

Abstract
L-selectin is a cell adhesion molecule that tethers leukocytes to the luminal walls of venules during inflammation and enables them to roll under the force of blood flow. Clustering of L-selectin during rolling is thought to promote outside-in signals that lead to integrin activation and chemokine receptor expression, ultimately contributing to leukocyte arrest. Several studies have underscored the importance of the L-selectin cytoplasmic tail in functionally regulating adhesion and signaling. Interestingly, the L-selectin tail comprises only 17 amino acids, and yet it is thought to bind simultaneously to several proteins. For example, constitutive association of calmodulin (CaM) and ezrin/radixin/moesin (ERM) to L-selectin confers resistance to proteolysis and microvillar positioning, respectively. In this report we found that recombinant purified CaM and ERM bound non-competitively to the same tail of L-selectin. Furthermore, molecular modeling supported the possibility that CaM, L-selectin, and moesin could form a heterotrimeric complex. Finally, using fluorescence lifetime imaging microscopy to measure fluorescence resonance energy transfer, it was shown that CaM, L-selectin, and ERM could interact simultaneously in vivo. Moreover, L-selectin clustering promoted CaM/ERM interaction in cis (i.e. derived from neighboring L-selectin tails). These results highlight a novel intracellular event that occurs as a consequence of L-selectin clustering, which could participate in transducing signals that promote the transition from rolling to arrest.
Transit of leukocytes from the bloodstream to the surrounding tissue is essential for inflammatory responses and is intricately coordinated by cell adhesion molecules (CAMs)7 on both leukocytes and endothelial cells. The selectins are a three-member family of CAMs originally identified in endothelial cells (E-selectin), platelets (P-selectin), and leukocytes (L-selectin) (1), which jointly execute leukocyte tethering and rolling along the luminal surface of venules (2, 3). The extracellular domain of the selectins harbor similar structural features, whereas the cytoplasmic tails of all three selectins are non-conserved, suggesting that the tails may be involved in regulating the function of each selectin uniquely. The cytoplasmic tail of L-selectin comprises only 17 amino acids, and yet a growing number of binding partners have been identified (4), including calmodulin (CaM) (5), the ezrin/radixin/moesin (ERM) family of membrane-cytoskeleton cross-linkers (6), α-actinin (7), and protein kinase C isoenzymes (8). Spatiotemporal regulation between L-selectin and its binding partners could justify how each protein may associate separately with the L-selectin tail. However, a number of these proteins are considered to interact constitutively, suggesting that the tail of L-selectin can accommodate multiple binding partners. For example, CaM associates constitutively with L-selectin in resting leukocytes and thereby protects the extracellular domain of L-selectin from proteolysis (5). Artificial activation of leukocytes with phorbol myristate acetate induces the release of CaM from L-selectin and shedding of the extracellular domain. ERMs are classically defined as membrane/cytoskeleton cross-linkers, because their N termini can bind to the tails of cell adhesion molecules and their C termini can bind to filamentous actin. The ERMs are also thought to be constitutively associated with L-selectin, because abrogating this interaction diminishes microvillar positioning, which in turn reduces tethering efficiency under flow (9). Additionally, phorbol myristate acetate-induced shedding of L-selectin is significantly decreased when ERM binding is abrogated (9). These observations suggest that ERM and CaM may have distinct and overlapping roles. The amino acid residues in the L-selectin tail that contribute to CaM and ERM binding are juxtaposed to one another (see Fig. 1A), which suggests that these proteins may either compete for the same binding site, or bind non-competitively to the same L-selectin tail. The fact that resting leukocytes express L-selectin, which is anchored to microvilli (10), suggests that both CaM and ERM are collectively involved in binding to the same L-selectin tail.
FIGURE 1.

Moesin FERM and CaM bind non-competitively with the cytoplasmic tail of l-selectin. A, single letter amino acid sequence of the cytoplasmic tail of L-selectin. Black letters define the polybasic, membrane proximal domain. The box and ovals depict amino acid residues that have been previously shown to contribute to binding CaM and ERM, respectively (see Refs. 5, 6, 9 for more detail). Underlined region of the L-selectin tail marks the epitope recognized by the CA21 monoclonal antibody. B, Coomassie-stained polyacrylamide gel showing the relative binding of moesin FERM or CaM to the L-selectin beads, which were pre-saturated with (lanes 1-5) or without (lanes 6-9) CaM. 5 μg/ml of CaM was used to preload the beads prior to incubation with increasing amounts of moesin FERM domain. Coomassie-stained gels are representative of three independent experiments. C, binding of moesin FERM domain and CaM to the cytoplasmic tail of L-selectin is calcium-independent. The L-selectin beads were preloaded with either moesin FERM (lanes 1 and 2) or CaM (lanes 3 and 4). Preloaded beads were then incubated with CaM and moesin FERM, respectively. Binding reaction was supplemented either with (lanes 2 and 4) or without (lanes 1 and 3) 5 mm EGTA. Bound proteins were resolved on polyacrylamide gels and subsequently stained with Coomassie Blue. Gel is representative of three independent experiments. D, biotin transfer of SBED from the L-selectin tail to either moesin FERM domain or CaM is equal and independent of pre-mixing (see supplemental Fig. S1 and “Materials and Methods” for more information of SBED biotin transfer procedure). In brief, 3.6 μm SBED-conjugated L-selectin tail was mixed with 4.6 μm CaM and/or moesin FERM at room temperature for 30 min. In mixing experiments, a 30-min gap was left between adding proteins, which was deemed ample time for the first protein to bind to the L-selectin tail. The left-hand top and bottom panels represent PVDF transfer membranes developed with 1 μg/ml streptavidin-horseradish peroxidase. The right-hand top and bottom panels represent the same PVDF membranes from the left-hand panels, which were subsequently stained with Coomassie Blue to show relative abundance of CaM and moesin FERM used in the experiment (loading control), and is representative of three independent experiments. E, equal concentrations of CaM (4.6 μm) or moesin FERM (4.6 μm) were mixed individually or together with increasing amounts of soluble L-selectin tail peptide (i.e. 0, 1.72, 3.44, 6.88, 13.75, 27.50, 55, 110, and 220 μm). Protein products were cross-linked with DSS, resolved on polyacrylamide gels, and transferred to a PVDF membrane for Western blotting with CA21 monoclonal anti-L-selectin tail antibody. Shifts in molecular masses of the L-selectin tail corresponded to the molecular mass of CaM (18 kDa), moesin FERM (30 kDa), or a mixture of the two (50 kDa). The arrow to the right of the molecular weight markers denotes the higher molecular weight complexes that likely correspond to a 1:1:1 stoichiometry between the tail of L-selectin, CaM, and moesin FERM. The Western blot is representative of three independent experiments.
L-selectin has also been described as a signaling receptor (11). For example, clustering L-selectin with either monoclonal antibody or multivalent physiological ligand has been shown to activate β1 (12) and β2 (13) integrins. Mobilization of the chemokine receptor, CXCR4, to the cell surface has also been shown to occur in response to L-selectin clustering (14). Collectively, these responses imply that L-selectin-dependent adhesion could be involved in facilitating the transition from rolling to arrest independently of, or in concert with, chemokines (15). Moreover, clustering of L-selectin is governed by the membrane-proximal polybasic region of the L-selectin tail (16), which includes the CaM and ERM binding sites (see Fig. 1A). This suggests that both CaM and ERM could potentially participate in mediating signals downstream of L-selectin engagement or clustering. A recent elegant study showed that clustering of P-selectin glycoprotein ligand-1 with monoclonal antibody elicits the binding of spleen tyrosine kinase to a cryptic immunoreceptor tyrosine-based activation motif resident within the N-terminal domain of moesin (17). This implies that ERMs act as signaling adaptors as well as structural linkers between the plasma membrane and the cortical actin-based cytoskeleton. Although clustering of L-selectin with monoclonal antibody has been shown to lead to a number of different cellular responses, little is known about the early intracellular events that are initiated in response to clustering L-selectin. The aim of this report was therefore to understand the molecular interactions that ensue in response to clustering L-selectin, with particular reference to CaM and ERM binding. Defining potentially unique interactions between CaM and ERM could lead to the identification of novel therapeutic targets for inhibiting L-selectin-dependent adhesion and signaling.
