

Abstract
Mammalian somatic growth is rapid in early postnatal life but then slows and eventually ceases in multiple tissues. We hypothesized that there exists a postnatal gene expression program that is common to multiple tissues and is responsible for this coordinate growth deceleration. Consistent with this hypothesis, microarray analysis identified more than 1600 genes that were regulated with age (1 vs. 4 wk) coordinately in kidney, lung, and heart of male mice, including many genes that regulate proliferation. As examples, we focused on three growth-promoting genes, Igf2, Mest, and Peg3, that were markedly down-regulated with age. In situ hybridization revealed that expression occurred in organ-specific parenchymal cells and suggested that the decreasing expression with age was due primarily to decreased expression per cell rather than a decreased number of expressing cells. The declining expression of these genes was slowed during hypothyroidism and growth inhibition (induced by propylthiouracil at 0–5 wk of age) in male rats, suggesting that the normal decline in expression is driven by growth rather than by age per se. We conclude that there exists an extensive genetic program occurring during postnatal life. Many of the involved genes are regulated coordinately in multiple organs, including many genes that regulate cell proliferation. At least some of these are themselves apparently regulated by growth, suggesting that, in the embryo, a gene expression pattern is established that allows for rapid somatic growth of multiple tissues, but then, during postnatal life, this growth leads to negative-feedback changes in gene expression that in turn slow and eventually halt somatic growth, thus imposing a fundamental limit on adult body size.
A complex genetic program occurs in multiple tissues postnatally and involves pathways that regulate cell proliferation, suggesting a role in postnatal somatic growth deceleration.
During mammalian embryogenesis, complex changes in gene expression are required for organ formation and development (1,2). These embryonic genetic programs have been studied intensively. However, mammalian development does not stop abruptly at birth; in postnatal life maturation continues in multiple organs (3,4). The genetic mechanisms required for postnatal development have received relatively little attention.
In particular, little is known about the mechanisms required to slow somatic growth postnatally. Mammalian body growth is rapid in early postnatal life, but then the growth rate slows, eventually declining toward zero, as the organism approaches its adult body size. This decline in growth rate is due primarily to a decrease in the rate of proliferation (5). One clue regarding the underlying mechanism is that the growth deceleration in different organs appears to be coordinated temporally, evolutionarily, and conditionally. Growth of different organs is coordinated temporally in that all major organs grow rapidly in prenatal and early postnatal life, but then the growth rates decline toward zero with age. Growth of different organs is also coordinated evolutionarily. In small mammals, such as the mouse, this decline occurs over weeks, whereas in larger mammals, such as the cow or human, the decline occurs over years, thus producing the greater body size. To maintain body proportions, growth of multiple organs must be modulated coordinately during evolution. Finally, growth of different organs is coordinated conditionally. Various growth-inhibiting conditions, such as malnutrition, hypothyroidism, and GH deficiency, each of which involve different mechanisms, appear to slow growth coordinately in multiple organs, although the magnitude of the growth inhibition in different organs is not uniform (6,7). Furthermore, if the growth-inhibiting condition resolves, then the growth of multiple organs does not just return to a normal rate but actually exceeds normal, a phenomenon known as catch-up growth (8).
One possible explanation for coordinated growth of different organs is that growth deceleration might be orchestrated by a hormonal or other systemic mechanism. However, there is evidence that the temporal coordination of growth is not due to a systemic mechanism. For example, in the late adolescent human, as the somatic growth rate approaches zero, GH levels and circulating IGF-I levels are higher than in the infant when growth is very rapid (9), nor do the levels of thyroid hormone, glucocorticoids, or other growth-regulating hormones change in a pattern that would account for this cessation of growth. Furthermore, transplantation experiments suggest that the growth rate of transplanted structures generally depends on the age of the donor animal, rather than the age of the recipient, again arguing against a hormonal or other systemic mechanism (10,11).
A second possible explanation for this coordination is that growth deceleration is directed by a program of gene expression that is common to multiple tissues. This putative, common mechanism might then be modulated by growth-regulating environmental conditions or genetically, through evolutionary changes, thus maintaining coordinate, proportional growth.
