

Abstract
Lack of functional dystrophin causes severe Duchenne muscular dystrophy. The subsarcolemmal location of dystrophin, as well as its association with both cytoskeleton and membrane, suggests a role in the mechanical regulation of muscular membrane stress. In particular, phenotype rescue in a Duchenne muscular dystrophy mice model has shown that some parts of the central rod domain of dystrophin, constituted by 24 spectrin-like repeats, are essential. In this study, we made use of rare missense pathogenic mutations in the dystrophin gene and analyzed the biochemical properties of the isolated repeat 23 bearing single or double mutations E2910V and N2912D found in muscle dystrophy with severity grading. No dramatic effect on secondary and tertiary structure of the repeat was found in mutants compared with wild type as revealed by circular dichroism and NMR. Thermal and chemical unfolding data from circular dichroism and tryptophan fluorescence show significant decrease of stability for the mutants, and stopped-flow spectroscopy shows decreased refolding rates. The most deleterious single mutation is the N2912D replacement, although we observe additive effects of the two mutations on repeat stability. Based on three-dimensional structures built by homology molecular modeling, we discuss the modifications of the mutation-induced repeat stability. We conclude that the main forces involved in repeat stability are electrostatic inter-helix interactions that are disrupted following mutations. This study represents the first analysis at the protein level of the consequences of missense mutations in the human dystrophin rod domain. Our results suggest that it may participate in mechanical weakening of dystrophin-deficient muscle.
Dystrophin-associated muscular dystrophies range from the severe Duchenne muscular dystrophy (DMD)3 to the milder Becker muscular dystrophy (BMD). Molecular genetic studies indicate that both disorders are the result of mutations in the huge gene that encodes dystrophin (1). Approximately two-thirds of the mutations in DMD and BMD are intragenic partial deletions (2). Indeed, deletion of exons disrupting the reading frame leads to premature translational termination and absence of the protein in DMD cases. On the other hand, mutations maintaining the translational reading frame can lead to semi-functional microdystrophins in BMD cases (3, 4). The remaining one-third of DMD/BMD patients have rare point mutations, such as micro-deletions/insertions or substitutions of one or more nucleotides (5-7). Despite the potential importance of this source of information on protein behavior in cells, the consequences of these mutations on biological properties of dystrophin are so far poorly documented, mainly because of the size and complexity of the cellular partnership of the protein. Dystrophin is a 427-kDa protein included within the dystrophin glycoprotein complex (DGC) (8). The N-terminal part of dystrophin is reported as a globular actin binding domain 1 (9). The C-terminal cysteine-rich domain of dystrophin is the binding region to the DGC by means to link to β-dystroglycan (10). The dystrophin central rod domain is made up of 24 weakly repeating units of 110 amino acids similar to the coiled-coil repeats of spectrin, and it is disrupted by four potential hinges that may ensure flexibility (11). These homologous sequence motifs have been proposed to form domains with a triple-helical bundle-type structure (12, 13).
To understand fully the biological role of the rod domain, further investigations are needed concerning the relationship between the sequences of the repeating units and their biochemical and biophysical properties. The study of cloned wild type or mutated repeating single units from spectrin-like proteins represents a first significant step forward in the investigation of the rod domain (14-20). Knowledge has been improved by work on micro- and minidystrophins lacking one or several repeats. The sub-domains R1-3 and R20-24, located between unstructured hinges 1-2 and 3-4, respectively, are present in the minidystrophin Δ17-48 found in a Becker patient (21). Although the distal R20-24 region of the rod domain lacks any known functional or binding activity (20), this region is thought to act as a passive linker or as a structural stabilizer of its adjacent DGC binding domain. Its proximity with the β-dystroglycan could contribute to proper mechanical behavior of the DGC in muscle.
Most of the rare BMD point mutations correspond to the following: (i) missense mutations in the N-terminal or C-terminal domains or (ii) splice-site mutations that likely act, like BMD deletions, via the production of deleted in-frame transcripts. Only few missense DMD- or BMD-linked point mutations have been reported in the dystrophin gene. 70 of such mutations were reviewed in the dystrophin gene encoding the rod domain (5). We focused our study on the rod domain repeat 23 (R23) encoded by exon 59 (22) and corresponding to residues 2800-2939 of the wild type full-length protein. In R23, a double mutation E2910V/N2912D is mentioned in six entries in the Leiden data base corresponding to seven patients (23). Three are referenced as DMD, one as intermediary muscular dystrophy, and one as BMD (22, 24, 25). Two are not assigned to a pathology but are observed in dilated cardiomyopathy. In this latter case, the authors do not consider that the mutation is the cause of the pathology but suggest that another as yet unobserved mutation may be involved (24). The single residue mutation E2910V is reported as a variant in a DMD case (26). Single mutation N2912D is mentioned in three cases from the Leiden data base with no mention of a pathology DMD, intermediary muscular dystrophy, or BMD pathology, but they are characterized as polymorphic variants (27). Interestingly, the Leiden data base (23) lists one example of a double mutation at the same site E2910G/N2912D, which is associated with a milder Becker muscular dystrophy. This indicates that, in association with the N2912D mutation, the E2910V mutation might have a more severe effect with respect to the disease state.
In this study, we aim to improve our understanding of the role of the rod domain in dystrophin and putative molecular basis of muscular dystrophy. We analyze in vitro the consequences of the natural single or double mutation E2910V/N2912D on isolated repeat 23. The main result is that the double mutant R23 E2910V/N2912D shows a substantially lower thermal and chemical stability as well as slower refolding than the wild type. The variation in the double mutant compared with the wild type is larger that those observed for the single mutants. We construct homology-based molecular model structures of the wild type and mutated repeat 23 of dystrophin. We propose that the changes in stability of a repeat from the rod domain could explain the loss of mechanical properties of dystrophin and thus the defects in muscle cells.
EXPERIMENTAL PROCEDURES
Materials—The pGEX-4T1 plasmid vector, GSTrap™ HP columns were purchased from GE Healthcare. The ER2566 bacteria were supplied by Ozyme (St. Quentin-en-Yvelines, France) and restriction enzymes by Promega (Charbonières, France).