Here, we show that CaM and ERM bind to non-competing regions of the same L-selectin tail to form a 1:1:1 complex. These observations were supported by molecular modeling of L-selectin with respective NMR and x-ray structures of CaM and the N-terminal domain of ERM proteins complexed with the cytoplasmic tails of other cell adhesion molecules, such as CD43 (18), intercellular adhesion molecule (ICAM)-2 (19), and P-selectin glycoprotein ligand-1 (20). Based on the predicted model of interaction, it appeared that CaM and ERM could interact independently of L-selectin, which was shown both in vitro and in vivo (using FRET-based microscopic techniques). Moreover, in vivo interaction between CaM and ezrin increased significantly when L-selectin was co-expressed. Interestingly, CaM-ezrin interaction was not observed when CD44 (known to bind ezrin, but not CaM) was co-expressed in place of L-selectin, confirming that the interaction between CaM and ERM was driven specifically by L-selectin. Collectively, these results demonstrate that CaM and ERM can bind non-competitively to a single cytoplasmic tail of L-selectin. Furthermore, binding between CaM and ezrin increased in response to L-selectin clustering, suggesting that CaM and ERM are involved in signaling downstream of L-selectin engagement.
MATERIALS AND METHODS
Chemicals and Antibodies—All chemicals were purchased from Sigma-Aldrich, unless otherwise stated. Anti-L-selectin (mouse monoclonal) antibody, CA21, was a kind gift from Julius Kahn (Boehringer-Ingelheim, Ridgefield, CT). DREG56 is an anti-human L-selectin antibody, which specifically recognizes the lectin domain (mouse IgG1 monoclonal, purchased from Santa Cruz Biotechnology, Santa Cruz, CA). Anti-CaM (IgG1 mouse monoclonal antibody) was purchased from Upstate Laboratories. Eurogentec (Belgium) was commissioned to generate rabbit polyclonal anti-moesin FERM domain antibody. DREG56 monoclonal antibody (IgG1) was purified on protein G columns using cell-free supernatants of hybridoma cell cultures (ATCC HB-300). Anti-ERM and anti-phospho-ERM affinity-purified rabbit antibodies were purchased from Cell Signaling Technologies.
Peptides and Plasmids—Peptides were synthesized by Will Mawby at the Department of Biochemistry, University of Bristol. Peptides were synthesized using an Applied Biosystems 430A peptide synthesizer and purified by reversed-phase high pressure liquid chromatography. Peptide length and purity were assessed using mass spectrometry.
WT cDNA of L-selectin was cloned in to pMT2 plasmid (kind gift from Thomas Tedder, Duke University) and had been used in our previous studies (9). The open reading frame of human CaM-GFP was amplified from template DNA kindly provided by Donald C. Chang (Hong Kong, China). XhoI and HindIII digestion sites were engineered at the 5′ and 3′ ends, respectively, and subcloned into eGFP-N1 vector (Clontech), which contained mCherry in place of GFP. The same procedure was undertaken to subclone the cDNA of WT L-selectin in to mCherry-N1 vector, using the same restriction sites.
Overexpression and Purification of Recombinant Moesin and Ezrin FERM Domains—Both recombinant human moesin and ezrin FERM domains were expressed in bacteria and purified as previously described (21). The expression plasmids coding for both proteins were a kind gift from Anthony Bretscher (Cornell University). Recombinant FERM domains were dialyzed in buffer A (150 mm NaCl, 25 mm HEPES, pH 7.4) prior to bead binding assay. Recombinant human CaM was purchased from BIOMOL international as a lyophilized powder and solubilized in buffer A at a final concentration of 2 mg ml-1.
L-selectin Tail Bead Binding Assay—N-Hydroxysuccinimide-coated Sepharose beads were purchased from Pierce and used to couple synthetic L-selectin cytoplasmic tail peptide (i.e. RRLKKGKKSKRSMNDPY), as outlined in the manufacturer's protocol. Approximately 1 μg of L-selectin tail was coupled to 10 μl of beads (settled volume). Residual non-reactive N-hydroxysuccinimide groups were blocked using 1 m Tris-HCl, pH 7.5. L-selectin tail beads were washed twice in buffer A prior to bead-binding assay. An excess amount of either moesin/ezrin FERM domain or CaM (at least 11.5 μm) was incubated with 10 μl of beads for 30 min at room temperature to allow binding. The “bead binding assay volume” was ∼1 ml in each case. Beads were continually agitated on a tube rotator. After 30 min of incubation at room temperature, beads were washed and pelleted by microcentrifugation at 3000 rpm for 1 min. The harvested beads were then either subjected to boiling in Laemmli buffer or were incubated with another protein to assess multiple binding of proteins to the L-selectin tail beads. In the latter case, the beads were incubated for a further 30 min at room temperature and then similarly washed prior to loading. Products were resolved by electrophoresis on 4-12% precast NuPAGE® gradient gels (Invitrogen). CaCl2 was included in some experiments at a final concentration of 1 mm. EGTA was added to a final concentration of 5 mm in other experiments.
Glycerol Gradient Sedimentation Analysis—Approximately 100 μg of moesin FERM domain and CaM were layered (individually or mixed) on top of discontinuous glycerol gradients in a final volume of 200 μl in buffer A containing no glycerol. Discontinuous glycerol gradients (4.5 ml) were generated in polyallomer centrifuge tubes (Beckman). A 200-μl cushion containing 60% glycerol (in buffer A) was added to the base of the tube followed by 700 μl of decreasing concentrations of glycerol mixed in buffer A (i.e. 50%, 40%, 35%, 30%, 20%, and 10%). Gradients were left to rest for ∼1-2 h at 4 °C prior to loading of protein samples. Tubes were then placed in a swing-out rotor (Beckman SW55) and centrifuged for 21 h at 55,000 rpm and 4 °C. Fractions (200 μl) were collected from the bottom of each centrifuge tube. Each fraction (40 μl) was boiled in Laemmli protein loading buffer and resolved on precast 4-12% NuPAGE® gradient gels. Visualization of resolved proteins was achieved by Coomassie blue (R250) staining of polyacrylamide gels.
DSS Chemical Cross-linking—Disuccinimidyl suberate (DSS, purchased from Pierce) is a homobifunctional chemical cross-linker. The final concentration of DSS used in each reaction was between 0.05 and 0.1 mm. Each cross-linking experiment was performed in a 25-μl final reaction volume containing 4.6 μm of CaM and/or moesin FERM domain, with or without a specified concentration of the L-selectin cytoplasmic tail peptide (see Fig. 5). The volume of cross-linker dissolved in DMSO was added to the reaction tube at a volume no greater than 0.625 μl. This equates to a maximum of 2.5% (v/v) final DMSO content in each reaction. Control experiments using carrier alone (i.e. 0.625 μl of DMSO) were conducted to verify that the amount of DMSO used had not affected the stability/electrophoretic mobility of moesin FERM domain, CaM, or the L-selectin tail.
FIGURE 5.

CaM and ERM interact independently of the l-selectin cytoplasmic tail. A, ribbon stereo side view of the CaM/L-selectin/moesin complex, which was adapted from the model shown in Fig. 2A. Amino acid residues belonging to the extended α-helix were removed, as GFP was tagged to lysine 296 of the ezrin FERM domain. The distance between the C-terminal ends of moesin FERM and CaM is ∼31 Å. The green and red dots represent the relative positions of where GFP and mCherry were tagged. B, individual sedimentation profiles of recombinant purified CaM, moesin FERM, and a mixture of the two are shown. LMW = low molecular weight (i.e. top of tube). HMW = high molecular weight (i.e. bottom of tube). Gels are representative of three independent experiments. C-E, a range of DSS concentrations (0.05, 0.1, 0.2, 0.4, and 0.8 mm) were incubated with fixed concentrations of CaM (4.6 μm) (C), moesin FERM (4.6 μm) (D), or a mixture of the two (E). Slight shifts in electrophoretic mobility of CaM and moesin FERM are seen, which is due to intramolecular cross-links caused by DSS treatment. Secondary structure of protein is expected to persist, even during boiling in protein loading buffer. Cross-linking was performed at room temperature for 45 min. Protein samples were resolved on 4-12% gradient gels and visualized by Coomassie Blue staining. The black arrowhead shows the position of dimeric moesin FERM domain. The bracket to the right of the polyacrylamide gel in D and E denotes the position of higher molecular weight complexes that formed upon cross-linking CaM with moesin FERM. These Coomassie-stained gels are representative of three independent experiments. Western blotting of CaM (F) and moesin FERM (G) confirmed that both CaM and moesin FERM were present in chemically cross-linked high ordered complexes. White asterisks correspond to relative positions of CaM/FERM dimer (D) and tetramer (T). Immunoblots are representative of three independent experiments. H, CaM-agarose beads were used to precipitate p-ERMs from whole cell lysates of U937 cells that were pre-treated with 25 nm of the phosphatase inhibitor, calyculin A, which increases p-ERM levels (36). The left lane in each panel represents the fraction of ERMs from unstimulated cell extracts. The right-hand lane in each panel is from calyculin A-treated cell extracts. The bottom panel shows that CaM-agarose beads selectively precipitated p-ERMs from calyculin A-treated extracts.