Based on this reasoning, we hypothesized that there would exist complex changes in gene expression during postnatal life. Some of these changes might be responsible for organ-specific maturation; these changes might be unique to each organ. In addition, we hypothesized that there exists a more general gene expression program, occurring during early postnatal life, which is common to multiple organs and is responsible for coordinate somatic growth deceleration. To test this hypothesis, we performed microarray analysis to identify changes in gene expression during early postnatal life. We particularly focused our attention on genes that were up- or down-regulated in multiple organs and thus are more likely to contribute to the putative common program of growth deceleration. Based on this analysis, we previously showed evidence that many of the genes in the Zac1-imprinted network (12), including Mest, Peg3, and Igf2, are down-regulated at the mRNA level postnatally (13). In the current study, we analyzed the microarray data more globally. We then performed in situ hybridization to determine which cells within the organs were expressing particular genes of interest. Finally, we asked whether the observed changes in gene expression were driven by time per se or growth of the organs by determining whether growth retardation induced by hypothyroidism slowed these changes in gene expression.
Materials and Methods
Animal care
All animals were maintained and used in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council 2003) and received standard rodent chow (Zeigler Bros, Gardners, PA) and water ad libitum.
Mice
C57BL/6 male mice (Charles River Laboratories, Wilmington, MA) were killed by carbon dioxide asphyxiation at the ages of 1, 4, or 8 wk (n = 10/group). These time points were selected to analyze the changes in gene expression that occur as somatic growth decelerates; growth in body mass is rapid at 1 wk, slower at 4 wk, and even slower at 8 wk of age (14). Because we wanted to focus on the mechanisms causing growth deceleration and not on the consequences of sex steroid action, mice killed at 4 and 8 wk were castrated at 3 wk of age (15). Tail length and total body and organ mass were measured after the animals were killed. The protocol was approved by the Animal Care and Use Committee, National Institute of Child Health and Human Development (NICHD), National Institutes of Health.
Tissue collection and processing
After the animals were killed, kidney, liver, heart, and lung were rapidly excised for processing. For RNA extraction, liver, heart, lung, and kidney were homogenized in TRIzol reagent (Invitrogen, Carlsbad, CA) on ice and stored at −80 C. For in situ hybridization, tissue were rapidly removed and frozen on dry ice. Lungs were inflated via trachea with 5 ml of optimal cutting temperature compound (Sakura Finetek USA, Torrance, CA) diluted 1:1 with PBS before freezing (16).
RNA extraction and purification
Total RNA was extracted using TRIzol reagent according to the manufacturer's instructions (Invitrogen) followed by RNeasy minikit purification (QIAGEN, Valencia, CA) according to the manufacturer's instructions. RNA concentration was determined by spectrophotometry at an absorbance of 260 nm. All RNA samples had a 260:280 nm ratio between 1.9 and 2.1. RNA integrity was determined using an Agilent 2100 bioanalyzer (Agilent Technologies, Santa Clara, CA) and only high-quality RNA (28S/18S >1.8) was used for further analysis.
Microarray analysis
Two micrograms of RNA per sample were processed and analyzed by the National Institute of Diabetes and Digestive and Kidney Diseases Core Facility at the National Institutes of Health. Affymetrix Mouse Genome 430 2.0 Array GeneChips (34,000 transcripts) were used (Affymetrix, Santa Clara, CA). Five chips were used for each organ (kidney, heart, and lung) at each time point (1, 4, and 8 wk of age), except that an 8-wk time point was not performed in heart because proliferation of cardiac myocytes has ceased by 4 wk (17). Each microarray chip was hybridized to labeled RNA derived from a single animal. Microarray signals were analyzed using the Affymetrix MAS5 algorithm. Up- and down-regulated genes were selected based on P < 0.01 as assessed by ANOVA using Partek Pro software (Partek, St. Charles, MO).
Bioinformatic analysis
Ingenuity Pathways Analysis Software 5.0 (Ingenuity Systems Inc., Redwood City, CA) was used to identify canonical pathways that were regulated similarly in the three tissues. These pathways represent sets of genes that participate in a common well-defined biological function. The analysis included genes that showed either an increase or decrease in expression with P < 0.01 by ANOVA. Heat maps were then constructed using JMP 7 software (SAS Institute Inc., Cary, NC) to visualize changes in expression of genes participating in specific canonical pathways.