Cloning—The plasmid pTG11025, kindly provided by Dr. S. Braun (Transgene, Strasbourg, France), is an Escherichia coli plasmid that carries the cDNA encoding the full-length Dp427m isoform of human dystrophin (NCBI Nucleotide Data Base code NM_004006). Recombinant dystrophin rod domain wild type and mutated single repeat R23 were cloned downstream of the sequence of glutathione S-transferase (GST) into pGEX-4T1 vector, using the BamHI restriction site of the pre-Scission thrombin recognition sequence and XhoI restriction site, as described previously for wild type single repeat R23 (20). Site-directed mutagenesis was carried out by PCR using complementary oligonucleotide primers containing single and double missense mutations (E2910V/N2912D) and primers previously used to clone single repeat R23. The PCR-amplified fragments encoding mutated single repeat R23 were subcloned into pGEM-T Easy vector (Promega, Charbonnieres, France), sequenced by the dideoxy chain termination method (Genome Express, Meylan, France), and directly cloned in the pGEX-4T1 vector using BamHI and XhoI restriction sites.
Boundaries of Wild Type and Mutated Single Repeat R23—Boundaries of recombinant R23 single repeats were extended by assuming the original alignment of spectrin-type triple-helical repeats in dystrophin as proposed previously (28). The four recombinant proteins were extended with six additional residues at the N-terminal end, including the N-terminal glycine-serine dipeptide resulting from thrombin cleavage of GST fusion proteins and seven additional residues at the C-terminal end according to Gratzer and co-workers (29, 30), who recommended extending the constructs by six to eight residues to improve solubility and stability.
Preparation of Wild Type and Mutated Single Repeat R23—Plasmid constructs were used to transform into the protease-deficient E. coli ER2566 strain, and cultures were then grown to an absorbance of 0.5 at 600 nm at 37 °C in LB broth supplemented with 100 μg/ml ampicillin. Expression of GST fusion proteins was induced by addition of 1 mm isopropyl β-d-thiogalactopyranoside. After 4 h, bacteria were harvested by centrifugation at 4000 × g for 15 min at 4 °C. As described previously (20), pellets from 500 ml of culture were resuspended in 20 ml of ice-cold lysis buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl) and incubated with 0.5 mg·ml-1 lysozyme on ice for 30 min before being broken up by sonication (U200S, UKA Labortechnic). After centrifugation at 10,000 × g for 20 min at 4 °C, filtered extracts were loaded onto a pre-equilibrated 5-ml GSTrap™ HP column at a flow rate of about 0.25 ml·min-1. The column was washed with 10 column volumes of lysis buffer at a flow rate of 1 ml·min-1, and 250 units thrombin were then added. On-column cleavage took place for 48 h at 4 °C, and fractions of 5 ml were collected in the washing buffer. Fractions were concentrated on 3K Centricon, and the final purification steps consisted of ion exchange chromatography on HiTrap Q FF (GE Healthcare) or fast protein liquid chromatography gel filtration on a Sephacryl S-200 column in lysis buffer containing 0.1 mm EDTA (TNE buffer). Protein purity was assessed by SDS-PAGE, and concentrations were determined by bicinchoninic acid (BCA) protein assay with bovine albumin as standard. Fast protein liquid chromatography analytical size exclusion chromatography was performed in TNE buffer on a Sephacryl S-100 column calibrated with alcohol dehydrogenase (RS = 4.6 nm, where RS indicates Stokes radius), bovine serum albumin (RS = 3.5 nm), ovalbumin (RS = 2.7 nm), and cytochrome c (RS = 1.7 nm).
Circular Dichroism Measurements—CD spectra were acquired at 20 °C with a path length of 0.2 cm on a Jasco J-715 or J-815 spectropolarimeter, equipped with Peltier devices for temperature control. From the molar ellipticity at 222 nm [θ222], the mean residue ellipticity [θ]MRW was calculated using the mean residue molar concentration of the proteins, and the percentage of α-helix was obtained using a 100% α-helix value of -36,000 degrees·cm2·dmol-1 at 222 nm according to Ref. 31. The thermal unfolding of each construct was monitored by CD at 222 nm at concentrations of 2.5 μm in TNE buffer with temperature increasing 1 °C/min from 15 to 85 °C. CD signals were fitted either to two-state or three-state transitions. For the two-state transition, the data are fitted to Equation 1,
 |
(Eq.1) |
where y is the CD signal at 222 nm at temperature T; αN and αD are the intercepts of the native and denatured states, respectively; and βN and βD are the slopes of the native and denatured states, respectively; ΔGUN is the free energy of unfolding, and R is the gas constant in cal·mol-1·K-1. As described previously (32), this equation can be rewritten as Equation 2 to facilitate nonlinear regression analysis by SigmaPlot 10.0 in Windows,
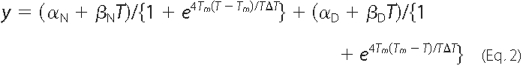
where Tm is the melting temperature, and ΔT is the width of the unfolding transition.
In the case of a three-state unfolding process, Equation 2 can be written as Equation 3,
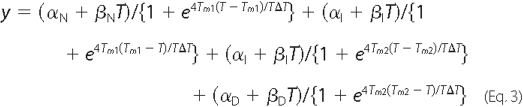
where αI is the intercept of the intermediate state; βI is the slope of the intermediate state; Tm1 is the melting temperature of the first transition between folded and intermediate state; and Tm2 is the melting temperature of the second transition between the intermediate and denatured state.
Steady-state Fluorescence Measurements and Urea Unfolding—Tryptophan fluorescence spectra of native or urea-treated proteins at 1 μm in TNE buffer were recorded at 295 nm excitation wavelength (bandwidth, 1 nm) on a Fluorolog 3 spectrofluorometer (Horiba Jobin-Yvon, France), using 10 × 4-mm quartz cuvettes at 20 °C. After appropriate correction, we obtained the emission spectra of nondenatured proteins. For denatured proteins previously incubated for 2 h in TNE with 0.25-8 m urea, we obtained the maximum wavelength (λmax) and intensity of fluorescence from the emission spectra. The unfolded fraction, y, was then plotted as a function of urea concentration, U, and fitted to a one-transition, two-state reversible process as described by Ref. 33 and as given in Equation 4,

where α is the intercept of the native (N) and denatured (D) states; β is the slope of the native (N) and denatured (D) states; U50% is the urea concentration at the mid-point of denaturation; m is the slope of the transition between the native and denatured states; R is the gas constant, 1.987 cal·mol-1·K-1; and T the temperature in K. The curves were fitted by nonlinear regression with SigmaPlot 10.0 in Windows. The term mU50% in Equation 4 is equal to the free energy of denaturation in the absence of urea, ΔGUN.
In the case of a three-state unfolding process, Equation 5 is as follows,
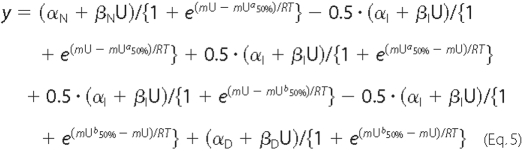
where αI is the intercept of the intermediate state, and βI is the slope of the intermediate state, with Ua50% and Ub50% representing the urea concentrations at mid-denaturation for the first and second transitions, respectively.