SBED Chemical Cross-linking—Sulfosuccinimidyl-2-[6-(biotinamido)-2-(p-azidobenzamido)hexanoamido]ethyl-1,3′-dithiopropionate) (SBED, Pierce) is a biotin-tagged, thiol-cleavable, heterobifunctional chemical cross-linking reagent. It is composed of a 14.3-Å spacer arm that holds a sulfonated N-hydroxysuccinimide active ester group at one end and a photoactivatable aryl azide reactive group at the other end (see supplemental Fig. S1). A thiol-cleavable bond is positioned centrally within the spacer arm. A biotin group is positioned between the thiol-cleavable bond and the aryl azide group (see supplemental Fig. S1).
To create the “bait” peptide, 0.1 mm of “No Weigh” SBED reagent was added to 220 μm of L-selectin tail peptide in 25 mm HEPES, 150 mm NaCl and incubated at room temperature for 1 h with occasional mixing. Excess uncoupled SBED reagent was separated from the biotinylated L-selectin tail peptide by loading the mixture on to a pre-equilibrated 5-ml D-salt™ polyacrylamide de-salting column (1800-Da exclusion limit, Pierce), and the eluent was collected. 10 × 0.5 ml volumes of 25 mm HEPES, 150 mm NaCl were then passed through the column, and each 0.5 ml of flow eluent was collected into separate Eppendorf tubes. The concentration of biotinylated L-selectin tail peptide within each fraction was then determined after measuring absorbance at 280 nm. The L-selectin tail peptide concentration was estimated to be ∼3.6 μm. The desired “prey” proteins (8 μg of FERM and/or 4 μg of calmodulin) were added to 3.6 μm bait peptide and allowed to bind at room temperature for 1 h with occasional mixing. For competition experiments, the first prey protein was added for 30 min before adding the second and incubating for a further 30 min. The bound proteins were then cross-linked to the bait peptide by UV exposure. Cross-linked products were then subjected to boiling in Laemmli buffer containing β-mercaptoethanol to break the thiol bond within the spacer arm. This resulted in the transfer of biotin from the L-selectin tail peptide to its bound partner. Boiled samples were resolved on polyacrylamide gels, transferred on to PVDF for Western blotting. Horseradish peroxidase-streptavidin (1:5000) was used to identify the biotinylated reactants. The difference in molecular mass between CaM (18 kDa) and moesin FERM (30 kDa) enabled easy identification of the biotinylated products.
Molecular Modeling of Moesin FERM/L-selectin/CaM Complex—Modeling was based on the known crystal structure of the moesin FERM domain and first long helix (PDB ID: 1E5W (22)), and the NMR structure of an extended form of CaM complexed a peptide belonging to the plasma membrane-associated calcium pump (PDB ID: 1CFF (23)).
Docking of moesin FERM on to the L-selectin tail was based on the previously solved radixin FERM/ICAM-2 crystal structure (PDB ID: 1J19). R357A of L-selectin was aligned with R405 of ICAM-2 and used to orient the L-selectin tail onto the known crystal structure of moesin FERM domain. The crystal structure of the ICAM-2/Radixin FERM domain complex (PDB ID: 1J19) (19) was also used to orient the moesin FERM domain (PDB ID: 1E5W). The extended NMR structure of CaM was then docked on to the L-selectin/moesin FERM complex, using the same domain of CaM that was bound to the membrane-associated calcium-pump peptide (as in the 1CFF structure). Manual adjustments were carried out to improve the fit between CaM, L-selectin, and moesin FERM. The whole complex was then subjected to molecular dynamics simulation and energy minimization, using various software that included AMBER99, YASARA, and YAMBER2 force field software (24), leaving the RRLKK part of the L-selectin tail fixed and the remaining 12 amino acids of the tail free to move. The resultant complex has been included as a supplemental pdb file. Minimization and dynamics cycles were performed until the conformations of all three proteins remain unchanged, and none of the proteins were “ejected.” A low energy of the total complex was obtained from the dynamics simulation (∼-28,858 kcal/mol). All modeling was performed using MolIDE (25) and Accelrys Discovery Studio and Yasra (26). A detailed breakdown of the hydrogen bonds can be found in supplemental Fig. S2.
Cell Culture, Transfection, Immunoblotting, and Immunofluorescence—COS-7 monkey fibroblast cell line was grown in Dulbecco's modified Eagle's medium (Invitrogen) containing 10% fetal calf serum (Helena Biosciences, Sunderland, UK), 100 units/ml penicillin, and 100 μg/ml streptomycin sulfate at 37 °C, 10% CO2. U937 human monocyte cell line was grown in RPMI 1640 medium (Invitrogen) containing 10% fetal calf serum and penicillin/streptomycin at 37 °C, 5% CO2.
For transfection, COS-7 cells were plated the night before to reach 70-80% confluency the following day. Cells were then collected from triple-vent 14-cm dishes using trypsin/EDTA (Invitrogen) and washed with 5 ml of cold electroporation buffer (120 mm KCl, 10 mm K2PO4·KH2PO4 (pH 7.6), 25 mm Hepes, 2 mm MgCl2, and 0.5% Ficoll). The buffer was removed, and cells were resuspended in 250 μl of cold electroporation buffer and electroporated at 250 V and 960 microfarads (Bio-Rad electroporator) with 2 μg of DNA for each construct. 1 × 106 U937 cells were transiently transfected using similar amounts of DNA, which was delivered using the MP-100 microporator (LabTech, UK) and following the manufacturer's instructions. Immunoblotting procedures have been previously described in detail (6).
COS-7 cells were seeded on to sterile 13-mm diameter (“0” thickness) glass coverslips at a density of 3 × 104 cells/ml and left to grow overnight before fixing and staining. The following day after microporation, U937 cells were washed and plated either onto poly-l-lysine (PLL)- or sialyl Lewisx (sLex)-coated glass coverslips. Binding to sLex-coated coverslips was performed at 37 °C for 5-10 min. PLL-coated coverslips were made by applying neat PLL to the coverslip for 15 min at room temperature prior to aspiration. Coverslips were left to dry overnight at room temperature. Coating of glass coverslips with sLex was achieved by adding 50 μg/ml NeutrAvidin™ (Pierce), dissolved in Ca2+/Mg2+-free PBS, to PLL and was left to bind under humidifying conditions for 1 h at room temperature. Excess NeutrAvidin was aspirated, the coverslip was washed in Ca2+/Mg2+-free PBS and then followed by a blocking step in 2% bovine serum albumin dissolved in Ca2+/Mg2+-free PBS. Multivalent sLex-PAA (Glycotech) was then applied to the coverslip and allowed to bind for 2 h under humidifying conditions. Excess sLex was aspirated and washed off prior to further blocking in 2% bovine serum albumin.
All cells were fixed in 4% paraformaldehyde (PFA) and permeabilized with 0.1% Nonidet P-40 substitute (Fluka), before blocking with bovine serum albumin and staining with appropriate antibodies for immunofluorescence. Coverslips that were processed for FRET were further treated with 1 mg/ml sodium borohydride dissolved in PBS for 10 min at room temperature before washing in PBS and mounting on to slides with fluorescent mounting medium (DakoCytomation). For confocal analysis, images were captured using a Zeiss LSM510 META confocal microscope (Carl Zeiss, Welwyn Garden City, UK) running version 3.2 of the LSM acquisition software. Image processing was performed with Adobe Photoshop CS (Adobe Systems, San Jose, CA).