Real-time quantitative RT-PCR for mouse tissues
Real-time RT-PCR was used to assess specific mRNA levels at 1, 4, and 8 wk in kidney, liver, lung, and heart (n = 5 animals per time point). Total RNA (100–200 ng) was reverse transcribed using Superscript III reverse transcriptase (Invitrogen) according to the manufacturer's instructions.
The resulting cDNA solution was diluted 10–25 times and stored at −20 C for later use. Quantitative real-time PCR was performed for paternally expressed 3 (Peg3), mesoderm-specific transcript (Mest), Igf2, SRY-box containing gene 4 (Sox4), and Igf2bp3 using the following assays containing primers and specific intron-spanning FAM or VIC-labeled (18S rRNA) TaqMan probes (Applied Biosystems, Foster City, CA): Peg3, Mm01337379_m1; Igf2, Mm00439564_m1; Mest, Mm00484993_m1; Sox4, Mm00486317_s1; Igf2bp3, Mm00502742_m1; and 18S rRNA, 4319413E. Reactions were performed in triplicate on cDNA derived from each animal according to the manufacturer's instructions, using the ABI Prism 7000 sequence detection system instrument (Applied Biosystems) according to the manufacturer's instructions. For Sox4, the primers did not span an intron. However, omission of reverse transcriptase from the procedure did not result in significant PCR amplification from genomic DNA (19). The relative quantity of each mRNA was calculated using the formula: relative expression = 2–ΔCT × 106, where CT represents the threshold cycle and ΔCT = (CT of gene of interest) − (CT of 18S rRNA). Values were multiplied by 106 for convenience of comparison (20).
Generation of riboprobes for in situ hybridization
In situ hybridization was performed for Peg3, Mest, Igf2bp3, Igf2, and Sox4 in kidney, liver, lung, and heart. To generate templates for transcription of RNA probes, cDNA from 1-wk-old mouse liver was amplified using Gene AmpliTaq Gold PCR reagent kit (Applied Biosystems) and primers (supplemental Table 1, published as supplemental data on The Endocrine Society's Journals Online web site at http://endo.endojournals.org), which contained either a T7 promoter or an SP6 promoter: T7 promoter for sense probes, TAATACGACTCACTATAGGGAG, or Sp6 promoter for antisense probes, TGGATTTAGGTGACACTATAGAAG (21).
The cDNA was PCR amplified using a 2720 thermal cycler (Applied Biosystems) at the following thermal cycling conditions: 95 C for 7 min, followed by 40 cycles of 95 C for 20 sec, 55 C for 20 sec, 72 C for 40 sec, followed by a final hold of 72 C for 7 min. A second amplification was performed with the same parameters using 35 instead of 40 cycles. The PCR product was confirmed by a single band of the expected size by agarose gel electrophoresis.
Single-stranded riboprobes for in situ hybridization were transcribed using an in vitro transcription systems (Promega, Madison, WI) incorporating α-35S-uridine 5-triphosphate (UTP) and α-35S CTP (800 Ci/mmol, GE HealthCare, Piscataway, NJ). T7 polymerase was used for sense probes and Sp6 polymerase for antisense probes. Riboprobes were purified by Micro Bio-Spin Columns P-30 in Tris buffer RNase-free (Bio-Rad Laboratories, Hercules, CA). 35S incorporation was assessed by comparing radioactivity before and after the column purification by liquid scintillation counting.
In situ hybridization
In situ hybridization was performed using frozen sections hybridized to 35S-labeled riboprobes as described by Zhou et al. (22). The sections were counterstained with hematoxylin and eosin. Silver grains were visualized by polarized epifluorescence microscopy combined with bright-field microscopy. The corresponding sense riboprobe was used as a negative control for each antisense probe.