NMR Spectroscopy—The NMR samples contained 0.5 mm uniformly 15N-labeled wild type R23 or mutated R23 dissolved in 90% H2O, 10% D2O, 10 mm potassium phosphate, pH 6.0. All the spectra were recorded at 303 K on a Bruker Avance 500 spectrometer equipped with a triple-resonance cryoprobe (1H, 13C, 15N). The heteronuclear experiments were conducted on 15N-labeled protein obtained by expression in minimum media in the presence of 15N-labeled ammonium chloride. Purification was performed in the same way as the unlabeled protein. Using standard Bruker sequence, HSQC spectra were recorded in phase-sensitive mode in both dimensions using echo-anti-echo TPPI gradient selection. 1H-15N HSQC spectra were acquired with 16 transients, with spectral windows of 16/32 ppm in the proton/nitrogen dimensions and the carrier set at the water frequency and 118 ppm, respectively. The relaxation delay used was 0.8 s. Spectra were processed with Topspin or NMRpipe/NMRdraw (34) and visualized with Topspin or NMRview (35) on a Linux station. A matrix of 2K × 256 points was acquired and zero-filled to 2K × 1K points after apodization by shifted sine-square multiplication and linear prediction in the F1 domain.
Refolding Kinetics by Stopped-flow Fluorescence Spectroscopy—Stopped-flow data were recorded on a BioLogic SFM-3, MOS-250 instrument (Grenoble, France). The dead time of the stopped-flow device was 2.2 ms. For refolding measurements, stock solutions of 8 m urea-treated proteins were prepared for 2 h in TNE buffer. The refolding reaction was initiated at 25 °C by 10-fold dilution of the urea-treated protein in TNE buffer. The final protein concentrations were 0.2 μm. The time-dependent fluorescence of tryptophan changes were monitored at excitation and emission wavelengths of 295 and 345 nm, respectively. Curves were derived from the averages of at least 10 individual kinetic data points after subtraction of the background urea buffer signal. By using Biokine software (BioLogic), the experimental results were fitted with exponential functions to obtain the rate constants.
Structure Calculation—Individual residues were assigned to the “a” to “g” positions within the heptads proposed by Winder (28) in predicted hydrophobic repeats. The fold recognition program mGenTHREADER (36, 37) was run to detect the best templates for comparative modeling. The alignment proposed by mGenTHREADER is consistent with the pattern proposed by Winder et al. (28) and Kusunoki et al. (38) with respect to the heptad motifs conserved in the spectrin repeats. Secondary structure predictions were performed by means of PSIPRED (39), incorporating four feed-forward neural networks that analyze the output obtained from PSI-BLAST (www.ncbi.nlm.nih.gov). Homology models of wild type and mutated R23 proteins were built using MODELLER 9.2 software (40). MODELLER is a program based on satisfaction of spatial restraints generated on the target sequence using its alignment with the three-dimensional structure of the templates. Additional restraints were fixed in compliance with the secondary structure prediction of PSIPRED. The loop modeling protocol of MODELLER 9.2 (41) was used to refine all loops of the selected model. Starting with random conformations, 20 structures were built up for each loop. The generated structures were assessed using MODELLER output as well as additional evaluations; the PROCHECK program was used to assess the stereochemical quality of the structures (42), supplemented by the following profile programs: ProSA-WEB (43); the web-based version of ProSA (44); VERIFY3D (45); ANOLEA (46, 47); and by examination on a graphic display. A representative model was selected and its energy minimized with the SYBYL software (TRIPOS Inc., St Louis, MO). The secondary structures of the model were analyzed and represented with the PyMOL program (Delano Scientific). Hydrophobic and electrostatic potentials on the molecular surfaces were calculated using the rTools (Kristian Rother) and Adaptive Poisson-Boltzmann Solver (APBS) (48) software packages, respectively, implemented in PyMOL.
RESULTS
Protein Characterization—Fig. 1 shows the amino acid sequence of the R23 repeat of human dystrophin, along with the predicted secondary structure and alignment with the sequence of the repeat 16 from chicken brain α-spectrin R16-17 (Protein Data Bank code 1cun) (49), which is the template selected for molecular modeling. According to Winder's heptad-based alignment (28), the Glu → Val and Asn → Asp mutations in positions 2910 and 2912 of human dystrophin in R23 are located in helix C of the triple helix coiled-coil, at positions “c” and “e” of the respective heptad. Wild type and mutated R23 were obtained in comparable purity, as verified by Coomassie Blue-stained SDS-polyacrylamide gels (Fig. 2A). Mass spectrometry analysis (INRA, Rennes, France) yields the expected molecular weight for wild type and mutated R23. Analytical size exclusion chromatography reveals that the four proteins are monomeric and monodisperse, with identical Stokes' radii of 2.4 nm (data not shown) (20). f/f0 values of 1.40 are compatible with the shape of coiled-coil single repeats. We conclude that neither the single nor the double mutation E2910V/N2912D induces self-aggregation or measurable variations in the frictional coefficient of the protein. Circular dichroism spectra (Fig. 2B) display the typical features of proteins with a predominant α-helix folding as expected for dystrophin repeats (16, 19, 30) as well as spectrin repeats (14, 50). A circular dichroism [θ222]/[θ208] value higher than 1 was observed for all proteins, indicating that the repeats are structured in coiled coils (51, 52). α-Helical contents derived from the molar ellipticity values at 222 nm are around 80% for the four proteins. The intrinsic fluorescence spectra of the four tryptophan residues do not show any significant difference in shape or maximum emission wavelength between wild type and double mutated R23 (Fig. 2C). Single mutants yield superposable fluorescence spectra (not shown). The one-dimensional 1H NMR spectra (Fig. 3A) of the wild type and the double mutant show differences in the tryptophan indole proton area around 10 ppm, suggesting a slight change in the chemical environment of one or several tryptophan residues. Modifications around the tryptophan residue(s) are also consistent with the changes observed in the strongly upfield-shifted methyl resonances. These shifts are compatible with the close proximity of methyl protons to the aromatic rings of the tryptophan. Overall, the proton spectra for both proteins are characteristic of structured protein species. Amide protons of the protein are spread out over 2 ppm, whereas the several methyl protons are shielded around 0 ppm. The two-dimensional 1H-15N spectra HSQC shown on Fig. 3B display a relatively good dispersion for both proteins. On both maps, many cross-peaks are found at positions compatible with a large structural similarity between the proteins. However, when comparing the two spectra we observe that about 30% of connectivities are strongly modified. HSQC spectra of the double mutated protein at 283 and 313 K do not display major variations that could have been associated with any partial unfolding of the protein.