FLIM and FRET Analysis—Time-domain fluorescence lifetime imaging microscopy (FLIM) was performed with a multiphoton microscope system as described (for details see Refs. 27, 28). Fluorescence lifetime imaging capability was provided by time-correlated single photon counting electronics (Becker & Hickl, SPC 700). A 40× objective was used throughout (Nikon, CFI60 Plan Fluor numerical aperture 1.3), and data were collected at 500 ± 20 nm through a bandpass filter (Coherent Inc., 35-5040). Acquisition times of the order of 300 s at low 890 nm excitation power were used to achieve sufficient photon statistics for fitting, while avoiding either pulse pile-up or significant photobleaching.
Data were analyzed as previously described (27, 29). The FRET efficiency is related to the molecular separation of donor and acceptor and the fluorescence lifetime of the interacting fraction by,
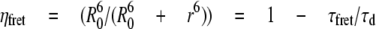 |
(Eq.1) |
where R0 is the Förster radius, R the molecular separation, ηfret is the lifetime of the interacting fraction, and τd the lifetime of the donor in the absence of acceptor. The donor-only control is used as the reference against which all other lifetimes are calculated in each experiment. ηfret and τd can also be taken to be the lifetime of the interacting fraction and non-interacting fraction, respectively. Quantification was made from all pixels within each cell when they were analyzed. All data were analyzed using TRI2 software (developed by Dr. Paul Barber, Gray Cancer Institute, London, UK). Histogram data presented here are plotted as mean FRET efficiency from the stated n number of cells over three experiments, ± S.E. Analysis of variance was used to test statistical significance between different populations of data. Each figure demonstrates the range of lifetime efficiencies per cell normalized for pixel intensity for each experimental condition.
CaM-Agarose Pulldown Assay—Approximately 5 × 107 U937 cells were used per CaM pulldown assay. Cells were treated with or without 500 μm cantharidin for 30 min at 37 °C. After treatment, the cells were washed once in 5 ml of PBS and subsequently harvested by centrifugation. The cell pellet was then lysed on ice for 15 min in 1 ml of lysis buffer (150 mm NaCl, 2 mm CaCl2, 20 mm Tris-HCl, pH 8, 10 mm NaF, 0.1 mm Na3VO4, 1 mm phenylmethylsulfonyl fluoride, protease inhibitor mixture, 1% Nonidet P-40, 1% ovalbumin, and 25 nm calyculin A). Lysates were clarified for 10 min at 14,000 rpm at 4 °C. 30 μl of the clarified lysate was retained for Western analysis after boiling with 70 μl of 2× Laemmli buffer. The remaining lysate was diluted to 10 ml using lysis buffer without Nonidet P-40 and added to 100 μl of CaM-agarose bead slurry (i.e. 50 μl of settled bead volume). Tubes were left to rotate for 2 h at 4 °C. Beads were washed at least four times to remove any unbound material. Beads were subsequently boiled in Laemmli buffer prior to gel electrophoresis and Western blotting as previously described (9).
RESULTS
Simultaneous Non-competitive Binding of CaM and Moesin FERM to the Tail of L-selectin—A peptide comprising the 17-amino acid cytoplasmic tail of L-selectin was synthesized, coupled to Sepharose beads (termed “L-selectin beads”), and used in pulldown assays to determine if CaM and ERM binding could occur simultaneously (Fig. 1A). L-selectin beads were saturated with or without recombinant purified CaM prior to incubation with increasing amounts of recombinant soluble moesin N-terminal domain (also termed band Four point one Ezrin/Radixin/Moesin (FERM)). Binding of moesin FERM to the L-selectin beads remained unchanged irrespective of CaM preloading (Fig. 1B), suggesting that moesin FERM binding was not compromised by CaM preloading. Similar results were obtained using ezrin FERM (data not shown), indicating that this interaction is relevant to other ERM family members. Reciprocal preloading of moesin FERM had little effect on the subsequent binding of CaM (Fig. 1C), and EGTA did not alter the binding of either CaM or moesin FERM to the L-selectin beads, indicating that Ca2+ was not required for binding (Fig. 1C). These data imply that, despite the short length of the L-selectin tail, both CaM and ERM can bind simultaneously with the L-selectin tail via distinct non-overlapping binding sites. However, this observation did not fully exclude the possibility that preloading of one protein could encourage the other to bind indirectly. We therefore used SBED, a thiol-cleavable, heterobifunctional chemical cross-linker, to determine if both CaM and moesin FERM were binding directly to the L-selectin tail. Successful transfer of biotin from the L-selectin tail to either CaM or moesin FERM can only occur as a consequence of direct binding (see supplemental Fig. S1 for details of the cross-linking method). Western blotting of biotin-labeled proteins, using horseradish peroxidase-conjugated streptavidin, revealed that both CaM and moesin FERM domain were labeled equally with biotin, irrespective of the order in which the proteins were mixed (Fig. 1D). Further evidence of a 1:1:1 complex was demonstrated using DSS, a non-cleavable homobifunctional chemical cross-linker. In this approach, CaM and/or moesin FERM were mixed with L-selectin tail peptide in the presence of the DSS cross-linker, resolved on polyacrylamide gels, and transferred to a PVDF membrane. The CA21 monoclonal antibody, which recognizes the membrane-distal portion of the L-selectin tail (see Fig. 1A), was used in Western blotting to detect increases in molecular weight after cross-linking L-selectin to its binding partner. Increasing the amount of L-selectin tail peptide to the cross-linking reaction (containing a fixed amount of DSS and either CaM or moesin FERM) led to a corresponding increase in molecular weight of the L-selectin tail peptide. This was suggestive of the L-selectin tail peptide and one of its binding partners forming a 1:1 complex (Fig. 1E). Furthermore, addition of increasing L-selectin tail peptide to fixed mixtures of DSS, CaM, and moesin FERM led to the emergence of a 50-kDa band, which was interpreted as a 1:1:1 complex between CaM (18 kDa), moesin FERM (30 kDa), and L-selectin tail peptide (∼2 kDa). Intriguingly, the CA21 monoclonal antibody readily detected the L-selectin peptide when complexed to both CaM and moesin FERM, suggesting that epitope recognition was not blocked. This suggested that binding of L-selectin to both CaM and ERM likely involved the membrane-proximal, and not the membrane-distal, region of the L-selectin tail (Fig. 1E).
Protein Structure Prediction of the Moesin FERM/L-selectin Tail/CaM Complex—Model building demonstrated that moesin FERM, CaM, and the tail of L-selectin are able to form a 1:1:1 complex (see “Materials and Methods” for details and Fig. 2). Minimization and dynamics cycles revealed that the complex was thermodynamically stable (∼-28,858 kcal/mol, see supplemental pdb file of the complex). Moreover, residues within the L-selectin tail that contributed to moesin and CaM binding were non-competing. Interestingly, top, side, and bottom views of the complex showed that significant regions of L-selectin tail are exposed in the complex (Fig. 2A). This configuration of binding could justify how the CA21 monoclonal antibody was able to recognize the membrane-distal portion of the L-selectin tail when simultaneously cross-linked to both moesin FERM and CaM (see Fig. 1E). Molecular modeling supported the possibility that both the moesin FERM and CaM made a significant contribution to the heterotrimeric complex (Fig. 2A). The model also revealed that regions A (or F1) and C (or F3) of the moesin FERM domain could make contact with CaM independently of L-selectin. Moreover, Lys-278, which resides within the first α-helix of moesin domain C (α1C), hydrogen bonded with Lys-148 and Met-144 of CaM (see supplemental Fig. S2 for a full list of hydrogen bonds, and pdb of the model provided in the supplemental material). This is of particular interest, as the structurally related α1C domain of band 4.1R has been previously shown to interact with CaM (30).
FIGURE 2.