Hypothyroid rat model
Male Sprague Dawley rats (Harlan, Indianapolis, IN) were made hypothyroid by introducing 1 g/liter propylthiouracil (PTU; Sigma-Aldrich, St. Louis, MO) at 1 d of age into the mother's drinking water (8,23). The PTU-containing water was changed twice per week. PTU treatment was discontinued at 5 wk of age. The PTU-treated animals were not weaned during the experiment because preliminary studies showed that they were not mature enough to support their own nutritional needs. Untreated animals served as concurrent controls and were weaned at the age of 3 wk. All rats were given depot leuprolide acetate (6 mg/kg, sc; Tap Pharmaceuticals, Deerfield, IL) every 3 wk, starting at 3 wk of age. Leuprolide acetate is a long-acting GnRH agonist that down-regulates LH and FSH secretion and thus gonadal steroid production (18). PTU-treated and control animals were killed at 5, 7, and 9 wk of age (n = 8–15/group). Additional control animals were killed at 3 wk of age. Heart, liver, and kidney were obtained at the time the animals were killed and frozen at −80 C for later processing.
We chose to study rats because there was a well-established model of growth retardation using PTU in the juvenile rat (8,23). We were unable to find an equivalent model that had been previously validated using juvenile mice. The time points used for rats (3, 5, 7, and 9 wk of age) are similar to the time points used for mice (1, 4, and 8 wk of age). Rats may mature slightly more slowly than mice in terms of age at birth (24,25) and sexual maturation in males (26,27), although not in females (28,29).
Real-time quantitative RT-PCR for rat tissues
Real-time RT-PCR was performed as described for mouse tissues except that the following assays (Applied Biosystems) were used: Igf2, Rn01454118_m1; Mest, Rn01500324_m1; Igf2bp3, Rn01410012_m1; and 18S rRNA, 4319413E. Custom assays (part no. 4332078 and sales order no. 2331931) were designed for Sox4 and Peg3. The assay IDs (Applied Biosystems) are: rat Peg3, CDS-5 × 6; rat Sox4, CD-ANY.
Results
Extensive changes in gene expression occur during the period of postnatal growth
In normal mice at 1, 4, and 8 wk of age microarray analysis was used to identify temporal changes in gene expression (defined by P < 0.01 at 1 vs. 4 wk of age, ANOVA) in kidney, lung, and heart.
More than 2000 genes were found to be significantly up-regulated with age in each organ and more than 2800 genes were found to be down-regulated (Fig. 1).
Figure 1.
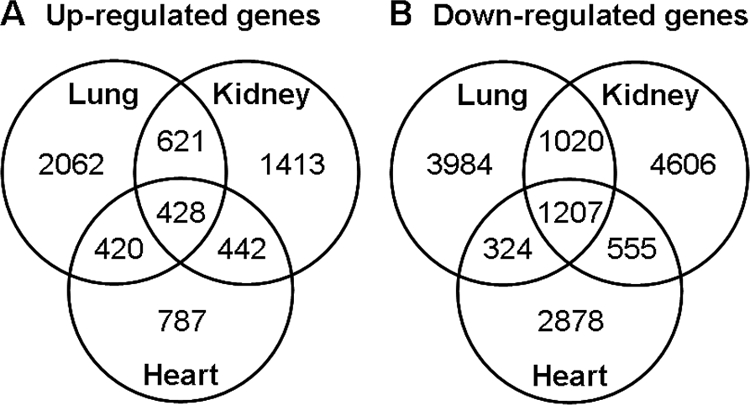
Venn diagrams showing the number of genes significantly up-regulated (A) and down-regulated (B) with age in mouse kidney, lung, and heart. RNA was isolated from mouse kidney and lung at 1, 4, and 8 wk of age and heart at 1 and 4 wk of age and analyzed by expression microarray to identify temporal changes in gene expression, defined by P < 0.01 (ANOVA).
Many genes show a temporal pattern of expression that is common to multiple organs
The number of genes that were up- or down-regulated in more than one organ was much higher than would be expected by chance if regulation in different organs were independent events. For each pair of organs, more than 800 genes were up-regulated in both and more than 1500 genes were down-regulated in both (P < 0.001, Pearson χ2 test, Fig. 1, A and B). There was an even more striking tendency for concordant regulation in all three organs studied; 1207 genes were down-regulated in all three organs (supplemental Table 2) and 428 were up-regulated in all three organs (supplemental Table 3), again far more than would be expected by chance (P < 0.001). These findings suggest that there is an extensive program of gene expression common to multiple organs during the period of postnatal growth, in addition to the expected organ-specific changes.