FIGURE 1.

Sequence alignment of the repeat R16 of chicken brain α-spectrin (CBα16) and the repeat R23 of human dystrophin (R23 WT). Alignment is represented using ESPript. Numbers above sequences correspond to CBα16, and numbers below sequences correspond to full-length dystrophin. The repeat 23 of dystrophin is extended from Leu2800 to Leu2939. Identical residues are indicated with white letters on a red background, whereas similar residues are indicated with red letters. The threeα-helices (HA, HB, and HC) for template CBα16 as well as those predicted by PSIPRED for R23 are represented as squiggles. Assignment of the heptad repeat is from Ref. 28. One heptad is enlarged in the inset, showing the location of the E2910V and N2912D mutations in helix C of R23 and their assignment within the heptad pattern.
FIGURE 2.

Comparison of WT and mutant R23 properties. A, SDS-PAGE analysis of purified WT, E2910V, N2912D, and E2910V/N2912D R23 revealed by Coomassie Blue staining. B, CD spectra of WT (circle) and E2910V/N2912D (triangle) R23 at 2.5 μm. CD spectra were obtained at 20 °C in TNE buffer, pH 7.5. C, tryptophan fluorescence spectra of WT (circle) and E2910V/N2912D (triangle) R23 at 1 μm in TNE buffer at 20 °C. Excitation wavelength is 295 nm with spectra recorded between 310 and 420 nm.
FIGURE 3.

NMR analysis. A, one-dimensional 1H NMR spectra of the WT R23 (bottom) and E2910V/N2912D R23 (top). B, overlay of 1H-15N two-dimensional HSQC spectra of the WT (black) and E2910V-N2912D (red) R23.
Unfolding-Refolding—Thermal transition curves of WT R23, E2910V R23, N2912D R23, and E2910V/N2912D R23 proteins were followed up by observing changes in CD intensity at 222 nm from 15 to 80 °C. The temperature was then set to 15 °C, and a CD spectrum was newly acquired. This shows that the four proteins recover their initial well folded structure after heating. The results show that WT R23 is denatured in a single-step process with a melting temperature of 66.6 °C (Fig. 4A). For the E2910V R23 protein, a single significant step-denaturing process is observed, with a melting temperature of 65.0 °C (Fig. 4B). By contrast, two-step process is clearly observed for thermal unfolding of E2910V/N2912D and N2912D mutants, which exhibit two apparent melting temperatures (Tm1 44.5 °C, Tm2 61.6 °C, and Tm1 41.8 °C and Tm2 62.5 °C, respectively) (Fig. 4, A and C). Nevertheless, the amplitude of the first step is about 40% for the double mutant, and it is only 20% in N2912D R23. At 80 °C, WT and all mutants are identically unfolded and do not exhibit a significant negative peak at 208 and 222 nm indicating that there is no remaining α-helix (data not shown).
FIGURE 4.

Unfolding and refolding of wild type and mutant R23. A-C, thermal unfolding as measured by CD spectroscopy at 222 nm. Unfolded fractions are plotted as described under “Experimental Procedures.” A, WT R23 (circle) follows a single step process (Tm 66.6 °C), and E2910V/N2912D R23 (triangle) follows a two-step process (Tm1 44.5 °C and Tm2 61.6 °C). B, E2910V R23 follows a single step process (Tm 65.0 °C). C, N2912D R23 follows a two-step process (Tm1 41.8 °C and Tm2 62.5 °C). D and E, urea unfolding as measured by variation of maximum emission wavelength in tryptophan fluorescence spectra. Unfolded fractions are plotted as described under “Experimental Procedures.” D, WT R23 (circle) follows a single step process (U50% 4.83 m), and E2910V/N2912D R23 (triangle) follows a two-step process (Ua50% 2.00 m and Ub50% 4.21 m). E, E2910V (triangle) and N2912D (circle) R23 mutants follow a single step process with U50% values of 4.80 and 4.62 m, respectively. F, rate constants of single exponential refolding kinetics observed by stopped-flow spectrofluorescence. WT or mutants R23 unfolded in 8.0 m urea were 10-fold diluted at 25 °C in urea free buffer. Measurements were performed by monitoring fluorescence emission at 345 nm after excitation at 295 nm. Final protein concentrations were 0.2 μm. Stopped-flow data used for rate constants calculation were the averages of at least 10 individual kinetics. Inset, refolding rate constants of WT R23 and E2910V/N2912D R23 at 12, 25, and 37 °C. Values are means ± S.E.
The tryptophan fluorescence data following urea denaturation (Fig. 4, D and E) indicates that wild type protein is unfolded in a single-step process with a urea concentration of 4.83 m at mid-denaturation. We observe a single step with mid-denaturation urea concentrations of 4.80 and 4.62 m for single mutants E2910V R23 and N2912D R23, respectively. Interestingly, the double mutant exhibits a weak first unfolding phase with a urea mid-denaturation value of 2 m, and then a second unfolding transition with a mid-denaturation urea concentration of 4.21 m, which is significantly lower that the mid-denaturation values determined for the three other proteins.
Refolding kinetics of the four proteins were then monitored by stopped-flow tryptophan fluorescence, with these proteins being first unfolded in 8.0 m urea and then rapidly diluted to a final urea concentration of 0.8 m. For all refolding experiments, the best fit for the curves is obtained with single exponentials. The calculated refolding rate constants are 53.4 ± 11.2 s-1 for WT R23, 41.7 ± 14.3 s-1 for E2910V R23, 20.0 ± 5.5 s-1 for N2912D R23, and 20.2 ± 5.2 s-1 for E2910V-N2912D R23 (Fig. 4F) (see supplemental material). Refolding analyses repeated at 12 and 37 °C for WT and E2910V-N2912D R23 also lead to single exponentials. Rate constants are, respectively, 13.9 ± 0.7 and 5.2 ± 0.1 s-1 at 12 °C and 70.1 ± 21.9 and 37.7 ± 5.8 s-1 at 37 °C (Fig. 4F, inset).
