

Abstract
Exposure to estrogens increases the risk of breast and endometrial cancer. It is proposed that the estrogen receptor (ER) may contribute to estrogen carcinogenesis by transduction of the hormonal signal and as a “Trojan horse” concentrating genotoxic estrogen metabolites in the nucleus to complex with DNA, enhancing DNA damage. 4-Hydroxyequilenin (4-OHEN), the major catechol metabolite of equine estrogens present in estrogen replacement formulations, autoxidizes to a redox-cycling quinone that has been shown to cause DNA damage. 4-OHEN was found to be an estrogen of nanomolar potency in cell culture using a luciferase reporter assay and, using a chromatin immunoprecipitation assay, was found to activate ERα binding to estrogen-responsive genes in MCF-7 cells. DNA damage was measured in cells by comparing ERα(+) versus ERα(-) cells and 4-OHEN versus menadione, a reactive oxygen species (ROS)-generating, but non-estrogenic, quinone. 4-OHEN selectively induced DNA damage in ERα(+) cells, whereas menadione-induced damage was not dependent on cellular ER status. The rate of 4-OHEN-induced DNA damage was significantly enhanced in ERα(+) cells, whereas ER status had no effect on the rate of menadione-induced damage. Imaging of ROS induced by 4-OHEN showed accumulation selective for the nucleus of ERα(+) cells within 5 min, whereas in ERα(-) or menadione-treated cells, no selectivity was observed. These data support ERα acting as a Trojan horse concentrating 4-OHEN in the nucleus to accelerate the rate of ROS generation and thereby amplify DNA damage. The Trojan horse mechanism may be of general importance beyond estrogen genotoxins.
An increased relative risk of breast cancer in postmenopausal women is strongly linked to several endocrine-related risk factors. One of these risk factors is long-term exposure to hormone or estrogen replacement therapy (HRT3 or ERT). The most widely prescribed formulations in the United States contain conjugated human estrogens and B-ring unsaturated conjugated equine estrogens, the latter constituting approximately half of the estrogen content of these formulations (1). Observations from various clinical trials and epidemiological studies collectively support the hypothesis that estrogen contributes to breast cancer and is probably causative (2–8). The large prospective Women's Health Initiative Study comparing postmenopausal women assigned HRT/ERT or placebo was terminated because of significant increases in breast cancer, stroke, and pulmonary embolism associated with therapy (9, 10). A recent analysis of data from the National Cancer Institute's Surveillance, Epidemiology, and End Results (SEER) registries showed that the age-adjusted incidence rate of breast cancer fell 6.7% in 2003 compared with 2002, which was linked to lowered use of HRT/ERT (11).
The collective evidence supports contributions to estrogen-sensitive breast cancer from both the proliferative and antiapoptotic hormonal effects of estrogen itself (12–16) and the genotoxic and mutagenic effects of estrogen metabolites (17, 18). In human breast tissue, estrogen metabolites have been detected (19), and DNA adducts of equine estrogens have been reported (20). Human estrogen 4-hydroxy catechol metabolites, formed by the action of cytochrome P450, are proposed to be genotoxic through formation of DNA-reactive, electrophilic o-quinones and by formation of DNA-damaging, reactive oxygen species (ROS) through redox cycling (21–26). The equine estrogen equilenin is of special interest as a carcinogen because: 1) it is selectively oxidized to the more genotoxic 4-hydroxy catechol isomer (4-OHEN); and 2) 4-OHEN autoxidizes to a redox-cycling o-quinone without need for cytochrome P450 catalysis (27, 28). 4-OHEN-o-quinone induces a variety of different types of DNA damage both in vitro and in vivo, including single strand breaks (29, 30), oxidized bases (31, 32), apurinic sites, and formation of cyclic DNA adducts (20, 33, 34).
The estrogen receptor (ER) may play two key roles in mediating estrogen carcinogenesis: transduction of the hormonal signal and transport of genotoxic estrogen metabolites to the nucleus to complex with DNA. In the latter role, ER acts as a “Trojan horse,” enhancing DNA damage. This hypothesis is persuasive for 4-OHEN because this catechol readily autoxidizes and redox cycles, theoretically generating large fluxes of ROS in the cell nucleus. However, 4-OHEN is autoxidized to a reactive quinone electrophile known to be trapped readily by glutathione and protein nucleophiles. It is not clear from previous work whether 4-OHEN represents a good ER ligand. Furthermore, the Trojan horse hypothesis has obvious weaknesses in that 4-OHEN may be trapped in the cytoplasm and may not be able to redox cycle as part of an ER-DNA complex.
To test the hypothesis, the hormonal estrogenicity of 4-OHEN was assayed using cellular reporters; a comparison was made between ERα-positive and -negative cells testing 4-OHEN-induced DNA damage; a comparison was made with the ROS-generating, but non-estrogenic, quinone, menadione; and finally, nuclear localization of 4-OHEN and generation of ROS was determined by chromatin immunoprecipitation (ChIP) assay and fluorescence confocal microscopy. The Trojan horse model may be of general importance in the study of environmental estrogen genotoxins and the nuclear concentration of genotoxins by other nuclear receptors and transcription factors.
EXPERIMENTAL PROCEDURES
The catechol estrogens were handled in accordance with NIH Guidelines for the Laboratory Use of Chemical Carcinogens (35).
Materials—All chemicals were purchased from Sigma or Fisher Scientific unless stated otherwise. 5-(and 6)-Chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate was purchased from Invitrogen. 4-OHEN was synthesized by treating equilin with Fremy's salt as described previously (36) with minor modifications (27). The comet assay and Fpg FLARE comet assay kits for detection of DNA single strand breaks and oxidized bases were purchased from Trevigen (Gaithersburg, MD). All buffers and reagents used in ChIP assay were from Upstate Biotechnology (Lake Placid, NY), and antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell Culture Conditions—MDA-MB-231 cells were obtained from the American Type Culture Collection (Manassas, VA) and maintained in minimum essential medium (MEM) supplemented with 1% penicillin-streptomycin-fungizome, 6 μg/liter insulin, 1% GlutaMAX (Invitrogen), 5% fetal bovine serum (Atlanta Biologicals, Atlanta), and 5% CO2 at 37 °C. The S30 cell line, a stable ERα transfectant of the MDA-MB-231 cell line, was a generous gift from Dr. V. C. Jordan (Fox Chase Cancer Center, Philadelphia). The S30 cells were maintained in phenol red-free MEM supplemented with the same solution as the MDA-MB-231 cells except for the addition of 5% charcoal-dextran-treated fetal bovine serum and 500 μg/ml Geneticin. The MCF-7 cells were maintained in RPMI 1640 containing 10% fetal bovine serum, 1% penicillin-streptomycin-fungizome, 6 μg/liter insulin, and 1% GlutaMAX. The MCF-7:K1 cell line, a clonal derivative of MCF-7 cells, was a kind gift from Dr. J. Frasor (University of Illinois at Chicago) and was maintained in MEM supplemented with 1% penicillin-streptomycin-fungizome, 25 μg/ml gentamicin, 2 mm glutamine, 5% calf bovine serum (Hyclone, Logan, Utah), and 2 mg/ml sodium bicarbonate solution. Estrogen-free medium for MCF-7:K1 cells was prepared by supplementing charcoal-dextran-treated fetal bovine serum to phenol red-free MEM, whereas other components remained the same except omitting gentamicin.