We next examined changes in gene expression within specific recognized functional pathways as defined by Ingenuity Pathways Analysis software. In some of these pathways, particularly the G1/S checkpoint regulation pathway, G2/M DNA damage checkpoint regulation pathway, Sonic Hedgehog signaling, and Wnt/β-catenin signaling, there was a similar pattern of temporal changes in the three organs studied, with a far greater number of genes that were down-regulated than up-regulated with age (Fig. 2).
Figure 2.

Temporal changes in expression of genes participating in specific functional pathways. Microarray analysis was used to evaluate changes in gene expression in lung and kidney of 1-, 4-, and 8-wk-old mice and in heart of 1- and 4-wk-old mice. Ingenuity Pathways Analysis software was used to analyze changes in functional pathways as defined by that software package. Changes within a pathway were then visualized using heat maps constructed using JMP software version 7 (SAS Institute). To depict temporal changes in gene expression, the microarray values for each organ were normalized to the values for the same organ at 1 wk of age. Black rectangles represent no change in gene expression. Green rectangles represent down-regulated genes, and red rectangles represent up-regulated genes compared with 1-wk-old animals. The color intensity corresponds to the magnitude of the change from baseline, expressed as log2 (value at time point/value at 1 wk). Genes showing significant changes in expression with age (P < 0.05) in all three organs are depicted. The canonical pathways shown here are: G1/S check point regulation, G2/M DNA damage check point regulation, Sonic Hedgehog signaling, and Wnt/β-catenin signaling.
Igf2, Mest, Peg3, Sox4, and Igf2bp3 mRNA expression declines with age in multiple organs
We next focused our attention on genes that were up- or down-regulated in all three organs and thus more likely to contribute to the putative common program of growth deceleration. Of the genes that were markedly down-regulated in all three organs, we selected 5 representative genes for more detailed characterization. Three of these, Peg3, Mest, and Igf2, have been implicated in somatic growth regulation; targeted ablation of each produces prenatal growth retardation (30,31,32,33,34,35,36). Thus, we reasoned that the observed decline in gene expression of these three genes during early postnatal life might contribute to the somatic growth deceleration that occurs postnatally. In contrast, the other two genes, Sox4 and Igf2bp3 have not been implicated in growth regulation. Sox4 is required for normal embryonic formation of the heart (32). The physiological function of Igf2bp3 is not known; however, its expression reaches a peak at embryonic d (E)12 and then decreases markedly (33), suggesting that it too functions primarily during embryonic development.
To better quantify the temporal changes in gene expression identified by microarray, we measured mRNA expression levels by real-time PCR in the three original organs studied and, additionally, in liver (Fig. 3). From 1 to 4 wk of age, Mest mRNA levels declined in kidney (23-fold, P < 0.001), lung (10-fold, P < 0.001), liver (13-fold, P < 0.001), and heart (13-fold, P < 0.002). From 4 to 8 wk of age, Mest expression did not change significantly.
Figure 3.

Age-related changes in mRNA expression of Mest, Peg3, Igf2, Igf2bp3, and Sox4 in kidney, lung, liver, and heart of 1-, 4-, and 8-wk-old mice. The relative expression of each mRNA was analyzed by real-time PCR, normalizing to 18S RNA. P values in upper right corner of each panel show overall change in expression with age (ANOVA).
Declines in expression of Peg3, Igf2bp3, Igf2, and Sox4 were also observed in multiple organs with age (Fig. 3). Thus, the real-time PCR results confirmed the marked declines in mRNA expression with age in kidney, lung, and heart. The fact that expression of all five genes also declined markedly in liver provides further evidence that the declines are part of a widespread genetic program occurring in many organs simultaneously.
Igf2, Mest, and Peg3 are expressed in parenchymal cells
We next considered the possibility that expression of these genes was declining in multiple organs because these genes were expressed primarily by a cell type common to multiple organs, e.g. endothelial cells or stromal fibroblasts. To test this possibility, we performed in situ hybridization for each of these genes in kidney, liver, heart, and lung from 1-wk-old mice.
We did not find that expression was restricted to cells that were common to multiple organs. Instead, expression was generally found in organ-specific parenchymal cells. For example, in liver, Mest was expressed by hepatocytes, showing a strong, inhomogeneous signal (signal intensity varied widely among different hepatocytes, Fig. 4). In kidney, Mest expression was particularly expressed in the glomeruli and at lower levels in the tubular epithelium. In heart, Mest mRNA signal was located within cardiac myocytes and in lung was found in pneumocytes (Fig. 4).