Molecular Modeling—Structural features of the repeat 23 of human dystrophin. As expected, mGenTHREADER reveals that the best templates are spectrin-repeat proteins, particularly repeats 16-17 of the chicken brain α-spectrin (CBα16-17, Protein Data Bank code 1cun) (49) and repeats 8-9 of the human erythroid β-spectrin (HEβ8-9, Protein Data Bank code 1s35) (38). R23 shares 16.2 and 12.7% of sequence identity with CBα16 and HEβ8, respectively. As described previously (38), despite a relatively low percentage of sequence identity, structures of individual spectrin-like repeats are nearly identical and provide good models for comparative modeling. The crystal structure of CBα16-17 provides the best three-dimensional structure template model for R23. Fig. 1 presents the sequence alignments of R23 with CBα16. Consistent with PSIPRED predictions and considering the helical continuity in the linker regions between the end of one repeat and the beginning of the adjacent one, we assumed for R23 that the extremities of the helices A and C are fully helical. As a consequence, we applied α-helix restraints on two segments of R23 that are not aligned with the CBα16 sequence, namely the N-terminal region (residues 2800-2806) and the N-terminal extremity of the helix C (residues 2893-2900), which is predicted as helical by PSIPRED. Forty homology models were built up in this way. The models with GA341 scores higher than 0.6 with the lowest target function and DOPE score were evaluated with ProSA-web and VERIFY3D. In the selected model, the loops are then refined, and all final structures were assessed using the strategy described under “Experimental Procedures.” For the most representative result shown in Fig. 5, VERIFY3D, ProSA, and ANOLEA profiles are indicative of a high quality model with a mean force potential that is positive for VERIFY3D, a negative one for ANOLEA, and ProSA with a ProSA global score of -6.32. After minimization, we obtain an excellent quality Ramachandran map, with no residue in the forbidden zones, 92.3% of residues in the most favored regions, and 6.9% in the allowed zone (see supplemental material). The triple helical coiled-coil fold of R23 is closely similar to those reported for spectrin repeats. Three helices, A (2800-28329), B (2838-2878), and C (2892-2935), are linked by two loops. We note that 82% of the residues are located in the helices, which is similar to the helical content calculated from the circular dichroism spectra. Helices A and C are slightly bent, whereas the longest helix B is kinked on the semi-conserved proline residue in position 2860. In addition to the canonical hydrogen bonds in the helices, side chains intra- and inter-helical hydrogen bonds stabilize the three helices bundle. Conserved hydrophobic residues on position a and d of the heptad pattern, lying at the interface of the three helices, have relatively little accessibility to the solvent and create hydrophobic clusters. The main differences with the previously described spectrin repeats arise from the 20-residue insertion specific to R23 (Leu2878 to Ala2897), between helices B and C. This BC loop is longer in R23 than in CBα16, HEβ8, and the other dystrophin repeats, so it does not align with the template. By performing extensive refinements using the loop module of MODELLER, we obtain optimal BC loop structures with an α-helix turn from residue Gly2880-Gln2886, which is not predicted by PSIPRED.
FIGURE 5.

Molecular modeling of WT R23. PyMOL representation of the WT R23 global fold. N and C termini are indicated as well as helices numbering (HA, HB, and HC). Main amino acids are colored and represented by stick representation as follows: Trp2807, Trp2820, Trp2915, and Trp2925 in yellow; Glu2910 and Asn2912 in green; and Pro2860 in red. The inset shows the enlarged and rotated region of the mutation with charged residues also in color (blue and red for positive and negative residues, respectively).
The inset in Fig. 5 shows an enlargement of the area containing the mutations. We used MODELLER to build the model of the R23 mutants from the WT R23 model, the conformation of the mutant side chain being optimized by a conjugate gradient and refined using molecular dynamics. The mutated residues are located in positions c and e in the third heptad hydrophobic repeat pattern of helix C (see Fig. 1). According to the selected model, a number of hydrophobic residues lie on the inward-facing surface of the helices and likely make up a stabilizing hydrophobic cluster. In the mutated area, these notably include Trp2820, a highly conserved tryptophan in a g position in helix A (28), and Trp2915 in an a position in helix C. The a and d residues in helix C, Ala2908 and Val2911, interact with the corresponding a and d residues in helices A, Leu2814 and Leu2817, respectively, and the a residue, Ile2862, in helix B.
The residues mutated in the dystrophic pattern are in heptad positions c (Glu2910) and e (Asn2912), and surround the stabilizing residue d (Val2911) in the heptad. As these two residues are in contact with the aqueous phase, we then used the rTools and the APBS software package to calculate hydrophobic and electrostatic surface scores for the WT and mutated R23 repeats.
Fig. 6A presents a ribbon plot of the chain fold. By examining the hydrophobic surface properties (Fig. 6B and supplemental material), we can see only a slight change between the WT and mutated repeat in the E2910V substitution area.
FIGURE 6.

Surface properties of the WT R23 and E2910V/N2912D R23. A, orientation of the molecule. The part of helix C containing the double mutation is in yellow. B, hydrophobic potentials at the molecular surface of WT R23 (left) and E2910V/N2912D R23 (right). Surface is colored with increasing hydrophobicity from green to red. C, electrostatic potentials at the molecular surface of WT R23 (left) and E2910V/N2912D R23 (right). Surface is colored with increasing electrostatic potentials from red to blue. The areas corresponding to the main differences between WT and E2910V/N2912D R23 proteins are delimited with dotted lines.
Fig. 6C shows the results of electrostatic surface analysis of WT and double mutant proteins. The blue and red coloring represents the positive and negative mean charge surfaces, respectively. As expected from primary structure analysis, the wild type repeat exhibits an overall negative charge. Following the Glu → Val substitution, only a slight change in the overall negative charge is observed in the electrostatic surface in the Glu2910 area. By contrast, a highly significant increase of the overall negative charge can be observed in the area corresponding to the location of the N2912D substitution (Fig. 6C, circled area).
To assess the features that might induce such variations, we then analyzed the surface properties of individual helices. Fig. 7 shows single side views of electrostatic surfaces of separated helices of R23. Helices are represented in their twisted structure from the model. Uncharged residues, indicated in Fig. 7 in white, are located on a line along each individual helix (dashed lines). They largely correspond to the a and d residues in heptads forming the hydrophobic interaction core between the three helices. Additionally, these models clearly show that the hydrophobic interaction core is surrounded by opposite electrostatic potentials from facing helices that contribute to the stability of the coiled-coil structure. Indeed, when examining each helix, dipolar electrostatic potentials appear. The N-terminal part of each helix is positively charged, and the C-terminal part is negatively charged, whereas the central part is more or less uncharged (Fig. 7). The mutated area of helix C corresponds to this central uncharged part (Fig. 7, circled area). Interestingly, in the N2912D R23, as well as in the double mutant, the dipolar electrostatic potential of helix C exhibits a significantly increased negative charge in the mutated area compared with the wild type.
FIGURE 7.