Transient Transfection and ERE-Luciferase Assays—Briefly, MCF-7 cells were cultured in estrogen-free media for 4 days before transfection. The cells were transfected with 2 μg of the pERE-luciferase plasmid, which contains three copies of the Xenopus laevis vitellogenin A2 ERE upstream of firefly luciferase. To normalize transfection efficiency, 1 μg of pRL-TK plasmid, which contained a cDNA encoding Renilla luciferase, was co-transfected with ERE-luciferase plasmid. Cells (4 × 105 cells/well) were transfected with ERE-luciferase, pretreated with or without 10 μm Ro-41-0960 (COMT inhibitor) for 24 h, and then treated with DMSO (0.05%), menadione (100 nm, 1 μm), E2 (1 nm), or 4-OHEN (100 nm) with or without 10 μm COMT inhibitor for 18 h. The luciferase activity in cell lysates was measured using the Dual-Luciferase assay system from Promega (Madison, WI) with a FLUOstar OPTIMA (BMG Labtech, Durham, NC). Data are reported as relative luciferase activity (firefly luciferase reading divided by the Renilla luciferase reading).
Chromatin Immunoprecipitation Assay—Cells were cultured in estrogen-free media for 4 days before treatment with compounds. Cells were treated with DMSO, E2, or 4-OHEN for 45 min, washed with phosphate-buffered saline (PBS), and cross-linked with 1.5% formaldehyde at room temperature for 15 min. Cells were rinsed twice with ice-cold PBS, collected into lysis buffer (1% SDS, 10 mm EDTA, 50 mm Tris-HCl, pH 8, and 1× protease inhibitor tablet (Roche Applied Science)), and sonicated 20 times for 10 s at 10% strength (Fisher Model 300 Sonic Dismembrator) followed by centrifugation at 21,000 × g for 10 min at 4 °C. Each sample was reserved for the inputs prior to immunoprecipitation. Supernatants were collected and diluted in immunoprecipitation buffer (1% Triton X-100, 2 mm EDTA, 150 mm NaCl, 20 mm Tris-HCl, pH 8.1) containing 1× protease inhibitor followed by immunoclearing with 8 μg of sheared salmon sperm DNA and protein A-agarose slurry (40 μl of 50% slurry in TE buffer (10 mm Tris-HCl, pH 8.1, 1 mm EDTA)) for 2 h at 4 °C. After centrifugation at 5,000 × g for 1 min, immunoprecipitation was performed by gently mixing the supernatants with the antibodies against ERα (3.3 μg/ml, HC-20) or IgG (3.3 μg/ml, normal rabbit IgG) overnight at 4 °C. After immunoprecipitation, protein A-agarose slurry (40 μl) containing 8 μg of salmon sperm DNA was added, and the incubation was continued for another 2 h at 4 °C. Precipitates were obtained by centrifugation at 5,000 × g for 1 min and washed sequentially for 5 min each in TSE I (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.1, 150 mm NaCl), TSE II (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.1, 500 mm NaCl), and LiCl buffer (0.25 m LiCl, 1% Nonidet P-40, 1% sodium deoxycholate, 1 mm EDTA, 10 mm Tris-HCl, pH 8.1). Precipitates were then washed three times with TE buffer, and DNA was extracted three times with 1% SDS, 0.1 m NaHCO3. Eluates were pooled and heated at 65 °C for 16 h to reverse the formaldehyde cross-linking. DNA fragments were purified with a QIAquick PCR purification kit (Qiagen, Valencia, CA). The subsequent PCR experiment was performed to detect DNA fragments of estrogen sensitive-gene, such as the ERE sequence. The pS2-ERE forward and reverse primer sequences, from -4919 to -4806 (5′ to 3′), were GACGGAATGGGCTTCATGA and AGTGAGAGATGGCCGGAAAA, respectively. The pS2 upstream forward and reverse primer sequences (5′ to 3′), spanning the -446 to -339 region of the pS2 promoter, were GGGTCTCAGTGGTGGCAGTA and ACCGCTCATACCATCCAGTC, respectively.
Single Cell Gel Electrophoresis Assay (Comet Assay)—The comet assay (37) was carried out as recommended by the manufacturer. Briefly, after cells (1.5 × 105 cells/ml) were treated with various concentrations of test compounds, the attached cells were trypsinized, combined with suspended cells, and washed twice with chilled PBS, pH 7.4 (Ca2+- and Mg2+-free; Trevigen). The cell suspension was combined with 0.5% low melting agarose, and the mixture was immediately placed onto a CometSlide. The slides were incubated at 4 °C in the dark for gelling before immersion in prechilled lysis solution for 30 min at 4 °C and then incubated in freshly prepared alkali (pH > 13) solution for 45 min. The assay was adapted for measurement at earlier time points by quenching with excess N-acetylcysteine (NAC; 10 mm) for 1 min. As described above, cells were washed with cold PBS, trypsinized, and collected by centrifugation. The cells were further washed with cold PBS and collected by centrifugation. PBS (2 ml) was added to produce a suspension that was mixed with low melting agarose, placed on glass slides, and then incubated at 4 °C for 10 min in the dark.
Alkali solution electrophoresis was performed for 10 min at 300 mA and 1 V/cm, and then the slides were immersed in prechilled 70% ethanol for 5 min and air-dried. The slides were then stained with SYBR Green and scored under a fluorescence microscope (Nikon Y-FL). The DNA damage from at least 100 cells/slide was scored in arbitrary units from 0 (intact DNA) to 4 (completely damaged DNA with tail only) (38). Scores were calculated using Equation 1 in which NA (intact DNA) and NB - NE (completely damaged DNA) were the number of different kinds of comets.
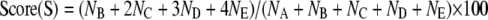 |
(Eq. 1) |
Modified Oxidative DNA Damage Comet Assay (Fpg FLARE Comet Assay)—Oxidized bases were determined using the Trevigen Fpg FLARE (fragment length analysis using repair enzymes) comet assay kit. Briefly, following treatment the cells were collected and washed as described previously and resuspended in PBS. The cell suspensions were combined with low melting agarose and transferred onto CometSlides. After fixing the cell/agar mixture to the slide at 4 °C, the cells were lysed at 4 °C in lysis buffer for 30 min. After equilibration with FLARE buffer (10 mm HEPES-KOH, pH 7.4, 0.1 m KCl) for 15 min, the slides were incubated at 37 °C for 60 min with the diluted Fpg enzyme, and appropriate buffer-only controls were included. Following equilibration with alkali solution, the slides underwent electrophoresis for 3 min at 300 mA. The slides were then fixed with 70% ethanol, stained with SYBR Green, and scored as described above using Equation 1. The difference between the scores of the Fpg-treated samples and buffer controls was proportional to the amount of oxidized bases in the cells.