Molecular modeling of the moesin FERM/l-selectin/CaM heterotrimeric complex. The respective crystal and NMR structures of the moesin FERM domain (cyan) and calmodulin (purple) were used to model interactions with the cytoplasmic tail of L-selectin (gold, see “Materials and Methods” for full explanation of the procedure). A, images were rendered using POV-ray software to show molecular surface (hydrogen atoms were removed prior to rendering of the image). Top, side, and bottom views of the heterotrimeric complex reveal that one side of the L-selectin tail is partly exposed. The model also reveals that interactions between CaM and the moesin FERM domain contribute substantially to the heterotrimeric complex. The cytoplasmic tail of L-selectin is marked with cyan and purple spots, which indicate the residues within the L-selectin tail that hydrogen-bond with moesin FERM and CaM, respectively (see supplemental Fig. S2 and supplemental pdb file for more detail). B, stereo view of the predicted L-selectin tail-moesin FERM interaction. Positions of the α1C, α5C, and α6C are indicated in white lettering. The contacting residues in the moesin FERM domain are very similar to those previously described for CD43, P-selectin glycoprotein ligand-1, and ICAM-2 (see Refs. 18-20). Predicted H-bonds between the L-selectin tail and moesin FERM are marked by red dashed lines, which are indicated by the red arrows. Blue and black lettering indicate the amino acid residues involved in forming putative H-bonds between moesin FERM and L-selectin, respectively.
Interestingly, specific amino acids within the moesin FERM domain that contacted the L-selectin tail had been previously characterized to form hydrogen bonds with the tails of other cell adhesion molecules such as P-selectin glycoprotein ligand-1, CD43, and ICAM-2 (18-20) (Fig. 2D). The most striking similarity between these interactions was the involvement of β5C in binding to different cytoplasmic tails, suggesting that the FERM domain may only bind to a single cytoplasmic tail of a cell adhesion molecule at any given time. Previously characterized tail-FERM complexes have been shown to interact within a region between α1C and β5C of the FERM domain (18-20), however the interaction of L-selectin with the moesin FERM domain involved the β5C and β6C, and one residue (His-288) from the α1C domain (Fig. 2B and supplemental Fig. 2). In summary, model building supports the notion that CaM, L-selectin, and moesin FERM can form a heterotrimeric complex. Although this model provides a good visual representation of how CaM and ERM might interact non-competitively with the L-selectin tail, more experiments would be required (such as mutagenesis studies) to verify that these interactions actually occur in vivo.
In Vivo Interaction of CaM, L-selectin, and Ezrin FERM—Based on our biochemical findings, we next sought to determine whether interactions between CaM, L-selectin, and ERMs could occur in vivo. Co-expression of CaM-GFP, full-length moesin (HA-tagged), and WT L-selectin in COS-7 cells revealed that all three proteins co-localized to membrane structures that resembled filopodial-like protrusions or microvilli (supplemental Fig. S3). Much of the expressed CaM-GFP appeared to be nuclear, consistent with previous findings (31), but also localized to membrane projections (supplemental Fig. S3). However, this approach could not address whether the three proteins were physically interacting with one another. To test whether FLIM could be used to study complex interactions between CaM, ERM, and L-selectin, COS-7 cells were transiently transfected with ezrin FERM-GFP together with WT L-selectin and CaM-monomeric cherry fluorescent protein (mCherry). FLIM was used to monitor FRET efficiency by measuring the fluorescence lifetime of the GFP (donor) molecule. The GFP fluorescence lifetime decreases when in close proximity (<10 nm or 100 Å) to an acceptor fluorochrome, such as mCherry (for details see “Materials and Methods”). Therefore, a reduction in fluorescence lifetime of the GFP donor is a consequence of direct interaction (FRET) of the fluorescently tagged proteins. In vivo interaction between CaM-mCherry and ezrin FERM-GFP was expressed as “FRET efficiency,” as outlined under “Materials and Methods.”
The FRET efficiency detected between CaM-mCherry and ezrin FERM-GFP in COS-7 cells did not increase significantly above the FRET efficiency seen with ezrin FERM-GFP (donor) alone (Fig. 3, A and B), clearly demonstrating that the two fluorescently tagged proteins could not interact in vivo. However, co-expression of L-selectin with CaM-mCherry and ezrin FERM-GFP led to statistically significant increases in FRET efficiency (Fig. 3C), suggesting that L-selectin was required for mediating the direct interaction between CaM-mCherry and ezrin FERM-GFP. In addition, expression of L-selectin induced a change in cell morphology, whereby transfected COS-7 cells adopted a “hairy cell” phenotype. This is in keeping with previous reports, which have observed increases in microvillar size and density following the ectopic expression of ERM-binding membrane proteins such as CD44, CD43, and ICAM-2 (32) (see lower panel and inset in Fig. 3A).
FIGURE 3.

FLIM reveals that FRET between CaM-mCherry and ezrin FERM-GFP is dependent on l-selectin clustering. A, upper panel, GFP donor lifetime of fluorescence was determined in COS-7 cells expressing ezrin FERM-GFP (termed N-ezrin-GFP) alone. Middle panel, expression of CaM-mCherry and ezrin FERM-GFP in COS-7 cells did not alter the FRET efficiency between the two fluorescently tagged proteins. Bottom panel, co-expression of CaM-mCherry, ezrin FERM-GFP, and L-selectin led to significant increases in FRET efficiency that was also detected at cell protrusions (inset). B, L-selectin-positive cells were identified as possessing a hairy phenotype. FRET analysis was performed on cells that were also positive for mCherry and GFP expression. Labeling of L-selectin with DREG56 was routinely performed on live cells, as DREG56 does not recognize L-selectin from fixed specimens. Cy5-conjugated donkey anti-mouse secondary antibody was subsequently used to locate DREG56 staining on 4% PFA-fixed cells. Cross-linking with Cy5-conjugated donkey anti-mouse secondary (2ary XL) antibody was performed on live cells immediately after removal of excess DREG56. C, histogram of data pooled from at least 15 cells over three different experiments. Statistical significance is based on FRET from transfectants expressing CaM and ezrin FERM-GFP (*, p < 0.01; **, p < 0.005). The lifetime is shown as a pseudocolor scale of blue (high lifetime) to red (low lifetime = FRET).
Live cell labeling of L-selectin was necessary, because the anti-L-selectin antibody, DREG56, cannot recognize L-selectin from PFA-fixed specimens. DREG56 labeling of L-selectin is known to promote clustering (33), which led us to determine whether clustering of L-selectin was contributing to the FRET between ezrin FERM-GFP and CaM-mCherry. Control experiments were performed without labeling COS-7 cells with DREG56, and triple transfectants were selected for FRET analysis on the basis of their transformed hairy cell phenotype (indicative of L-selectin expression). No significant increase in FRET efficiency was observed above background in the absence of labeling with DREG56 (Fig. 3, B (top panel) and C). This strongly implied that DREG56-dependent clustering of L-selectin was promoting the observed increase in FRET efficiency between CaM-mCherry and ezrin FERM-GFP. Furthermore, cross-linking of DREG56 with anti-mouse secondary antibody led to further increases in FRET efficiency (Fig. 3, B and C). The extent of clustering L-selectin was therefore directly correlated to the extent of FRET efficiency between CaM and ezrin FERM. Interestingly, replacing L-selectin with CD44 (which is known to bind ezrin but not CaM) failed to induce any increases in FRET efficiency, irrespective of clustering, suggesting that the interaction between CaM and ezrin FERM was facilitated specifically by L-selectin (supplemental Fig. S4).
Expression of ezrin FERM-GFP and CaM-mCherry in U937 cells did not yield any significant increases in FRET efficiency above basal levels (Fig. 4). In contrast, labeling U937 cells with DREG56 led to significant increases in FRET efficiency, which again rose in response to further cross-linking with anti-mouse secondary antibody (Fig. 4). These results confirm that CaM-ERM interaction is driven by clustering of L-selectin in leukocytes and is due to the continued binding of CaM and ERM to L-selectin. In contrast to COS-7 cells, little change in the overall phenotype of U937 cells was seen in response to overexpressing L-selectin.
FIGURE 4.