Electrostatic potentials of each individual helix HA, HB, and HC for WT and R23 mutants. Surface is colored with increasing electrostatic potentials from red to blue. Numbering corresponds to first and last amino acids of each helix. Dashed lines indicate the location of the hydrophobic residues a and d by which the helices interact. The areas corresponding to the main differences between WT R23 and single or double R23 mutants (E2910V/N2912D) are delimited with dotted lines. For mutants, location of residues 2910, 2911, and 2912 is indicated by solid line and arrows. Residue 2910 is on the opposite face and indicated by a dotted line.
DISCUSSION
The aim of our work was to understand how point mutations might affect the properties of one repeat of the dystrophin rod domain and lead to a severe Duchenne muscular dystrophy. We focus on the R23 repeat of the rod domain, which contained a double mutation encountered in DMD (22, 24, 25). We find evidence for a decreased stability of the R23 dystrophin repeat containing the double mutation. Although the effects of single mutations indicate that both E2910 and N2912 contribute to protein stability, the N2912D mutation induces a more dramatic destabilization than the E2910V mutation.
All wild type and mutant R23 were expressed as fusion proteins in E. coli and further purified with flanking peptide extensions at the end of the canonical motifs. Following removal of the N-terminal GST tag during the purification step, the four proteins were obtained as soluble monomers. The amount of α-helix as measured by circular dichroism is in good agreement with a proper spectrin-like repeat folding. As reported by other authors (29, 30), the small sequence extensions likely prevent fraying of the helices at the termini of the isolated repeats, but they are unlikely to interact with other residues. At room temperature, we find nearly identical α-helicities for the wild type and mutant proteins. This indicates that the valine and aspartic acid side chains present in the mutants (V2910 and D2912) are compatible with proper folding of the triple α-helix coiled-coil repeat. From hydrodynamic behavior in size exclusion chromatography, we also conclude that the slight changes observed in hydrophobic and electrostatic surface properties between wild type and mutated whole repeat R23 do not induce in vitro self-aggregation or measurable changes in shape. We clearly demonstrate that the single and double mutations induce a reduction of thermal and chemical stability. Nevertheless, according to comparative refolding velocity analysis in vitro, we can infer that the R23 repeat containing either the single or the double mutations might follow a folding process that leads to a canonical coiled-coil structure but with a reduced stability and the appearance of a stable intermediary state in the unfolding process of the N2912D R23 and the double mutated repeat.
The amino acid substitutions analyzed in this study do not directly involve the hydrophobic stabilizing residues of coiled coils in positions a and d of the characteristic heptads spectrin-like repeats. If mutated, these residues are known to induce a dramatic loss of stability of a single spectrin repeat. However, our study is concerned with residues located at the c (Glu2910) and e (Asn2912) positions flanking the stabilizing valine residue in a d heptad position. According to our data, these mutations have a limited effect on the global fold of R23.
To investigate the structural features of the R23 repeat and the determinants involved in the loss of stability of mutated R23, we performed NMR experiments and built homology models. Sufficient yields were obtained for the production of highly stable 15N-labeled R23, which enabled us to carry out NMR experiments to gather information on the wild type and double-mutated R23. At this stage, we observed that about 30% of the signals show significant chemical shift variations on the 15N HSQC spectrum, which means that about a third of the double mutant R23 residues exhibit modification to their local environment compared with the WT R23. It is interesting to note the impact of mutations on the NMR spectra while the secondary structures are conserved. To gain a deeper insight into the mutation effect, we hoped to characterize the assignment of the R23 repeat to identify the shifted residues in double mutant R23 and if possible to determine the structures of both proteins. However, the assignment of the coiled-coil proteins is more difficult than in the case of globular proteins, because of a poorer dispersion of the resonances and the small number of long range constraints (53). Indeed, preliminary studies (data not shown) on a double-labeled 15N-13C WT R23 show that the Cα and Cβ signals are particularly degenerated, making it difficult to assign the resonance of the R23 backbone.
Molecular modeling of the R23 repeat is based on the homology with the chicken brain α-spectrin repeat 16 with respect to the “nested model” (29). As described under “Results,” we obtained satisfactory scores and profiles from the validation software. By examination of the model with the PyMOL software, we can verify that the global features of a spectrin single repeat are satisfied. The residues a and d undergo hydrophobic interactions lying on the inward facing surface of the helices. Two buried tryptophans, Trp2820 in helix A and Trp2915 in helix C, make up a notable hydrophobic cluster of the R23 coiled-coil structure. These observations fit well with the previously reported interactions between two conserved tryptophan residues, which represent a stabilizing feature through most of the spectrin-like repeats (38, 50). Among these canonical conserved tryptophans, the Trp2915 under a heptad position appears in the model as the residue closest to the mutated area. More precisely, it lies close to the residue Asn2912 in the e heptad position, whereas the residue Glu2910 in the c position is located on the opposite face of the coiled-coil R23 repeat structure. Hence, the N2912D mutation is likely to have a predominant influence on the behavior of Trp2915. Indeed, the NMR data showed that the chemical environment of one of the four tryptophans is modified in the double mutant compared with WT. The two other tryptophan residues of R23 (Trp2807 in helix A and Trp2925 in helix C) are each located at the extremities of the triple helical coiled-coil structure and thus are unlikely to have an environment that could be significantly affected by the mutations. Consequently, we propose first that the double mutation could indirectly destabilize the hydrophobic cluster comprising the two conserved tryptophans facing each other.
Several studies have shown that intra- or inter-helix electrostatic interactions contribute to the structural stability of coiled-coil spectrin single repeats (49, 54). It is noteworthy that, when analyzed individually, the anti-parallel helices A, B, and C from the R23 repeat exhibit opposite electrostatic potentials. This highlights the existence of interhelical stabilizing electrostatic forces between these charged helices. We observe that electrostatic potentials at the molecular surfaces of WT and mutant R23 are markedly different at the mutation site of helix C, thus leading to differences in the whole triple helical repeat. The electrostatic potentials are slightly modified by the E2910V mutation, which changes a charged residue to an uncharged one. In contrast, the electrostatic potential modification appears to be more prominent with the N2912D mutation, which changes a neutral residue to a negatively charged one. Indeed, the Asn2912 residue in the WT R23 helix C is surrounded by two negatively charged residues, Glu2909 and Glu2916. The occurrence of an aspartic acid following mutation in this area induces anomalous intra-helical repulsive forces. In addition, according to the electrostatic potential surfaces, the charge complementarities between the three helices appear to be greatly affected around the mutation site.
The two mutants bearing the N2912D mutation, namely N2912D and the double mutant, show a two-step unfolding process. However, the extent of the remaining α-helix content at 50 °C is higher in the single mutant than in the double mutant. These results point out the additive destabilizing effect of the E2910V mutation on the N2912D mutation. Therefore, the origin of the unfolding process could arise from a loss of structure of helix C because of the local unfavorable charges or from a more complex process involving part of the three interacting helices with the disruption of the hydrophobic cluster inducing the complete unfolding of the R23 mutants.