Analysis of 8-Oxo-dG in Breast Cancer Cells by LC-MS/MS—8-Oxo-dG analysis was carried out as described previously (31) with minor modifications. After incubation with test compounds, the floating cells were collected by centrifugation, and the attached cells were trypsinized and then harvested by centrifugation. The cells were combined and washed with 10 ml of PBS. After centrifugation, the cell pellets were homogenized in 3.5 ml of lysis buffer (320 mm sucrose, 10 mm Tris, pH 7.4, 5 mm MgCl2, 10 mm Triton X-100, and 50 mm mannitol). The nuclei pellets were treated for 30 min at 37 °C with RNase T1 (1000 units) and RNase A (0.2 mg) in solution buffer (1% SDS, 1 mm EDTA, 10 mm Tris, pH 7.4, 0.45 m NaCl) and further incubated with proteinase K (0.8 mg) for 30 min at 37 °C. NaCl and Tris were added to achieve final concentrations of 0.62 m and 20 mm, respectively. An equal volume of 1-butanol was added, the samples were thoroughly mixed and centrifuged, and the bottom aqueous layer was isolated. After isopropanol precipitation, the DNA was washed twice with 70% ethanol. The DNA was dissolved in 100 μl of buffer (25 mm ammonium acetate, pH 5.3, 0.1 mm ZnCl2), mixed with 2 μl of 10 mm Desferral, and hydrolyzed using nuclease P1 (4 units) and alkaline phosphatase (8 units) for 30 min at 37 °C. The enzymes were removed by ultrafiltration using a Microcon YM-30 centrifugal filter (Millipore). Stable isotopically labeled [15N5]8-oxo-dG was added to the ultrafiltrate as the surrogate standard, and the mixture was analyzed using LC-MS/MS on a Thermo (San Jose, CA) TSQ Quantum triple quadrupole mass spectrometer coupled with a Surveyor HPLC System and photodiode array detector. The samples were separated using a YMC (YMC Co., Wilmington, NC) AQ C18 column (2.0 × 250 mm) and guard column (4.0 × 20 mm) at a flow rate of 0.2 ml/min with a gradient mobile phase starting at 5% methanol/water and increasing to 10% methanol over 6 min, increasing to 20% methanol/water over another 6 min, increasing to 90% methanol/water for 5 min, and then equilibrium with 5% methanol/water for 15 min in one step. The native dG was determined by UV scanning from 240 nm to 290 nm. The 8-oxo-dG was detected using selected reaction monitoring and collision-induced dissociation for the fragmentation pathway of m/z 282 → 192 with a dwell time of 0.5 s/ion using negative ion electrospray (39).
Determination of ROS by CM-H2DCFDA—Both S30 and MDA-MB-231 cells were grown (106 cells/ml) on each of eight wells on a sterile Nunc™ chambered coverglass and incubated for 48 h at 37 °C with 5% CO2 in phenol red-free MEM supplemented with 10% stripped fetal bovine serum medium. S30 and MDA-MB-231 cells were labeled with 10 μm CM-H2DCFDA for 30 min at 37 °C, 5% CO2 (40, 41). CM-H2DCFDA-treated cells were rinsed twice with PBS to remove the unincorporated dye, and 0.2 μg/ml Hoechst stain was added to the cells to detect nuclear staining. The cells were then treated with 4-OHEN (1 μm), menadione (1 μm), or DMSO (0.5%) for 5 min, and imaging was performed with a Zeiss LSM 510 laser-scanning confocal microscope with the detector gain adjusted to eliminate the background autofluorescence. The fluorescence signal from CM-H2DCFDA was monitored with a 488 nm argon/krypton laser and a 530 nm band pass filter. The Hoechst nuclear staining signal was monitored with a 345 nm UV laser and 420 nm band pass filter. A ×63 (1.2 numerical aperture) water immersion objective was used for all experiments. Images were analyzed using the analysis tool provided in the Zeiss biophysical software package.
Statistical Analyses—ERE-luciferase assays and comet and FLARE comet studies were performed three times, and the results of 8-oxo-dG measurement using LC-MS/MS were obtained from two separate experiments. All data were expressed as the mean ± S.D. The statistical analysis of these results consisted of one-way analysis of variance with Dunnett's or Tukey's multiple comparison tests using GraphPad Prism version 4 for Windows.
RESULTS
4-OHEN Is a Poor Ligand for the Isolated ER but an Estrogen of Nanomolar Potency in Cell Culture—The first question to address was the estrogenicity of 4-OHEN in ERα-positive cells. Previous reports have shown low affinity in the radioligand competitive binding assay to recombinant full-length ERα, with significant variance probably because of the instability of 4-OHEN: IC50 = 1.5 ± 0.2 μm (31, 42). Furthermore, binding to ER by 4-methoxy equilenin (4-MeOEN), the product of COMT action on 4-OHEN, was not even detectable under the same assay conditions (43). Nevertheless, 4-MeOEN was observed to be an estrogen agonist in both ERα(+) Ishikawa (EC50 = 0.16 ± 0.1 nm) and MCF-7 cells (EC50 = 6.5 ± 0.6 nm) (43). Similarly, 4-OHEN was observed to be an agonist with nanomolar potency (EC50 = 5.7 ± 2.8 nm) as measured in ERα(+) MCF-7 cells transiently transfected with an ERE-luciferase reporter vector and treated for 18 h with test compound. As expected, co-administration of the selective estrogen receptor modulator 4-hydroxytamoxifen (4-OHTAM) or the pure estrogen antagonist ICI 182,780 inhibited the estrogenic activity of 4-OHEN (Fig. 1). As 4-MeOEN has been shown to be an estrogen of nanomolar potency, it was necessary to rule out COMT-mediated methylation of 4-OHEN as the cause of estrogenicity using the COMT inhibitor Ro-41-0960 (Fig. 1). Finally, menadione was confirmed to be devoid of estrogenic or antiestrogenic activity.
FIGURE 1.

ERα-mediated activation of ERE-luciferase activity in MCF-7 cells. Shown are control (0.05% DMSO), E2 (1 nm), 4-OHEN (0.1 μm), 4-OHTAM (10 nm), ICI 182,780 (ICI, 1 μm), menadione (1 μm), Ro-41-0960 (Ro, 10 μm) for 18 h. Experimental details are described under “Experimental Procedures.” Data represent the mean ± S.D. (n = 3).
4-OHEN Binding Causes Translocation of ERα to the Nucleus of MCF-7 Cells—Although 4-OHEN is a full and potent estrogen, multiple mechanisms of nonclassical estrogenic signaling are being revealed involving membrane-associated ER (44). Both 4-MeOEN and 4-OHEN are reported to activate extracellular signal-regulated kinase at 5 min in MCF-7 cells, a target for rapid membrane-associated ER-mediated response (43). Therefore it was deemed essential to demonstrate that activity resulted from nuclear localization and binding to DNA of 4-OHEN-liganded ERα. The status of the ligand-activated ERα present on the estrogen-responsive regions of the pS2 promoter was determined using ChIP and analyzed by semiquantitative PCR (Fig. 2). In this assay, MCF-7:K-1 cells were used because they contain high levels of ERα and very low levels of Her2/neu expression (45). In addition, pS2 mRNA and the transcription rate of the pS2 gene were shown to be markedly increased by treatment with E2 (46). These features of MCF-7: K-1 cells facilitated the analysis of the promoter region of the pS2 gene occupied by ligand-activating ERα using the ChIP assay. After estrogen stimulation, the maximal protein recruitment to the promoters of pS2 and cathepsin D were observed by ChIP at 45 min (47, 48). Based on these results, MCF-7:K1 cells were treated with 4-OHEN for 45 min, and chromatin was cross-linked by formaldehyde treatment.
FIGURE 2.

PCR products for pS2 ERE and pS2 upstream (negative control) in the ChIP assay. Cells were cultured in estrogen-free media for 4 days before treatment. Cells were treated with DMSO, E2 (10 nm), or 4-OHEN for 45 min and then washed and cross-linked using 1.5% formaldehyde for 15 min. All samples were pulled down by antibodies (Ab) against ERα and IgG and then incubated with protein A/G agarose slurry. 1, Input, positive control, all DNA fragments without any antibody treatment; 2, rAb ERα, ERα antibody; 3, rIgG, negative control, illustrating the specificity of the ERα antibody.