CaM-ezrin interaction in response to l-selectin clustering is readily observed in monocytes. A, control experiments revealed that CaM-mCherry/N-Ezrin-GFP interaction was not observed in U937 cells in the absence of WT L-selectin, which is similar to that seen in COS-7 cells (Fig. 3). B, cells were microporated with plasmids encoding ezrin FERM-GFP (N-Ezrin-GFP), CaM-mCherry, along with WT L-selectin as stated under “Materials and Methods.” Cells were labeled in suspension (on ice) with or without DREG56 antibody, followed by further cross-linking with or without Cy5-conjugated donkey anti-mouse secondary antibody. Cells were then plated on to PLL-coated coverslips and placed in a 37 °C incubator for ∼10 min. Cells were subsequently fixed in 4% PFA and processed for FRET using FLIM. C, histogram depicts data from experiment in B. Data were pooled from a total of at least 15 cells. Statistical significance is based on cells expressing ezrin-GFP and CaM-mCherry (*, p < 0.05; **, p < 0.005). Lifetime is shown as a pseudocolor scale of blue (high lifetime) to red (low lifetime = FRET).
CaM and ERM Interact in Vitro—CaM has been previously shown to interact with at least two distinct regions of the band 4.1R FERM domain (30). Moreover, these regions are defined as Ca2+-dependent and Ca2+-independent binding sites. Sequence alignment revealed that these two regions had poor amino acid conservation with the ERMs, and it was concluded that CaM binding was limited specifically to the FERM domain of band 4.1R. However, because the crystal structures of all the ERM FERM domains have been solved, it is clear that these two CaM-binding sites are structurally conserved with band 4.1R. It was to this end that we explored whether recombinant purified moesin FERM could interact with recombinant purified CaM independently of the L-selectin tail. Non-linear glycerol gradient sedimentation analysis was used to determine if CaM and moesin FERM could interact in solution (Fig. 5B). The relative sedimentation profiles of CaM and moesin FERM altered dramatically when mixed together (Fig. 5B), suggesting that the purified proteins could interact in solution independently of the L-selectin tail. Molecular weight assignment proved difficult, because the CaM-moesin FERM complexes formed broad peaks in solution. DSS cross-linking followed by PAGE was therefore used to determine more precisely the size of complexes formed between moesin FERM and CaM. Equal concentrations (4.6 μm) of CaM, moesin FERM, or a mixture of the two, were subjected to increasing amounts of DSS. Incubation of DSS with CaM alone did not result in the formation of CaM multimers (Fig. 5C), whereas trace amounts of moesin FERM dimers were observed upon treatment with DSS (arrowhead in Fig. 5D). In contrast, higher ordered complexes readily formed when moesin FERM and CaM were incubated with increasing amounts of DSS (Fig. 5E). The emergence of a 50-kDa band strongly correlated with CaM-moesin FERM dimerization (i.e. 18-kDa CaM plus 32-kDa moesin FERM). Similar results were obtained using the ezrin FERM domain (data not shown), again suggesting that the interaction could occur with all ERM members. Western blotting confirmed that the higher molecular mass complexes, in particular at 50 kDa, contained both CaM and moesin FERM (Fig. 5, F and G), suggesting that CaM and ERMs formed complexes independently of L-selectin.
CaM-agarose beads were used to establish if endogenous ERMs could be precipitated from whole cell lysates of U937 cells. In leukocytes, ERM proteins have been shown to shuttle between active and inactive states, which can be regulated by chemokines (34). ERM activation is induced by one of two ways: binding of the FERM domain to phosphatidylinositol 4,5-bisphosphate or by phosphorylation of a conserved threonine residue present in the C terminus of all ERMs (35). Interestingly, activation of ERMs with calyculin A (a phosphatase inhibitor that dramatically increases C-terminal threonine phosphorylation of ERMs; see Ref. 36) was required for effective precipitation of ERM from U937 cell lysates (Fig. 5H). This suggests that the CaM-binding site lies within a region of the FERM domain that can be potentially masked by binding of the C-terminal ERM tail. Interestingly, this observation would be in keeping with the molecular model that predicts the involvement of moesin α1C binding to CaM, which is masked by the C-terminal tail of moesin in the folded conformation (21).
Activation of Ezrin Increases Binding to CaM, Both in the Presence and Absence of L-selectin Clustering—FRET and FLIM were used to assess whether constitutive activation of ezrin could lead to increased binding with CaM in vivo. U937 cells were transfected with L-selectin, CaM-mCherry, and either full-length (WT) ezrin-GFP or full-length constitutively active (TD) ezrin-GFP (where the C-terminal threonine at position 567 is mutated to an aspartate). Additionally, using full-length ezrin-GFP allowed us to determine if ezrin FERM-GFP was responsible for blocking interaction with CaM-mCherry, as seen in COS-7 and U937 cells (Figs. 3 and 4). Triple transfectants were seeded onto either sLex- or PLL-coated coverslips, which would promote L-selectin clustering, or not. Samples were then fixed in 4% PFA and analyzed for FRET efficiency. The FRET efficiency between CaM-mCherry and full-length ezrin-GFP was modest (but significantly increased above the 2% basal level) in U937 cells plated on PLL. More importantly, under these conditions, FRET occurred independently of L-selectin clustering (Fig. 6, A and B), suggesting that full-length ezrin-GFP could partially restore FRET with CaM-mCherry in vivo. Moreover, co-expression of full-length TD ezrin-GFP in place of full-length WT ezrin-GFP led to a further significant increase in FRET efficiency (Fig. 6, A and B), which corroborated with our in vitro findings that activation of ERMs increased binding to CaM (Fig. 5H). Plating triply transfected U937 cells onto sLex-coated coverslips led to even higher FRET efficiencies between full-length ezrin-GFP and CaM-mCherry (Fig. 6, C and D), suggesting that clustering significantly increased the ezrin-CaM interaction. These results confirm that activation of ezrin increases binding to CaM. These results also confirm that distancing GFP from the FERM domain facilitates interaction between CaM and ezrin, both dependently and independently of L-selectin clustering.
FIGURE 6.

Binding between CaM and full-length ezrin occurs in vivo, which increases upon l-selectin clustering. U937 monocytes were microporated with plasmids encoding full-length ezrin-GFP (either WT or TD ezrin-GFP), CaM-mCherry, and L-selectin. Cells were then split and seeded on to either PLL (A and B) or sLex (C and D) for 5 min under static conditions at 37 °C, fixed in 4% PFA, and subsequently processed for FRET analysis. The histograms to the right in B and D are representative of 15 cells analyzed over three different experiments. Statistical significance is based on comparison with cells expressing GFP alone (*, p < 0.01; **, p < 0.005). Lifetime is shown as a pseudocolor scale of blue (high lifetime) to red (low lifetime = FRET).
DISCUSSION
Understanding the dynamic interplay between the tails of cell adhesion molecules and their binding partners is of great importance, particularly within the context of understanding how adhesion and signaling are regulated during leukocyte recruitment. These data outlined in this report highlight the complex interactions that occur between L-selectin and associated binding partners under “resting” conditions, and when L-selectin is clustered with monoclonal antibody or multivalent ligand (Fig. 7). Using a range of in vitro and in vivo approaches, the evidence provided in this report leads to the hypothesis that both CaM and ERM are associated with L-selectin in resting cells and co-exist in a heterotrimeric complex. We have characterized an early intracellular event that occurs specifically in response to ligand-induced clustering of L-selectin. The fact that clustering of CD44 (in place of L-selectin) failed to induce FRET between CaM and ezrin suggests that this interaction could be unique to L-selectin, or to other cell adhesion molecules that bind both CaM and ERM. To date, L-selectin is the only cell adhesion molecule that has been characterized to bind both CaM and ERM. Given that clustering of L-selectin is known to promote integrin activation and increase surface expression of chemokine receptor, it is possible that the cis interaction between CaM and ERM may provide a necessary step in this mechanism. It is conceivable that abrogating CaM and ERM interaction in cis could represent a novel therapeutic target to block L-selectin-dependent leukocyte recruitment during chronic inflammation (37-39).
FIGURE 7.

Clustering of l-selectin induces interaction between CaM and ERM in cis. Based on our in vitro and in vivo experiments, it appears that CaM, L-selectin, and ERM likely form a heterotrimeric complex under “resting” conditions. Molecular modeling has also supported the possibility of this configuration (see Fig. 2). In addition, CaM and ERM may function to hold K-Ras and SOS sufficiently apart, so that K-Ras is not activated. It should be appreciated that GFP tagging of the ezrin FERM domain likely disrupts the heterotrimeric complex from forming. In contrast, GFP tagging of full-length ezrin partially restored the heterotrimeric complex, as shown in Fig. 6. Engagement of L-selectin with multivalent ligand induces clustering of L-selectin. The persistent binding of CaM and ERM with the L-selectin tail suggests that Ras and SOS could potentially unite in cis and mediate downstream signaling. Examples include the activation of β1 and β2 integrins, mobilization of the chemokine receptor CXCR4 to the plasma membrane and the generation of reactive oxygen species (ROS).