In summary, even if each mutation does not modify the folding of the repeat, they induce gradual faster thermal or chemical unfolding. If the N2912D is much more deleterious than the E2910V mutation, the synergy of the two effects of the E2910V/N2912D mutation is necessary to induce a major thermal and chemical instability. According to our data, we assume that the hydrophobic cluster near the mutation area is only indirectly affected by the mutations and that the more striking effect of the mutations is the impact on the electrostatic potentials stabilizing the coiled coil.
To conclude, this study for the first time describes at the biochemical level the analysis of some of the rare pathogenic missense mutations in the dystrophin gene (4, 5, 7, 23). Our data support a critical role for the rod domain in different aspects of dystrophin function, in particular a reduced stability of the R23 repeat. If these modifications were transposed to the whole R20-24 sub-domain or to the whole rod domain, this might contribute to the mechanical weakening of dystrophin-deficient muscle, leading to dramatic differences in muscle cell life. The interactions between the C-terminal part of dystrophin and the DGC have been characterized at the residue level (55). These interactions could also be regulated by the other parts of dystrophin close to the C terminus, namely the R20-24 sub-domain. This latter includes the R23 repeat studied here, which has been shown to natively exhibit a remarkably high stability and no lipid binding properties (20). We can hypothesize that β-dystroglycan binding of dystrophin through its C terminus requires an adjacent domain highly stable by itself or stabilized through interaction with a yet unknown partner.
Supplementary Material
Acknowledgments
We thank C. Rocher (UMR CNRS 6026), M. R. Allo, and S. Pastezeur (UMR CNRS 6061) for technical assistance, D. Mollé (INRA STLO) for providing mass spectrometry facilities, and PRISM-IFR140 Rennes for NMR and spectroscopy facilities. M. S. N. Carpenter post-edited the English style.
This work was supported in part by the Association Française Contre les Myopathies. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1-S6.
Footnotes
The abbreviations used are: DMD, Duchenne muscular dystrophy; R23, rod domain 23; WT, wild type; BMD, Becker muscular dystrophy; DGC, dystrophin glycoprotein complex; GST, glutathione S-transferase.
References
- 1.Hoffman, E. P., Brown, R. H., and Kunkel, L. M. (1987) Cell 51 919-928 [DOI] [PubMed] [Google Scholar]
- 2.Darras, B. T., Blattner, P., Harper, J. F., Spiro, A. J., Alter, S., and Francke, U. (1988) Am. J. Hum. Genet. 43 620-629 [PMC free article] [PubMed] [Google Scholar]
- 3.Koenig, M., Beggs, A. H., Moyer, M., Scherpf, S., Heindrich, K., Bettecken, T., Meng, G., Muller, C. R., Lindlof, M., Kaariainen, H., de la Chapelle, A., Kiuru, A., Savontaus, M.-L., Gilgenkrantz, H., Recan, D., et al. (1989) Am. J. Hum. Genet. 45 498-506 [PMC free article] [PubMed] [Google Scholar]
- 4.White, S., Kalf, M., Liu, Q., Villerius, M., Engelsma, D., Kriek, M., Vollebregt, E., Bakker, B., van Ommen, G. J., Breuning, M. H., and den Dunnen, J. T. (2002) Am. J. Hum. Genet. 71 365-374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roberts, R. G., Gardner, R. J., and Bobrow, M. (1994) Hum. Mutat. 4 1-11 [DOI] [PubMed] [Google Scholar]
- 6.Chaturvedi, L. S., Mukherjee, M., Srivastava, S., Mittal, R. D., and Mittal, B. (2001) Exp. Mol. Med. 33 251-256 [DOI] [PubMed] [Google Scholar]
- 7.Beroud, C., Tuffery-Giraud, S., Matsuo, M., Hamroun, D., Humbertclaude, V., Monnier, N., Moizard, M. P., Voelckel, M. A., Calemard, L. M., Boisseau, P., Blayau, M., Philippe, C., Cossee, M., Pages, M., Rivier, F., Danos, O., Garcia, L., and Claustres, M. (2007) Hum. Mutat. 28 196-202 [DOI] [PubMed] [Google Scholar]
- 8.Ervasti, J. M., and Campbell, K. P. (1993) Curr. Opin. Cell Biol. 5 82-87 [DOI] [PubMed] [Google Scholar]
- 9.Norwood, F. L., Sutherland-Smith, A. J., Keep, N. H., and Kendrick-Jones, J. (2000) Structure (Lond.) 8 481-491 [DOI] [PubMed] [Google Scholar]
- 10.Huang, X., Poy, F., Zhang, R., Joachimiak, A., Sudol, M., and Eck, M. J. (2000) Nat. Struct. Biol. 7 634-638 [DOI] [PubMed] [Google Scholar]
- 11.Koenig, M., and Kunkel, L. M. (1990) J. Biol. Chem. 265 4560-4566 [PubMed] [Google Scholar]
- 12.Parry, D. A., Dixon, T. W., and Cohen, C. (1992) Biophys. J. 61 858-867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Speicher, D. W., and Marchesi, V. T. (1984) Nature 311 177-180 [DOI] [PubMed] [Google Scholar]
- 14.Winograd, E., Hume, D., and Branton, D. (1991) Proc. Natl. Acad. Sci. U. S. A. 88 10788-10791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kahana, E., Marsh, P. J., Henry, A. J., Way, M., and Gratzer, W. B. (1994) J. Mol. Biol. 235 1271-1277 [DOI] [PubMed] [Google Scholar]
- 16.Le Rumeur, E., Fichou, Y., Pottier, S., Gaboriau, F., Rondeau-Mouro, C., Vincent, M., Gallay, J., and Bondon, A. (2003) J. Biol. Chem. 278 5993-6001 [DOI] [PubMed] [Google Scholar]
- 17.Batey, S., Randles, L. G., Steward, A., and Clarke, J. (2005) J. Mol. Biol. 349 1045-1059 [DOI] [PubMed] [Google Scholar]