As shown in Fig. 2, treatment of MCF-7:K-1 cells with 4-OHEN induced a significant increase in binding of the pS2 gene promoter by ERα. Given the ∼500–2000-bp size of the DNA fragments produced by sonication in these experiments, PCR analysis did not detect any significant increase in ERα occupancy of a region corresponding to 4,400 bp upstream of the pS2 ERE gene. No PCR amplification product was detected in the chromatin immunoprecipitation pulled down with IgG. These results showed that 4-OHEN activates ERα to bind to the promoter region of estrogen-responsive genes in cells.
4-OHEN-induced DNA Single Strand Breaks and 8-Oxo-dG Formation Are Enhanced by ERα in Breast Cancer Cells—The comet assay and measurement of 8-oxo-dG provide two assays of oxidative DNA damage. 4-OHEN was observed to cause concentration-dependent DNA single strand breaks in both S30 ERα(+) and MDA-MB-231 ER(-) cell lines (Fig. 3A). In addition, ERα(+) cells were more sensitive to 4-OHEN-induced DNA single strand breaks, confirming previously published data (31). After pretreatment with 4-OHTAM for 1 h or with ICI 182,780 for 24 h, DNA damage induced by 4-OHEN was significantly inhibited in ERα(+) cells (Fig. 3B), whereas no protection was observed in ER(-) cells (Fig. 3C). It should be noted that the low concentration of 4-OHTAM used is compatible with ER-mediated antiestrogenic activity and not with the putative antioxidant activity of 4-OHTAM associated with much higher concentrations. Similar results were obtained using LC-MS/MS to quantitate the formation of 8-oxo-dG (Table 1) in the presence of 4-OHEN and in the presence or absence of 4-OHTAM. The data show that ERα(+) cells are more sensitive to oxidative DNA damage induced by 4-OHEN.
FIGURE 3.

Induction of DNA single strand breaks by 4-OHEN measured using the comet assay. Cells were treated with various concentrations of 4-OHEN for 90 min in S30 and MDA-MB-231 cells. A, squares, 4-OHEN in S30 cells; circles, 4-OHEN in MDA-MB-231 cells. B, closed squares, 4-OHEN in S30 cells; open squares, 4-OHEN + 4-OHTAM (10 nm, 1 h) in S30 cells; closed circles, 4-OHEN + ICI 182,780 (1 μm, 24 h) in S30 cells. C, closed circles, 4-OHEN in MDA-MB-231 cells; open circles, 4-OHEN + 4-OHTAM (10 nm, 1 h) in MDA-MB-231 cells. Data represent the mean ± S.E. (n = 3).
TABLE 1.
LC-MS/MS analysis of 8-oxo-dG in S30 and MDA-MB-231 cells treated with 4-OHEN or menadione Cells were treated with 4-OHEN or menadione (10 μm) for 90 min with or without 4-OHTAM (10 nm) pretreatment for 60 min. Experimental details are described under “Experimental Procedures.” Data represent the mean ± S.D. (n = 2). *, significant difference (p < 0.05) compared with the S30 sample treated with 4-OHEN only.
|
Treatment
|
8-Oxo-dG/dG × 105
|
|||
|---|---|---|---|---|
|
4-OHEN
|
Menadione
|
|||
| S30 | MDA-MB-231 | S30 | MDA-MB-231 | |
| DMSO | 1.06 ± 0.04 | 0.98 ± 0.04 | 1.20 ± 0.25 | 1.10 ± 0.21 |
| 10 μm | 1.65 ± 0.16 | 1.37 ± 0.08* | 2.26 ± 0.41 | 2.23 ± 0.07 |
| 10 μm + 4-OHTAM | 1.29 ± 0.01* | 1.34 ± 0.13 | 2.40 ± 0.51 | 2.62 ± 0.14 |
Menadione-induced Oxidative DNA Damage Is ER-independent in Breast Cancer Cells—Using both the comet assay and Fpg FLARE comet assay, the induction of DNA single strand breaks and oxidized bases by menadione was assayed in ERα(+) and ER(-) cells under the same conditions as described for 4-OHEN-treated cells. The expected concentration-dependent increase in DNA single strand breaks (Fig. 4A) and oxidized bases (Fig. 4B) was observed in both cell lines. The extent of DNA damage in ER(-) cells was comparable with 4-OHEN at the same concentration, compatible with similar redox-cycling mechanisms for ROS generation induced by menadione and 4-OHEN (Scheme 1). In contrast to 4-OHEN, no effect of cellular ER status was seen upon menadione-induced oxidative damage. In accord with this observation, 4-OHTAM pretreatment had no effect on the level of oxidative damage in response to menadione (Fig. 4). Similar results were obtained in LC-MS/MS experiments (Table 1). These data support the hypothesis that menadione-induced DNA damage is ER-independent.
FIGURE 4.

Induction of DNA single strand breaks and oxidized bases by menadione measured using the comet assay (single strand breaks) (A) and Fpg FLARE comet assay (oxidized bases) (B). Cells were treated with various concentrations of menadione for 90 min in S30 and MDA-MB-231 cells. Closed diamonds, menadione in S30 cells; open diamonds, menadione + 4-OHTAM (10 nm, 1 h) in S30 cells; open circles, menadione in MDA-MB-231 cells. Data represent the mean ± S.E. (n = 3).
SCHEME 1.

Generation of ROS by redox cycling of 4-OHEN and menadione.
ERα Accelerates the Rate of 4-OHEN-induced DNA Damage—The results presented above show that 4-OHEN-induced DNA damage is modestly enhanced by ERα. This generally supports the Trojan horse hypothesis that posits the liganded ER complex as an active nuclear transporter of the small molecule genotoxin (Scheme 2). Unassisted, passive diffusion of genotoxins to the nucleus is a parallel process; one might predict that the active process would lead to more rapid induction of oxidative DNA damage after cells are exposed to genotoxin. To address this hypothesis, the time dependence of 4-OHEN-induced DNA damage was assayed in ERα(+) versus ER(-) cells using the comet assay (Fig. 5A). In ERα-positive cells, induction of DNA damage by 4-OHEN was more rapid. Furthermore, the ER antagonist 4-OHTAM at low concentrations was able to reduce significantly the rate of accumulation of oxidative DNA damage in ERα(+) cells, essentially attenuating DNA damage to the level observed in the ER(-) cell line. Finally, no significant effect of 4-OHTAM was observed in ER-negative cells. These results suggest that active transport by ERα accelerates genotoxin-induced DNA damage in the nucleus of ERα-positive cells.
SCHEME 2.

Proposed mechanisms of induction of DNA damage by 4-OHEN or menadione in breast cancer cells. Active transport of estrogenic genotoxins by the ER-enhanced (“Trojan horse”) pathway is contrasted with passive diffusion by ER-independent pathways for estrogenic and non-estrogenic genotoxins.
FIGURE 5.