Figure 4.

mRNA expression of Igf2, Mest, and Peg3 in kidney, liver, heart, and lung of 1-wk-old mice by in situ hybridization. Frozen sections were hybridized to 35S-labeled riboprobes. The corresponding sense riboprobe was used as a negative control for each antisense probe. Silver grains were visualized by polarized epifluorescence microscopy combined with bright-field microscopy and appear as green fluorescent dots on a hematoxylin- and eosin-stained background: G, Glomerulus; V, cardiac valve; M, myocyte.
Similarly, Igf2 signal was observed in all organs studied with the following distribution: in kidney, signal was found in the glomeruli, in liver in hepatocytes with a homogeneous pattern, in heart expression was primarily in endocardial cells lining cardiac valves (37), and in lung, expression was observed in pneumocytes (Fig. 4).
Peg3 mRNA expression showed a similar distribution with signal observed in kidney, mainly in the glomeruli; in liver, mainly in hepatocytes; in heart in myocytes and endocardial cells covering cardiac valves; and in lung the signal was inhomogeneous and localized in pneumocytes (Fig. 4). For each gene studied, a corresponding sense probe was used as negative control and showed only a low-level background pattern (Fig. 4).
For Sox4 and Igf2bp3, we were unable to obtain a convincing signal by in situ hybridization, despite trying multiple different probes, presumably because mRNA expression levels are so much lower for these genes (data not shown).
The decrease in Peg3, Mest, and Igf2 expression involves a decrease in expression per cell
We next asked whether the declines in expression levels for these genes represented a decrease in the average expression per cell or a decrease in the number of cells expressing the gene. If the second hypothesis were correct, then in situ hybridization at 4 wk of age should have revealed a small number of cells that express the genes at high levels. However, this possibility was not supported by the data; at 4 wk of age, mRNA expression by in situ hybridization was uniformly low for all genes in all organs (data not shown). Therefore, the findings suggest that average expression per cell declines with age.
Hypothyroidism slows the decline in expression of Igf2, Peg3, and Mest
We next asked whether the observed changes in gene expression are a function of time per se or a function of growth. For growth-regulating genes in particular, we hypothesized that the changes in expression might be driven by growth, not age. This hypothesis predicts that a period of growth inhibition would slow the observed changes in gene expression.
To test that prediction, we studied rats that had undergone a period of growth retardation caused by hypothyroidism, which had been induced by adding propylthiouracil to the drinking water of the mother beginning on the first day of life. At the end of the 5-wk treatment period, organ weight was decreased in animals that had received PTU compared with control animals (kidney, 160 ± 3 vs. 552 ± 17 mg, mean ± sem, P < 0.001; liver, 1230 ± 25 vs. 6630 ± 330 mg, P < 0.001; heart, 158 ± 4 vs. 598 ± 26 mg, P < 0.001).
As expected, in control animals, which did not receive PTU, Igf2 mRNA levels declined significantly with age in all the organs studied: kidney, liver, and heart (each P < 0.001, Fig. 5). In animals that had previously received PTU, Igf2 mRNA expression during the recovery period (7–9 wk of age) was increased relative to controls in all three organs: kidney (P < 0.001), liver (P = 0.02), and heart (P < 0.001, Fig. 5), consistent with the prediction that growth retardation by PTU slowed the programed decline in gene expression. At 5 wk of age, the mRNA levels might reflect not only this maturational delay but also the effects of concurrent hypothyroidism, and, indeed, the net effect on expression, compared with control animals, was variable (Fig. 5).
Figure 5.

Effect of hypothyroidism on the decline in gene expression of Igf2, Mest, Peg3, Igf2bp3, and Sox4. Male rats were made hypothyroid by adding PTU to the drinking water of the mother from 1 d until 5 wk of age and then were allowed to recover from the hypothyroidism and from the resulting growth inhibition. Untreated animals (open symbols) served as concurrent controls. For most of the genes, in most organs, mRNA expression declined with age in control animals. After the treatment period, the animals that had previously received PTU (closed symbols) showed an apparent delay in many of these declines, particularly for Igf2, Mest, and Peg3. a, Effect of age on expression in control animals; b, effect of previous treatment on expression during recovery period (wk 7–9). NS, Not significant.