Our in vivo data suggest that GFP tagging of the ezrin FERM domain potentially destabilizes the CaM/L-selectin/ERM heterotrimeric complex, forcing L-selectin to interact with either CaM-mCherry or ezrin FERM-GFP but not both. This would explain why FRET between CaM-mCherry and ezrin FERM-GFP was observed exclusively as a consequence of clustering L-selectin. In support of this, C-terminal tagging of full-length ezrin with GFP restored FRET with CaM-m-Cherry in the absence of clustering L-selectin (Fig. 6A).
We had previously shown that phorbol myristate acetate stimulation of primary murine lymphocytes leads to the differential activation of moesin and ezrin (6). Interestingly, ezrin activity appeared to be constitutive, whereas binding of moesin was regulated by phorbol myristate acetate stimulation. Indeed, others have shown more recently that ezrin and moesin can behave non-redundantly in intact T cells (40). We have not fully excluded the possibility that ezrin and moesin could behave non-redundantly in respect of binding to L-selectin. However, L-selectin-independent binding of moesin or ezrin to CaM appears to be redundant.8 Further investigation is required to determine if ezrin and moesin compete for binding to L-selectin and/or CaM in vivo. Other factors such as the relative abundance of ERM and post-translational modification could dictate redundancy in binding.
Molecular modeling of the CaM/L-selectin/moesin FERM complex revealed that regions of the L-selectin tail were exposed. This approach has provided a very useful insight into how the heterotrimeric complex might be arranged in vivo, although further studies are required to validate the model. Furthermore, it reveals how the CA21 monoclonal is able to recognize the chemically cross-linked CaM/L-selectin/moesin FERM complex on Western blots (Fig. 1). It is possible that the exposed face of the L-selectin tail seen in our molecular model could provide an available binding site for α-actinin, which is thought to also bind constitutively with the membrane distal portion of the L-selectin tail. Understanding how and if α-actinin forms a tertiary complex with CaM, L-selectin, and ERM will be the focus of future studies.
The biological significance of CaM/ERM interaction during L-selectin-dependent adhesion could be 2-fold. Firstly, CaM-ERM interaction could provide structural support for L-selectin-dependent capture under flow. Binding of CaM to ERM in cis could stabilize activated ERM and increase association of L-selectin with the cortical actin cytoskeleton. This could restrict the lateral mobility of L-selectin within the plasma membrane to stabilize tether formation (41). Indeed, dimerization/clustering of L-selectin enhances tethering under flow (42), reduces the detergent extractability of L-selectin (43), and reduces the net rolling velocity (44). Secondly, CaM-ERM binding in cis could promote signaling downstream of L-selectin engagement for subsequent integrin activation, or mobilization of chemokine receptors (such as CXCR4) to the plasma membrane. Monoclonal antibody or ligand-induced clustering of L-selectin has been shown to activate the Ras/SOS pathway (45), which acts upstream of Rap1A to activate β1 and β2 integrins (46). Interestingly, CaM has been shown to interact with K-Ras (47), and activated ezrin is known to interact with guanine nucleotide exchange factors, such as SOS (48). Recent proteomic analyses of leukocyte microvilli showed that K-Ras and Rap1A are enriched in microvilli and would therefore be in the correct vicinity to receive signals downstream of L-selectin (49). Based on these findings, it is tempting to speculate that CaM and ERM, when bound to the same tail of L-selectin, could hold Ras and SOS sufficiently apart so that one cannot activate the other. Upon ligand-induced clustering, Ras and SOS could be brought together via the coalescence of neighboring tails, which could trigger the pathway specifically during adhesion under flow (Fig. 7). Furthermore, it is likely that activation of β1 or β2 integrins occur downstream of the Ras/SOS pathway, via Rap1A (50). The exchange factor CalDAG-guanine nucleotide exchange factor has received recent attention in regulating leukocyte arrest (3), although its involvement in L-selectin-dependent signaling has not been investigated. It is possible that L-selectin-CaM interaction may serve as a physical link between L-selectin and CalDAG-guanine nucleotide exchange factor, which could potentially facilitate integrin activation via Rap1A. Identifying proteins that can interact directly with a CaM/ERM dimer, or a CaM/L-selectin/ERM trimer, may hold promise for future attempts to isolate downstream signaling targets. Finally, a recent elegant study has shown that CD44/E-selectin-dependent leukocyte rolling along inflamed cremasteric venules induces clustering of L-selectin (51). However, it is not clear as to whether this outcome is due to outside-in signaling or inside-out signaling. Nonetheless, it is possible that clustering of L-selectin during CD44-dependent rolling provides one or more important signals to trigger the transition from leukocyte rolling to arrest, which could be driven, in part, by L-selectin-dependent CaM-ERM interaction in cis.
Supplementary Material
Acknowledgments
We are grateful to Sue Cotterill for her helpful advice with SBED cross-linking experiments. We thank Julius Kahn for generously providing CA21 antibody, Guillaume Charras for ezrin FERM-GFP, full-length WT, and TD ezrin-GFP constructs, and Donald C. Chang for providing human CaM-GFP construct. We thank Ann Ager for critical reading of the manuscript.
This work was supported in part by the Wellcome Trust, the Ludwig Institute for Cancer Research, and the European Union (FP6 contract LHSG-CT-2003-502935). All the authors declare that they have no financially competing interests. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1-S4.
Author's Choice—Final version full access.
Footnotes
The abbreviations used are: CAM, cell adhesion molecule; CaM, calmodulin; ERM, ezrin/radixin/moesin; ICAM, intercellular adhesion molecule; WT, wild type; GFP, green fluorescent protein; eGFP, enhanced GFP; DSS, disuccinimidyl suberate; SBED, sulfosuccinimidyl-2-[6-(biotinamido)-2-(p-azidobenzamido)hexanoamido]ethyl-1,3′-dithiopropionate); PVDF, polyvinylidene difluoride; PLL, poly-l-lysine; sLex, sialyl Lewisx; PBS, phosphate-buffered saline; PFA, paraformaldehyde; mCherry, CaM-monomeric cherry fluorescent protein; FLIM, fluorescence lifetime imaging microscopy; FRET, fluorescence resonance energy transfer.