Time dependence of 4-OHEN and menadione-induced DNA single strand breaks in breast cancer cells using the comet assay. Cells were treated with 4-OHEN (4 μm) or menadione (4 μm) for various incubation times. A, closed squares, 4-OHEN in S30 cells; open squares, 4-OHEN + 4-OHTAM (10 nm, 1 h) in S30 cells; closed circles (dashed lines), 4-OHEN in MDA-MB-231 cells; open circles, 4-OHEN + 4-OHTAM (10 nm, 1 h) in MDA-MB-231 cells. Data were fitted to a first order equation for ease of visualization. B, time dependence was assayed using NAC to quench the reaction. NAC (10 mm) was added at the indicated time points after 4-OHEN or menadione treatment, and then the cells were incubated for a further 1 min. Closed squares, 4-OHEN in S30 cells; closed circles (dashed lines), 4-OHEN in MDA-MB-231 cells; closed diamonds, menadione in S30 cells; open circles, menadione in MDA-MB-231 cells. Data were fitted to a linear equation. Data represent the mean ± S.E. (n = 3).
ERα Enhances the Initial Rate of 4-OHEN-induced DNA Damage—To strengthen the hypothesis that ERα active transport rapidly enhances genotoxin-induced DNA damage, it was informative to examine very early time points after genotoxin application. Unfortunately, most common methods of quantitation are not readily suited to be used at early time points. A recent study on ER-mediated gene transcription reported, remarkably, that early and rapid formation of 8-oxo-dG, upon binding of the ER complex to DNA, was required to trigger chromatin and DNA conformational changes essential for estrogen-induced transcription in MCF-7 cells (49). The fluorescence method used for rapid detection of 8-oxo-dG required rapid fixing of cells with paraformaldehyde, washing, and permeabilization followed by a 15-h incubation with a test kit.
The comet assay was adapted for early time point measurements using an excess of the cell-permeable quinone and ROS scavenging agent NAC. Control experiments were conducted using different concentrations of NAC and different incubations times. Incubation with NAC (10 mm) for 1 min was chosen followed by rapid cooling and an accelerated work-up of no more than 50 min before cell lysis. The work-up period could not be reduced further without increasing variance; however, in control experiments, extended work-up times did not lead to increased amounts of DNA damage using the NAC protocol. In the absence of NAC, extended work-up time was observed to lead to increased comet scores. The major advantage of NAC quenching is the scavenging of ROS, which might continue to be generated during the comet assay work-up. The method is therefore not seen as inferior to alternative procedures reported.
4-OHEN did not significantly induce DNA single strand breaks in ER(-) cells within 8 min, suggesting that in the absence of ER, DNA damage occurs relatively slowly (Fig. 5B). However, in ERα(+) cells, DNA damage was dramatically increased at these early time points, with an almost 10-fold higher initial rate of single strand breakage, confirming that ERα enhances the rate of DNA damage. This conclusion was supported by similar experiments with menadione treatment; equivalent and minor amounts of oxidative DNA damage were observed in cells regardless of ER status. These results indicate that ERα plays a role in dramatically enhancing the initial rate of 4-OHEN-induced DNA damage.
4-OHEN Rapidly Generates ROS in the Nucleus of ERα-positive Breast Cancer Cells—In contrast to measurements of oxidative DNA damage (Comet assay, LC-MS/MS), the rapid and real-time detection of ROS by confocal fluorescence microscopy is reliably able to measure and localize the cellular generation of ROS. Cells were treated with CM-H2DCFDA, a cell-permeable dichlorofluorescein ROS indicator that is sensitive to oxidizing oxygen radicals, including hydroxyl radicals formed from H2O2 (40). Cells were treated with 4-OHEN (1 μm), menadione (1 μm), or DMSO vehicle (0.5%) (Fig. 6). In ERα(+) cells, within 5 min generation of ROS induced by 4-OHEN was selectively localized in the nucleus (Fig. 6A), whereas in ER(-) cells, ROS were equally distributed in both the nucleus and the cytoplasm (Fig. 6B). ROS generation induced by menadione was detected, but in contrast, no differences were observed between the two cell lines based upon ER status.
FIGURE 6.

Detection of ROS in S30 (A) and MDA-MB-231 cells (B) using CM-H2DCFDA. Cells were labeled with CM-H2DCFDA and treated separately with 4-OHEN (1 μm), menadione (1 μm), or DMSO vehicle control. Confocal images were taken before and after a 5-min treatment with each compound or vehicle control. Ex/Em (488 nm/530 nm), describes the fluorescent CM-DCF excitation/emission signal; Ex/Em (345 nm/420 nm), describes excitation and emission wavelengths of the nuclear staining signal; Overlay, describes both the CM-DCF and nuclear signals. The images shown are representative of at least three independent experiments.
Application of confocal fluorescence microscopy to the actions of E2 itself (1 μm) in S30 and MDA-MB-231 cells did not reveal generation of comparable levels of ROS to those observed for the redox-cycling quinones menadione and 4-OHEN-o-quinone, nor was dependence on ER status observed (Fig. 7). The results of this control experiment are not compatible with generation of ROS in the nucleus resulting from hormonal activity of 4-OHEN. The data are in accord with the ability of ERα to bind, actively translocate, and concentrate 4-OHEN in the nucleus, leading to redox cycling-enhanced ROS generation in the nucleus and, in turn, amplified DNA damage.
FIGURE 7.

Detection of ROS in S30 and MDA-MB-231 cells using CM-H2DCFDA. Cells were labeled with CM-H2DCFDA and treated with E2 (1 μm). Confocal images were taken before and after a 5-min treatment with each compound or vehicle control. Similar data (not shown) were obtained with E2 (10 nm). Ex/Em (488 nm/530 nm), describes the fluorescent CM-DCF excitation/emission signal; Ex/Em (345 nm/420 nm), describes the excitation and emission wavelengths of the nuclear staining signal; Overlay, describes both the CM-DCF and nuclear signals. The images shown are representative of at least three independent experiments.
DISCUSSION
It is well accepted that the ER plays a major role in estrogen-induced carcinogenesis (13, 16). ER binding of estrogen leads to cell proliferation and anti-apoptotic signaling in hormone-sensitive target tissues such as the breast and endometrium. However, a contributor to the hormonal carcinogenesis mechanism must be the incorporation, in rapidly dividing cells, of DNA mutations that lead to initiation and/or promotion of the carcinogenic process. Yager and Davidson (16), in particular, have made the case for the contribution to carcinogenesis from oxidative metabolism of estrogens to genotoxic, mutagenic metabolites. Metabolism of estrogens to catechols and further oxidation to reactive o-quinones has been correlated with the genotoxicity of estrogens (17, 18). Estrogens that are potent ER agonists and are oxidized to electrophilic and redox-cycling metabolites have the potential to contribute to the initiation, promotion, and progression of hormone-sensitive cancers as dual mechanism carcinogens.
The equine estrogens contained in HRT and ERT formulations that are proven to increase breast cancer risk in postmenopausal women are potent estrogens; the ER binding affinity, uterotrophic potency, and cell proliferation potency of equilenin and equilin approach that of E2 (50). Furthermore, their oxidative metabolites are argued to have more genotoxic potential than those of endogenous estrogens (17). In contrast to endogenous estrogens that are selectively oxidized to 2-hydroxy catechols rather than the carcinogenic 4-hydroxy isomers, equine estrogens are predominantly hydroxylated at the 4-position to give 4-OHEN and 4,17β-dihydroxyequilenin (27, 34, 51). Furthermore, unlike endogenous catechol estrogens, 4-OHEN autoxidizes to an o-quinone that is genotoxic in breast cancer cells; it is thus a redox-cycling genotoxin (Scheme 1).
