PubMed] [Google Scholar]
- 23.Jourd'heuil, D., Jourd'heuil, F. L., and Feelisch, M. (2003) J. Biol. Chem. 278 15720-15726 [DOI] [PubMed] [Google Scholar]
- 24.Nakatani, K., Okuyama, H., Shimahara, Y., Saeki, S., Kim, D. H., Nakajima, Y., Seki, S., Kawada, N., and Yoshizato, K. (2004) Lab. Investig. 84 91-101 [DOI] [PubMed] [Google Scholar]
- 25.Schmidt, M., Laufs, T., Reuss, S., Hankeln, T., and Burmester, T. (2005) Neurosci. Lett. 374 207-211 [DOI] [PubMed] [Google Scholar]
- 26.Lu, Q., Jourd'heuil, F. L., and Jourd'heuil, D. (2007) J. Cell. Physiol. 212 827-839 [DOI] [PubMed] [Google Scholar]
- 27.Shivapurkar, N., Stastny, V., Okumura, N., Girard, L., Xie, Y., Prinsen, C., Thunnissen, F. B., Wistuba, I. I., Czerniak, B., Frenkel, E., Roth, J. A., Liloglou, T., Xinarianos, G., Field, J. K., Minna, J. D., and Gazdar, A. F. (2008) Cancer Res. 68 7448-7456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu, R., Harrison, P. M., Chen, M., Li, L., Tsui, T. Y., Fung, P. C., Cheung, P. T., Wang, G., Li, H., Diao, Y., Krissansen, G. W., Xu, S., and Farzaneh, F. (2006) Mol. Ther. 13 1093-1100 [DOI] [PubMed] [Google Scholar]
- 29.Hamdane, D., Kiger, L., Dewilde, S., Green, B. N., Pesce, A., Uzan, J., Burmester, T., Hankeln, T., Bolognesi, M., Moens, L., and Marden, M. C. (2003) J. Biol. Chem. 278 51713-51721 [DOI] [PubMed] [Google Scholar]
- 30.Fago, A., Hundahl, C., Dewilde, S., Gilany, K., Moens, L., and Weber, R. E. (2004) J. Biol. Chem. 279 44417-44426 [DOI] [PubMed] [Google Scholar]
- 31.Griffiths, C., and Garthwaite, J. (2001) J. Physiol. 536 855-862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clarkson, R. B., Norby, S. W., Smirnov, A., Boyer, S., Vahidi, N., Nims, R. W., and Wink, D. A. (1995) Biochim. Biophys. Acta 1243 496-502 [DOI] [PubMed] [Google Scholar]
- 33.Kim-Shapiro, D. B., Schechter, A. N., and Gladwin, M. T. (2006) Arterioscler. Thromb. Vasc. Biol. 26 697-705 [DOI] [PubMed] [Google Scholar]
- 34.Owens, G. K., Kumar, M. S., and Wamhoff, B. R. (2004) Physiol. Rev. 84 767-801 [DOI] [PubMed] [Google Scholar]
- 35.Thomas, D. D., Espey, M. G., Ridnour, L. A., Hofseth, L. J., Mancardi, D., Harris, C. C., and Wink, D. A. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 8894-8899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bove, P. F., and Van der Vliet, A. (2006) Free Radic. Biol. Med. 41 515-527 [DOI] [PubMed] [Google Scholar]
- 37.Borutaite, V., Matthias, A., Harris, H., Moncada, S., and Brown, G. C. (2001) Am. J. Physiol. 281 H2256-H2260 [DOI] [PubMed] [Google Scholar]
- 38.Bellamy, T. C., Griffiths, C., and Garthwaite, J. (2002) J. Biol. Chem. 277 31801-31807 [DOI] [PubMed] [Google Scholar]
- 39.Rodriguez-Juarez, F., Aguirre, E., and Cadenas, S. (2007) Biochem. J. 405 223-231 [DOI] [PMC free article] [PubMed] [Google Scholar]