Similarly, Mest mRNA levels in control animals declined significantly with age in the kidney (P < 0.001) and liver (P < 0.001) (Fig. 5). In heart, mRNA levels changed significantly with age but did not show a simple, progressive decline. Although this difference from the findings in the mouse heart could be related to species differences between rats and mice, we think it more likely reflects the fact that the mouse findings included data from 1 wk of age, which may have helped detect early declines in expression. For the two organs in which Mest expression declined with age in control rats, PTU caused an apparent delay in this decline: kidney (P < 0.001) and liver (P = 0.001) (Fig. 5).
Peg3 mRNA levels showed a similar pattern of regulation. In control animals, mRNA levels declined significantly with age in kidney (P < 0.001) and heart (P < 0.001) but not liver (Fig. 5). For the two organs in which Peg3 expression declined with age in control animals, PTU caused an apparent delay in this decline: kidney (P = 0.01) and heart (P = 0.03) (Fig. 5).
This general pattern of decline with age in control animals and delayed decline in animals that previously received PTU was not observed consistently for Igf2bp3 or Sox4 (Fig. 5), the two genes that we studied but have not been implicated in growth regulation. For Igf2bp3, this pattern was observed only in kidney and for Sox4 in none of the organs studied.
Discussion
Using expression microarray analysis, we found that extensive changes in gene expression occur with age in mice during the period of postnatal growth. In each of the organs studied, kidney, lung, and heart, more than 2000 genes were found to be significantly up-regulated with age and more than 2800 genes down-regulated. The number of genes that were regulated coordinately in all three organs was strikingly high, with 1207 genes down-regulated in all three organs and 428 up-regulated in all three organs, far more overlap than would be expected by chance, suggesting that there is an extensive program of gene expression common to multiple organs during postnatal growth, in addition to the expected tissue-specific changes. The common program included genes involved in regulating G1/S and G2/M checkpoints, Hedgehog signaling, and Wnt/β-catenin signaling. There were more genes that were down-regulated with age than were up-regulated in these pathways. The common program of gene expression was observed in organs of widely differing embryonic origin, specifically, kidney and heart, which are derived from mesoderm, and lung and liver, which are derived from endoderm.
We next focused our attention on genes that were up- or down-regulated in all three organs and thus more likely to contribute to the putative common program of growth deceleration. We selected five genes, Igf2, Mest, Peg3, Sox4, and Igf2bp3, that were markedly down-regulated in all three organs for more detailed characterization. First, to better quantify the temporal changes in gene expression identified by microarray, we measured mRNA expression levels by real-time PCR in the same three organs studied by microarray. The real-time PCR results confirmed the marked declines in mRNA expression with age. We also measured mRNA levels in a fourth organ, liver, and found dramatic declines with age, very similar in pattern to the declines found in heart, kidney, and lung, further confirming that this gene expression program is occurring globally in multiple organs. Our findings using real-time PCR are consistent with previous evidence that expression of some of these genes declines postnatally in specific tissues (13,20,38,39).
We chose to study lung, heart, and kidney by microarray and, in addition, liver by real-time PCR as examples of organs in which proliferation decreases with age, as opposed to other tissues, such as intestinal epithelium, epidermis, and hematopoietic tissue, in which proliferation continues into adulthood. Even in the organs studied, the decline in proliferation rate is not completely uniform. In renal tubular epithelial cells and hepatocytes, proliferation declines with a similar pattern after 2 wk of age (5). In cardiomyocytes, earlier studies in rats suggested that proliferation continues beyond 3 wk of age, although at a decreasing rate (40,41). However, more recent studies in rats and mice suggest that proliferation ceases close to the time of birth and is followed by approximately 2 wk of binucleation and hypertrophy without cytokinesis (17,40). In lung, the decline in proliferation appears to occur earlier than in kidney or liver (41,42).
The observed expression patterns may differ in different species. For example, in the human, IGF2 is expressed in many adult tissues, although, at least in kidney and liver, its expression is considerably lower than in fetal tissues (43,44). Similarly, in this study, only male rats were studied to reduce the number of variables. Consequently, we do not know whether the results generalize to females.