Questions remain as to the relevance of equine estrogens and the 4-OHEN metabolite as human carcinogens. Although relatively stable in the absence of reductant, 4-OHEN-o-quinone is readily trapped by nucleophiles including GSH. In addition, the concentration of oxidative metabolites in target tissues may be low, although 4-OHEN-DNA adducts have been reported in human breast tissue (20). If 4-OHEN or its o-quinone represents a good estrogenic ligand, the ER would be capable of translocation of this genotoxin to the nucleus where oxidative DNA damage would be amplified or accelerated, even at lower concentrations. The ER would act as a Trojan horse, and ER-positive cells would be highly sensitive to DNA damage. The Trojan horse model would also hold for alternative nuclear receptors and transcription factors and other ligands, for example, environmental estrogen genotoxins and polyaromatic hydrocarbon metabolites in the case of the arylhydrocarbon receptor (52).
4-OHEN causes covalent adduction and oxidation of DNA (17, 25, 30, 31). For the Trojan horse mechanism to hold for 4-OHEN, the first requirement is that the catechol or o-quinone must be a ligand for the ER and must trigger translocation to the nucleus and binding to an ER-related DNA response element. 4-OHEN has previously been reported to have low affinity for recombinant ER; however, 4-MeOEN, which had even lower ER affinity in the radioligand displacement assay, has been reported to satisfy all criteria for a classical estrogen of nanomolar potency acting via ERα binding (43). Similarly, 4-OHEN was observed herein to function as an estrogen with nanomolar potency in ERα(+) breast cancer cell lines, the activity of which was blocked by the antiestrogen ICI 182,780 and the selective estrogen receptor modulator 4-OHTAM. Moreover, using the ChIP assay of chromatin-protein mixtures, the binding of the pS2 estrogen target gene promoter by ERα in MCF-7 cells was shown to be dependent upon 4-OHEN; that is, 4-OHEN triggers ERα nuclear translocation and DNA binding in breast cancer cells.
To test the relevance of the Trojan horse mechanism to oxidative DNA damage, comparison is required between ERα(+) and ERα(-) cell lines but also between 4-OHEN and a redox-cycling quinone that is not an ER ligand. Thus, MDA-MB-321 breast cancer cells were compared with S30 cells, the variant of the same breast cancer cell line stably transfected with ERα. The non-estrogenic p-quinone, menadione, well known to produce oxidative DNA damage and to redox cycle, provided the comparator redox-cycling genotoxin (53). Oxidative DNA damage was concentration-dependent for both 4-OHEN and menadione. DNA damage was observed to be ∼ 1.3-fold higher in S30 cells than in MDA-MB-231 cells after 90 min of incubation with 4-OHEN, but this was not the case for menadione. Furthermore, DNA damage due to 4-OHEN was uniquely attenuated in ERα(+) cells in response to 4-OHTAM or ICI 182,780. No effects of antiestrogens were seen in menadione-treated cells.
These results strongly support the Trojan horse hypothesis. However, the absolute difference between DNA damage produced by passive diffusion (4-OHEN in ER(-) cells and menadione) versus ER-assisted active transport (4-OHEN in ER(+) cells) is modest. In accord with this observation, Essigmann and colleagues (54, 55) have reported that a nitrogen mustard conjugated to estrogen showed no more than a 2.5-fold higher potency toward killing ERα(+) cells. The rate of oxidative DNA damage caused by agents such as 4-OHEN and menadione is expected to be amplified by high nuclear concentrations of the redox-cycling quinones generating high local concentrations of ROS, in particular the more reactive ROS, such as hydroxyl and alkoxyl radicals. Therefore, it was important to assay the time course of DNA damage in response to 4-OHEN and menadione. Using the standard comet assay in which DNA damage is quenched by cooling in the dark, formation of DNA single strand breaks was indeed seen to be time-dependent. To measure earlier time points, the comet assay was adapted by adding NAC to quench quinones and ROS in addition to cooling. Oxidative DNA damage, at early time points induced by 4-OHEN in ERα(+) breast cancer cells, was observed uniquely and dramatically to be enhanced.
In contrast to DNA damage, localization of ROS generation can readily be measured in real-time employing confocal microscopy with a cell-permeable, ROS-sensitive reporter dye. Again comparing 4-OHEN with menadione in the two different breast cancer cell environments (ERα(+) versus ERα(-)) the prediction based upon the Trojan horse hypothesis is straight-forward: 1) 4-OHEN and menadione are predicted to generate ROS in a time-dependent manner in all cell compartments by passive diffusion and 2) ER-mediated active transport of 4-OHEN to the nucleus is predicted to increase the rate of ROS generation in the nucleus relative to other cell compartments. Using co-administration of a nucleus-specific dye, these predictions were fully borne out by the data. At 5 min after 4-OHEN administration, generation of ROS was selectively elevated in the nucleus, fully in accord with data from the comet assay.
Breast cancer risk is associated with estrogen receptor status. Hormone-dependent genomic instability during cell proliferation and regulation of antioxidant genes have been proposed as purely hormonal ER-dependent contributors to DNA damage (56–58). Catechol estrogen metabolites have been proposed to contribute to DNA damage (16), although these genotoxic effects, taken alone, are thought to be ER-independent (21). Mammary tumors were reported in ERα knock-out mice expressing the Wnt-1 oncogene; however, tumor formation was delayed compared with mice with wild type ERα-positive animals (59, 60). The Trojan horse mechanism could also amplify the ER-dependent genotoxicity of the endogenous 4-hydroxy catechol estrogens, although in contrast to equine estrogen catechols, these do not undergo autoxidation.
Clinical use of HRT in women appears to be correlated with increased risk of ER-positive breast cancer but not with ER-negative breast cancer (61, 62). Recent clinical studies have been interpreted as showing a significant decline in the incidence of breast cancer for women with ER-positive tumors after discontinuation of HRT (11, 63, 64). One of the major oxidative metabolites in HRT, 4-OHEN, is autoxidized to an o-quinone that causes DNA damage in breast cancer cells. The data presented herein show that ERα acts as a Trojan horse, rapidly amplifying the rate of oxidative DNA damage caused by 4-OHEN in breast cancer cells and potentially enhancing the mutagenicity of equine estrogens in ER-positive cells. This genotoxic metabolite mechanism, coupled with the potent proliferative estrogenicity of equine estrogens and their metabolites, could contribute to hormone-dependent breast cancer in women. The nuclear translocation and localization of 4-OHEN by ERα will be selective for binding to DNA at estrogen-responsive genes, implying sequence-selective DNA damage and contributing to mutagenicity, which requires further study. The recent proposal that rapid and transient formation of 8-oxo-dG is required to trigger estrogen-induced transcription in MCF-7 cells should also be examined in light of the actions of 4-OHEN in ER-positive cells (49). Finally, it should be noted that the Trojan horse mechanism is applicable to other nuclear receptors.
Acknowledgments
We thank Dr. Zhihui Qin for the synthesis of 4-OHEN. In addition, we greatly appreciate Dr. V. C. Jordan (Fox Chase Cancer Center, Philadelphia, PA), Dr. Debra A. Tonetti (University of Illinois at Chicago) for the gift of the S30 cell line and Dr. J. Frasor (University of Illinois at Chicago) for the gift of the MCF-7:K1 cell line.