D. J. Killock and A. Ivetic, unpublished data.
References
- 1.Gonzalez-Amaro, R., and Sanchez-Madrid, F. (1999) Crit. Rev. Immunol. 19 389-429 [PubMed] [Google Scholar]
- 2.Ley, K., and Kansas, G. S. (2004) Nat. Rev. Immunol. 4 325-335 [DOI] [PubMed] [Google Scholar]
- 3.Ley, K., Laudanna, C., Cybulsky, M. I., and Nourshargh, S. (2007) Nat. Rev. Immunol. 7 678-689 [DOI] [PubMed] [Google Scholar]
- 4.Ivetic, A., and Ridley, A. J. (2004) Biochem. Soc. Trans. 32 1118-1121 [DOI] [PubMed] [Google Scholar]
- 5.Kahn, J., Walcheck, B., Migaki, G. I., Jutila, M. A., and Kishimoto, T. K. (1998) Cell 92 809-818 [DOI] [PubMed] [Google Scholar]
- 6.Ivetic, A., Deka, J., Ridley, A., and Ager, A. (2002) J. Biol. Chem. 277 2321-2329 [DOI] [PubMed] [Google Scholar]
- 7.Pavalko, F. M., Walker, D. M., Graham, L., Goheen, M., Doerschuk, C. M., and Kansas, G. S. (1995) J. Cell Biol. 129 1155-1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kilian, K., Dernedde, J., Mueller, E. C., Bahr, I., and Tauber, R. (2004) J. Biol. Chem. 279 34472-34480 [DOI] [PubMed] [Google Scholar]
- 9.Ivetic, A., Florey, O., Deka, J., Haskard, D. O., Ager, A., and Ridley, A. J. (2004) J. Biol. Chem. 279 33263-33272 [DOI] [PubMed] [Google Scholar]
- 10.Erlandsen, S. L., Hasslen, S. R., and Nelson, R. D. (1993) J. Histochem. Cytochem. 41 327-333 [DOI] [PubMed] [Google Scholar]
- 11.Green, C. E., Pearson, D. N., Camphausen, R. T., Staunton, D. E., and Simon, S. I. (2004) J. Immunol. 172 7780-7790 [DOI] [PubMed] [Google Scholar]
- 12.Giblin, P. A., Hwang, S. T., Katsumoto, T. R., and Rosen, S. D. (1997) J. Immunol. 159 3498-3507 [PubMed] [Google Scholar]
- 13.Hwang, S. T., Singer, M. S., Giblin, P. A., Yednock, T. A., Bacon, K. B., Simon, S. I., and Rosen, S. D. (1996) J. Exp. Med. 184 1343-1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ding, Z., Issekutz, T. B., Downey, G. P., and Waddell, T. K. (2003) Blood 101 4245-4252 [DOI] [PubMed] [Google Scholar]
- 15.Zarbock, A., and Ley, K. (2008) Am. J. Pathol. 172 1-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mattila, P. E., Green, C. E., Schaff, U., Simon, S. I., and Walcheck, B. (2005) Am. J. Physiol. 289 C323-C332 [DOI] [PubMed] [Google Scholar]
- 17.Urzainqui, A., Serrador, J. M., Viedma, F., Yanez-Mo, M., Rodriguez, A., Corbi, A. L., Alonso-Lebrero, J. L., Luque, A., Deckert, M., Vazquez, J., and Sanchez-Madrid, F. (2002) Immunity 17 401-412 [DOI] [PubMed] [Google Scholar]
- 18.Takai, Y., Kitano, K., Terawaki, S., Maesaki, R., and Hakoshima, T. (2008) J. Mol. Biol. 381 634-644 [DOI] [PubMed] [Google Scholar]
- 19.Hamada, K., Shimizu, T., Yonemura, S., Tsukita, S., and Hakoshima, T. (2003) EMBO J. 22 502-514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takai, Y., Kitano, K., Terawaki, S., Maesaki, R., and Hakoshima, T. (2007) Genes Cells 12 1329-1338 [DOI] [PubMed] [Google Scholar]
- 21.Pearson, M. A., Reczek, D., Bretscher, A., and Karplus, P. A. (2000) Cell 101 259-270 [DOI] [PubMed] [Google Scholar]
- 22.Edwards, S. D., and Keep, N. H. (2001) Biochemistry 40 7061-7068 [DOI] [PubMed] [Google Scholar]
- 23.Elshorst, B., Hennig, M., Forsterling, H., Diener, A., Maurer, M., Schulte, P., Schwalbe, H., Griesinger, C., Krebs, J., Schmid, H., Vorherr, T., and Carafoli, E. (1999) Biochemistry 38 12320-12332 [DOI] [PubMed] [Google Scholar]
- 24.Krieger, E., Darden, T., Nabuurs, S. B., Finkelstein, A., and Vriend, G. (2004) Proteins 57 678-683 [DOI] [PubMed] [Google Scholar]
- 25.Krieger, E., Nabuurs, S. B., and Vriend, G. (2003) Methods Biochem. Anal 44 509-523 [DOI] [PubMed] [Google Scholar]
- 26.Canutescu, A. A., and Dunbrack, R. L., Jr. (2005) Bioinformatics 21 2914-2916 [DOI] [PubMed] [Google Scholar]
- 27.Parsons, M., Monypenny, J., Ameer-Beg, S. M., Millard, T. H., Machesky, L. M., Peter, M., Keppler, M. D., Schiavo, G., Watson, R., Chernoff, J., Zicha, D., Vojnovic, B., and Ng, T. (2005) Mol. Cell. Biol. 25 1680-1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parsons, M., Messent, A. J., Humphries, J. D., Deakin, N. O., and Humphries, M. J. (2008) J. Cell Sci. 121 265-271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prag, S., Parsons, M., Keppler, M. D., Ameer-Beg, S. M., Barber, P., Hunt, J., Beavil, A. J., Calvert, R., Arpin, M., Vojnovic, B., and Ng, T. (2007) Mol. Biol. Cell 18 2935-2948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nunomura, W., Takakuwa, Y., Parra, M., Conboy, J. G., and Mohandas, N. (2000) J. Biol. Chem. 275 6360-6367 [DOI] [PubMed] [Google Scholar]
- 31.Deisseroth, K., Heist, E. K., and Tsien, R. W. (1998) Nature 392 198-202 [DOI] [PubMed] [Google Scholar]
- 32.Yonemura, S., and Tsukita, S. (1999) J. Cell Biol. 145 1497-1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen, C., Ba, X., Xu, T., Cui, L., Hao, S., and Zeng, X. (2006) J. Biochem. 140 229-235 [DOI] [PubMed] [Google Scholar]
- 34.Brown, M. J., Nijhara, R., Hallam, J. A., Gignac, M., Yamada, K. M., Erlandsen, S. L., Delon, J., Kruhlak, M., and Shaw, S. (2003) Blood 102 3890-3899 [DOI] [PubMed] [Google Scholar]
- 35.Ivetic, A., and Ridley, A. J. (2004) Immunology 112 165-176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hishiya, A., Ohnishi, M., Tamura, S., and Nakamura, F. (1999) J. Biol. Chem. 274 26705-26712 [DOI] [PubMed] [Google Scholar]
- 37.Renkonen, J., Tynninen, O., Hayry, P., Paavonen, T., and Renkonen, R. (2002) Am. J. Pathol. 161 543-550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosen, S. D. (2004) Annu. Rev. Immunol. 22 129-156 [DOI] [PubMed] [Google Scholar]
- 39.Galkina, E., Kadl, A., Sanders, J., Varughese, D., Sarembock, I. J., and Ley, K. (2006) J. Exp. Med. 203 1273-1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ilani, T., Khanna, C., Zhou, M., Veenstra, T. D., and Bretscher, A. (2007) J. Cell Biol. 179 733-746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dwir, O., Kansas, G. S., and Alon, R. (2001) J. Cell Biol. 155 145-156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dwir, O., Steeber, D. A., Schwarz, U. S., Camphausen, R. T., Kansas, G. S., Tedder, T. F., and Alon, R. (2002) J. Biol. Chem. 277 21130-21139 [DOI] [PubMed] [Google Scholar]
- 43.Evans, S. S., Schleider, D. M., Bowman, L. A., Francis, M. L., Kansas, G. S., and Black, J. D. (1999) J. Immunol. 162 3615-3624 [PubMed] [Google Scholar]
- 44.Li, X., Steeber, D. A., Tang, M. L., Farrar, M. A., Perlmutter, R. M., and Tedder, T. F. (1998) J. Exp. Med. 188 1385-1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brenner, B., Gulbins, E., Schlottmann, K., Koppenhoefer, U., Busch, G. L., Walzog, B., Steinhausen, M., Coggeshall, K. M., Linderkamp, O., and Lang, F. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 15376-15381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bos, J. L. (2005) Curr. Opin. Cell Biol. 17 123-128 [DOI] [PubMed] [Google Scholar]
- 47.Villalonga, P., Lopez-Alcala, C., Bosch, M., Chiloeches, A., Rocamora, N., Gil, J., Marais, R., Marshall, C. J., Bachs, O., and Agell, N. (2001) Mol. Cell. Biol. 21 7345-7354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Orian-Rousseau, V., Morrison, H., Matzke, A., Kastilan, T., Pace, G., Herrlich, P., and Ponta, H. (2007) Mol. Biol. Cell 18 76-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hao, J. J., Wang, G., Pisitkun, T., Patino-Lopez, G., Nagashima, K., Knepper, M. A., Shen, R. F., and Shaw, S. (2008) J. Proteome Res. 7 2911-2927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sebzda, E., Bracke, M., Tugal, T., Hogg, N., and Cantrell, D. A. (2002) Nat. Immunol. 3 251-258 [DOI] [PubMed] [Google Scholar]
- 51.Hidalgo, A., Peired, A. J., Wild, M. K., Vestweber, D., and Frenette, P. S. (2007) Immunity 26 477-489 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.