- 18.An, X., Guo, X., Zhang, X., Baines, A. J., Debnath, G., Moyo, D., Salomao, M., Bhasin, N., Johnson, C., Discher, D., Gratzer, W. B., and Mohandas, N. (2006) J. Biol. Chem. 281 10527-10532 [DOI] [PubMed] [Google Scholar]
- 19.Saadat, L., Pittman, L., and Menhart, N. (2006) Biochim. Biophys. Acta 1764 943-954 [DOI] [PubMed] [Google Scholar]
- 20.Legardinier, S., Hubert, J. F., Le Bihan, O., Tascon, C., Rocher, C., Raguenes-Nicol, C., Bondon, A., Hardy, S., and Le Rumeur, E. (2008) Biochim. Biophys. Acta 1784 672-682 [DOI] [PubMed] [Google Scholar]
- 21.England, S. B., Nicholson, L. V., Johnson, M. A., Forrest, S. M., Love, D. R., Zubrzycka-Gaarn, E. E., Bulman, D. E., Harris, J. B., and Davies, K. E. (1990) Nature 343 180-182 [DOI] [PubMed] [Google Scholar]
- 22.Lenk, U., Hanke, R., and Speer, A. (1994) Neuromuscul. Disord. 4 411-418 [DOI] [PubMed] [Google Scholar]
- 23.Aartsma-Rus, A., Van Deutekom, J. C., Fokkema, I. F., Van Ommen, G. J., and Den Dunnen, J. T. (2006) Muscle Nerve 34 135-144 [DOI] [PubMed] [Google Scholar]
- 24.Feng, J., Yan, J., Buzin, C. H., Towbin, J. A., and Sommer, S. S. (2002) Mol. Genet. Metab. 77 119-126 [DOI] [PubMed] [Google Scholar]
- 25.Hofstra, R. M., Mulder, I. M., Vossen, R., de Koning-Gans, P. A., Kraak, M., Ginjaar, I. B., van der Hout, A. H., Bakker, E., Buys, C. H., van Ommen, G. J., van Essen, A. J., and den Dunnen, J. T. (2004) Hum. Mutat. 23 57-66 [DOI] [PubMed] [Google Scholar]
- 26.Mendell, J. R., Buzin, C. H., Feng, J., Yan, J., Serrano, C., Sangani, D. S., Wall, C., Prior, T. W., and Sommer, S. S. (2001) Neurology 57 645-650 [DOI] [PubMed] [Google Scholar]
- 27.Prior, T. W., Wenger, G. D., Papp, A. C., Snyder, P. J., Sedra, M. S., Bartolo, C., Moore, J. W., and Highsmith, W. E. (1995) Hum. Mutat. 5 263-268 [DOI] [PubMed] [Google Scholar]
- 28.Winder, S. J., Gibson, T. J., and Kendrick-Jones, J. (1995) FEBS Lett. 369 27-33 [DOI] [PubMed] [Google Scholar]
- 29.Calvert, R., Kahana, E., and Gratzer, W. B. (1996) Biophys. J. 71 1605-1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kahana, E., and Gratzer, W. B. (1995) Biochemistry 34 8110-8114 [DOI] [PubMed] [Google Scholar]
- 31.Greenfield, N., and Fasman, G. D. (1969) Biochemistry 8 4108-4116 [DOI] [PubMed] [Google Scholar]
- 32.Kusunoki, H., Minasov, G., MacDonald, R., and Mondragon, A. (2004) J. Mol. Biol. 344 495-511 [DOI] [PubMed] [Google Scholar]
- 33.MacDonald, R., and Pozharski, E. (2001) Biochemistry 40 3974-3984 [DOI] [PubMed] [Google Scholar]
- 34.Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J., and Bax, A. (1995) J. Biomol. NMR 6 277-293 [DOI] [PubMed] [Google Scholar]
- 35.Johnson, B. A., and Blevins, R. A. (1994) J. Biomol. NMR 4 603-614 [DOI] [PubMed] [Google Scholar]
- 36.Jones, D. T. (1999) J. Mol. Biol. 287 797-815 [DOI] [PubMed] [Google Scholar]
- 37.McGuffin, L. J., Bryson, K., and Jones, D. T. (2000) Bioinformatics (Oxf.) 16 404-405 [DOI] [PubMed] [Google Scholar]
- 38.Kusunoki, H., MacDonald, R., and Mondragon, A. (2004) Structure (Lond.) 12 645-656 [DOI] [PubMed] [Google Scholar]
- 39.Jones, D. T. (1999) J. Mol. Biol. 292 195-202 [DOI] [PubMed] [Google Scholar]
- 40.Sali, A., and Blundell, T. L. (1993) J. Mol. Biol. 234 779-815 [DOI] [PubMed] [Google Scholar]
- 41.Fiser, A., Do, R. K., and Sali, A. (2000) Protein Sci. 9 1753-1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laskowski, R. A., Rullmannn, J. A., MacArthur, M. W., Kaptein, R., and Thornton, J. M. (1996) J. Biomol. NMR 8 477-486 [DOI] [PubMed] [Google Scholar]
- 43.Wiederstein, M., and Sippl, M. J. (2007) Nucleic Acids Res. 35 W407-W410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sippl, M. J. (1993) J. Comput. Aided Mol. Des. 7 473-501 [DOI] [PubMed] [Google Scholar]
- 45.Luthy, R., Bowie, J. U., and Eisenberg, D. (1992) Nature 356 83-85 [DOI] [PubMed] [Google Scholar]
- 46.Sippl, M. J. (1990) J. Mol. Biol. 213 859-883 [DOI] [PubMed] [Google Scholar]
- 47.Melo, F., and Feytmans, E. (1998) J. Mol. Biol. 277 1141-1152 [DOI] [PubMed] [Google Scholar]
- 48.Baker, R. D. (2001) Lifetime Data Anal. 7 65-83 [DOI] [PubMed] [Google Scholar]
- 49.Grum, V. L., Li, D., MacDonald, R. I., and Mondragon, A. (1999) Cell 98 523-535 [DOI] [PubMed] [Google Scholar]
- 50.MacDonald, R. I., Musacchio, A., Holmgren, R. A., and Saraste, M. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 1299-1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou, N. E., Kay, C. M., and Hodges, R. S. (1992) J. Biol. Chem. 267 2664-2670 [PubMed] [Google Scholar]
- 52.Long, F., McElheny, D., Jiang, S., Park, S., Caffrey, M. S., and Fung, L. W. (2007) Protein Sci. 16 2519-2530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schnell, J. R., Zhou, G. P., Zweckstetter, M., Rigby, A. C., and Chou, J. J. (2005) Protein Sci. 14 2421-2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Djinovic-Carugo, K., Gautel, M., Ylanne, J., and Young, P. (2002) FEBS Lett. 513 119-123 [DOI] [PubMed] [Google Scholar]
- 55.Hnia, K., Zouiten, D., Cantel, S., Chazalette, D., Hugon, G., Fehrentz, J. A., Masmoudi, A., Diment, A., Bramham, J., Mornet, D., and Winder, S. J. (2007) Biochem. J. 401 667-677 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.