This work was supported, in whole or in part, by National Institutes of Health Grant CA130037 (to J. L. B.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: HRT, hormone replacement therapy; ERT, estrogen replacement therapy; ChIP, chromatin immunoprecipitation; CM-DCF, 5-(and 6)-chloromethyl-2′,7′-dichlorofluorescein; CM-H2DCFDA, 5-(and 6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester; COMT, catechol-O-methyltransferase; DMSO, dimethyl sulfoxide; E2, 17β-estradiol; ER, estrogen receptor; ERE, estrogen-responsive element; 4-MeOEN, 4-methoxy equilenin; NAC, N-acteylcysteine; 4-OHTAM, 4-hydroxytamoxifen; 4-OHEN, 4-hydroxyequilenin; 8-oxo-dG, 8-oxo-7,8-dihydro-2′-deoxyguanosine; ROS, reactive oxygen species; MEM, minimum essential medium; PBS, phosphate-buffered saline; LC-MS/MS, liquid chromatography-tandem mass spectroscopy.
References
- 1.Li, J. J., Li, S. A., Oberley, T. D., and Parsons, J. A. (1995) Cancer Res. 55 4347-4351 [PubMed] [Google Scholar]
- 2.Colditz, G. A., Hankinson, S. E., Hunter, D. J., Willett, W. C., Manson, J. E., Stampfer, M. J., Hennekens, C., Rosner, B., and Speizer, F. E. (1995) N. Engl. J. Med. 332 1589-1593 [DOI] [PubMed] [Google Scholar]
- 3.Key, T., Appleby, P., Barnes, I., and Reeves, G. (2002) J. Natl. Cancer Inst. 94 606-616 [DOI] [PubMed] [Google Scholar]
- 4.Grodstein, F., Stampfer, M. J., Colditz, G. A., Willett, W. C., Manson, J. E., Joffe, M., Rosner, B., Fuchs, C., Hankinson, S. E., Hunter, D. J., Hennekens, C. H., and Speizer, F. E. (1997) N. Engl. J. Med. 336 1769-1775 [DOI] [PubMed] [Google Scholar]
- 5.Lupulescu, A. (1995) Cancer Investig. 13 287-295 [DOI] [PubMed] [Google Scholar]
- 6.Service, R. F. (1998) Science 279 1631-1633 [DOI] [PubMed] [Google Scholar]
- 7.Vogel, V. G., Yeomans, A., and Higginbotham, E. (1993) Breast Cancer Res. 28 195-210 [DOI] [PubMed] [Google Scholar]
- 8.Zumoff, B. (1998) Proc. Soc. Exp. Biol. Med. 217 30-37 [DOI] [PubMed] [Google Scholar]
- 9.Rossouw, J. E., Anderson, G. L., Prentice, R. L., LaCroix, A. Z., Kooperberg, C., Stefanick, M. L., Jackson, R. D., Beresford, S. A., Howard, B. V., Johnson, K. C., Kotchen, J. M., and Ockene, J. (2002) J. Am. Med. Assoc. 288 321-333 [DOI] [PubMed] [Google Scholar]
- 10.Shumaker, S. A., Legault, C., Rapp, S. R., Thal, L., Wallace, R. B., Ockene, J. K., Hendrix, S. L., Jones, B. N., III, Assaf, A. R., Jackson, R. D., Kotchen, J. M., Wassertheil-Smoller, S., and Wactawski-Wende, J. (2003) J. Am. Med. Assoc. 289 2651-2662 [DOI] [PubMed] [Google Scholar]
- 11.Ravdin, P. M., Cronin, K. A., Howlader, N., Berg, C. D., Chlebowski, R. T., Feuer, E. J., Edwards, B. K., and Berry, D. A. (2007) N. Engl. J. Med. 356 1670-1674 [DOI] [PubMed] [Google Scholar]
- 12.Feigelson, H. S., and Henderson, B. E. (1996) Carcinogenesis 17 2279-2284 [DOI] [PubMed] [Google Scholar]
- 13.Henderson, B. E., and Feigelson, H. S. (2000) Carcinogenesis 21 427-433 [DOI] [PubMed] [Google Scholar]
- 14.Nandi, S., Guzman, R., and Yang, J. (1995) Proc. Natl. Acad. Sci. U. S. A. 92 3650-3657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flototto, T., Djahansouzi, S., Glaser, M., Hanstein, B., Niederacher, D., Brumm, C., and Beckmann, M. W. (2001) Horm. Metab. Res. 33 451-457 [DOI] [PubMed] [Google Scholar]
- 16.Yager, J. D., and Davidson, N. E. (2006) N. Engl. J. Med. 354 270-282 [DOI] [PubMed] [Google Scholar]
- 17.Bolton, J. L., and Thatcher, G. R. J. (2008) Chem. Res. Toxicol. 21 93-101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaikwad, N. W., Yang, L., Muti, P., Meza, J. L., Pruthi, S., Ingle, J. N., Rogan, E. G., and Cavalieri, E. L. (2008) Int. J. Cancer 122 1949-1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang, Q., Aft, R. L., and Gross, M. L. (2008) Chem. Res. Toxicol. 21 1509-1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Embrechts, J., Lemiere, F., Van Dongen, W., Esmans, E. L., Buytaert, P., Van Marck, E., Kockx, M., and Makar, A. (2003) J. Am. Soc. Mass Spectrom. 14 482-491 [DOI] [PubMed] [Google Scholar]
- 21.Cavalieri, E., Chakravarti, D., Guttenplan, J., Hart, E., Ingle, J., Jankowiak, R., Muti, P., Rogan, E., Russo, J., Santen, R., and Sutter, T. (2006) Biochim. Biophys. Acta 1766 63-78 [DOI] [PubMed] [Google Scholar]
- 22.Bolton, J. L. (2002) Toxicology 177 55-65 [DOI] [PubMed] [Google Scholar]
- 23.Jefcoate, C. R., Liehr, J. G., Santen, R. J., Sutter, T. R., Yager, J. D., Yue, W., Santner, S. J., Tekmal, R., Demers, L., Pauley, R., Naftolin, F., Mor, G., and Berstein, L. (2000) J. Natl. Cancer Inst. Monogr. 95-112 [DOI] [PubMed]
- 24.Liehr, J. G. (2000) Endocr. Rev. 21 40-54 [DOI] [PubMed] [Google Scholar]
- 25.Bolton, J. L., Pisha, E., Zhang, F., and Qiu, S. (1998) Chem. Res. Toxicol. 11 1113-1127 [DOI] [PubMed] [Google Scholar]
- 26.Cavalieri, E. L., Stack, D. E., Devanesan, P. D., Todorovic, R., Dwivedy, I., Higginbotham, S., Johansson, S. L., Patil, K. D., Gross, M. L., Gooden, J. K., Ramanathan, R., Cerny, R. L., and Rogan, E. G. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 10937-10942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang, F., Chen, Y., Pisha, E., Shen, L., Xiong, Y., van Breemen, R. B., and Bolton, J. L. (1999) Chem. Res. Toxicol. 12 204-213 [DOI] [PubMed] [Google Scholar]
- 28.Shen, L., Pisha, E., Huang, Z., Pezzuto, J., Krol, E., Alam, Z., van Breemen, R., and Bolton, J. (1997) Carcinogenesis 18 1093-1101 [DOI] [PubMed] [Google Scholar]
- 29.Chen, Y., Shen, L., Zhang, F., Lau, S. S., van Breemen, R. B., Nikolic, D., and Bolton, J. L. (1998) Chem. Res. Toxicol. 11 1105-1111 [DOI] [PubMed] [Google Scholar]
- 30.Chen, Y., Liu, X., Pisha, E., Constantinou, A. I., Hua, Y., Shen, L., van Breemen, R. B., Elguindi, E. C., Blond, S. Y., Zhang, F., and Bolton, J. L. (2000) Chem. Res. Toxicol. 13 342-350 [DOI] [PubMed] [Google Scholar]
- 31.Liu, X., Yao, J., Pisha, E., Yang, Y., Hua, Y., van Breemen, R. B., and Bolton, J. L. (2002) Chem. Res. Toxicol. 15 512-519 [DOI] [PubMed] [Google Scholar]
- 32.Okamoto, Y., Chou, P. H., Kim, S. Y., Suzuki, N., Laxmi, Y. R., Okamoto, K., Liu, X., Matsuda, T., and Shibutani, S. (2008) Chem. Res. Toxicol. 21 1120-1124 [DOI] [PubMed] [Google Scholar]
- 33.Shen, L., Qiu, S., Chen, Y., Zhang, F., van Breemen, R. B., Nikolic, D., and Bolton, J. L. (1998) Chem. Res. Toxicol. 11 94-101 [DOI] [PubMed] [Google Scholar]
- 34.Zhang, F., Swanson, S. M., van Breemen, R. B., Liu, X., Yang, Y., Gu, C., and Bolton, J. L. (2001) Chem. Res. Toxicol. 14 1654-1659 [DOI] [PubMed] [Google Scholar]
- 35.National Institutes of Health (1981) NIH Guidelines for the Laboratory Use of Chemical Carcinogens, U. S. Government Printing Office, Washington, D. C.
- 36.Han, X., and Liehr, J. G. (1995) Carcinogenesis 16 2571-2574 [DOI] [PubMed] [Google Scholar]
- 37.Singh, N. P., McCoy, M. T., Tice, R. R., and Schneider, E. L. (1988) Exp. Cell Res. 175 184-191 [DOI] [PubMed] [Google Scholar]
- 38.Collins, A. R., Dobson, V. L., Dusinska, M., Kennedy, G., and Stetina, R. (1997) Mutat. Res. 375 183-193 [DOI] [PubMed] [Google Scholar]
- 39.Hua, Y., Wainhaus, S. B., Yang, Y., Shen, L., Xiong, Y., Xu, X., Zhang, F., Bolton, J. L., and van Breemen, R. B. (2001) J. Am. Soc. Mass Spectrom. 12 80-87 [DOI] [PubMed] [Google Scholar]
- 40.LeBel, C. P., Ischiropoulos, H., and Bondy, S. C. (1992) Chem. Res. Toxicol. 5 227-231 [DOI] [PubMed] [Google Scholar]
- 41.Ubezio, P., and Civoli, F. (1994) Free Radic. Biol. Med. 16 509-516 [DOI] [PubMed] [Google Scholar]
- 42.Liu, X., Pisha, E., Tonetti, D. A., Yao, D., Li, Y., Yao, J., Burdette, J. E., and Bolton, J. L. (2003) Chem. Res. Toxicol. 16 832-837 [DOI] [PubMed] [Google Scholar]
- 43.Chang, M., Peng, K. W., Kastrati, I., Overk, C. R., Qin, Z. H., Yao, P., Bolton, J. L., and Thatcher, G. R. (2007) Endocrinology 148 4793-4802 [DOI] [PubMed] [Google Scholar]
- 44.Pedram, A., Razandi, M., and Levin, E. R. (2006) Mol. Endocrinol. 20 1996-2009 [DOI] [PubMed] [Google Scholar]
- 45.Cho, H. S., Ng, P. A., and Katzenellenbogen, B. S. (1991) Mol. Endocrinol. 5 1323-1330 [DOI] [PubMed] [Google Scholar]
- 46.Harrington, W. R., Kim, S. H., Funk, C. C., Madak-Erdogan, Z., Schiff, R., Katzenellenbogen, J. A., and Katzenellenbogen, B. S. (2006) Mol. Endocrinol. 20 491-502 [DOI] [PubMed] [Google Scholar]
- 47.Carroll, J. S., and Brown, M. (2006) Mol. Endocrinol. 20 1707-1714 [DOI] [PubMed] [Google Scholar]
- 48.Shang, Y., Hu, X., DiRenzo, J., Lazar, M. A., and Brown, M. (2000) Cell 103 843-852 [DOI] [PubMed] [Google Scholar]
- 49.Perillo, B., Ombra, M. N., Bertoni, A., Cuozzo, C., Sacchetti, S., Sasso, A., Chiariotti, L., Malorni, A., Abbondanza, C., and Avvedimento, E. V. (2008) Science 319 202-206 [DOI] [PubMed] [Google Scholar]
- 50.Mueck, A. O., Seeger, H., and Wallwiener, D. (2003) Climacteric 6 221-227 [PubMed] [Google Scholar]
- 51.Pisha, E., Lui, X., Constantinou, A. I., and Bolton, J. L. (2001) Chem. Res. Toxicol. 14 82-90 [DOI] [PubMed] [Google Scholar]
- 52.Burczynski, M. E., and Penning, T. M. (2000) Cancer Res. 60 908-915 [PubMed] [Google Scholar]
- 53.Nutter, L. M., Ngo, E. O., Fisher, G. R., and Gutierrez, P. L. (1992) J. Biol. Chem. 267 2474-2479 [PubMed] [Google Scholar]
- 54.Mitra, K., Marquis, J. C., Hillier, S. M., Rye, P. T., Zayas, B., Lee, A. S., Essigmann, J. M., and Croy, R. G. (2002) J. Am. Chem. Soc. 124 1862-1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sharma, U., Marquis, J. C., Nicole Dinaut, A., Hillier, S. M., Fedeles, B., Rye, P. T., Essigmann, J. M., and Croy, R. G. (2004) Bioorg. Med. Chem. Lett. 14 3829-3833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mobley, J. A., and Brueggemeier, R. W. (2004) Carcinogenesis 25 3-9 [DOI] [PubMed] [Google Scholar]
- 57.Stopper, H., Schmitt, E., Gregor, C., Mueller, S. O., and Fischer, W. H. (2003) Mutagenesis 18 243-247 [DOI] [PubMed] [Google Scholar]
- 58.Fischer, W. H., Keiwan, A., Schmitt, E., and Stopper, H. (2001) Mutagenesis 16 209-212 [DOI] [PubMed] [Google Scholar]
- 59.Bocchinfuso, W. P., Hively, W. P., Couse, J. F., Varmus, H. E., and Korach, K. S. (1999) Cancer Res. 59 1869-1876 [PubMed] [Google Scholar]
- 60.Bocchinfuso, W. P., and Korach, K. S. (1997) J. Mammary Gland Biol. Neoplasia 2 323-334 [DOI] [PubMed] [Google Scholar]
- 61.Cobleigh, M. A., Norlock, F. E., Oleske, D. M., and Starr, A. (1999) J. Am. Med. Assoc. 281 1528-1530 [DOI] [PubMed] [Google Scholar]
- 62.Rosenberg, L. U., Einarsdottir, K., Friman, E. I., Wedren, S., Dickman, P. W., Hall, P., and Magnusson, C. (2006) Cancer Epidemiol. Biomark. Prev. 15 2482-2488 [DOI] [PubMed] [Google Scholar]
- 63.Glass, A. G., Lacey, J. V., Jr., Carreon, J. D., and Hoover, R. N. (2007) J. Natl. Cancer Inst. 99 1152-1161 [DOI] [PubMed] [Google Scholar]
- 64.Kerlikowske, K., Miglioretti, D. L., Buist, D. S., Walker, R., and Carney, P. A. (2007) J. Natl. Cancer Inst. 99 1335-1339 [DOI] [PubMed] [Google Scholar]