We next used in situ hybridization to determine which cell types in each organ expressed Igf2, Mest, and Peg3 in the 1-wk-old mouse. In general, we found expression in organ-specific parenchymal cells. These findings suggest that the coordinate decline in expression in multiple organs is not occurring simply because gene expression is restricted to cells that are common to multiple organs, such as endothelial cells or stromal fibroblasts. Instead, expression of these genes appears to be declining coordinately in multiple different cell types throughout the body, suggesting that a common genetic program is occurring in many tissues simultaneously.
In situ hybridization was next performed in tissues from 4-wk-old mice. We had considered the possibility that some cells might continue to show high levels of expression at 4 wk of age but that the number of these expressing cells might be smaller than at 1 wk of age, thus explaining the overall declines in expression observed by microarray and real-time PCR. Such a finding would support the hypothesis that, with age, many cells stop expressing these growth-promoting genes and thus perhaps drop out of the cell cycle, whereas a dwindling number continues to express these genes and continue to proliferate. However, our findings did not support that hypothesis. Instead, we found that, at 4 wk of age, mRNA expression was uniformly low for all genes studied in all organs, indicating that the overall decrease in Igf2, Mest, and Peg3 expression was due in large part to decreased expression per cell rather than simply a decrease in the number of expressing cells.
Igf2, Mest, and Peg3 are particularly good candidates for genes that might be responsible for postnatal growth deceleration. Igf2 encodes an IGF. IGF-II acts primarily in a paracrine fashion, binding to the type 1 IGF receptor. The liganded receptor acts as a tyrosine kinase to potently stimulate proliferation in many cell types, in vivo and in vitro (45,46). Targeted ablation of Igf2 in mice leads to marked growth retardation with a body mass decreased to 60% of normal at birth. Postnatal growth maintains the body mass at 60% of normal (31,36). Thus, ablation primarily affects somatic growth in early life, which fits the finding that Igf2 expression occurs only during early life and declines thereafter. Conversely, overexpression of Igf2 in vivo leads to increased somatic growth (47). Mest encodes a presumptive α/β-fold hydrolase, with unknown substrates (48,49). Targeted ablation causes embryonic growth retardation associated with reduced postnatal survival rates and abnormal maternal behavior (48). Conversely, in mouse interspecies hybrids, loss of imprinting of Mest is associated with increased fetal somatic growth (34). Peg3 encodes a nuclear protein that contains 12 Kruppel-type zinc finger domains and two proline-rich repeat domains and that is thought to participate in apoptotic pathways. Targeted ablation of Peg3 produces mice with body mass 85% that of wild-type at E17.5, 81% at birth, and 65% by 4 wk of age. The major organs in the mutants, although proportionally smaller, are morphologically normal. These mice also exhibited abnormal maternal behavior (50). Thus, Igf2, Mest, and Peg3 are each required for normal embryonic growth, suggesting that the decline in expression during postnatal life may contribute to postnatal growth deceleration. Three of the genes studied, Mest, Peg3, and Igf2, reportedly belong to the Zac1 imprinted gene network (12). Based on the expression microarray data described in the current study, we previously showed evidence that many of the genes in this network, including Mest, Peg3, and Igf2, are down-regulated at the mRNA level postnatally (13). In the current study, we analyzed the microarray data more globally, and we also examined representative genes more extensively using in situ hybridization and the hypothyroid model of growth retardation.
In contrast, Sox4 and Igf2bp3 have not been implicated in somatic growth regulation but rather appear to be involved in embryogenesis. Sox4 is a member of the SOX family of transcription factors, which contain an HMG domain (51). Sox4 is expressed in the mouse embryo in the mesenchyme of the branchial arches, trachea and esophagus (32,52), nervous system (53), and embryonic growth plate (54). Targeted ablation of Sox4 results in embryos that die at E14 of cardiac valvular insufficiency (32). Sox4 also appears to be required for lymphocyte differentiation (51), and it also may play a role in chondrogenesis (55). Igf2bp3 is a member of a family of proteins that bind specific mRNAs including the 5′ untranslated region of Igf2 mRNA. Igf2bp3 seems to be involved in tumorigenesis and embryonic development. Peak expression occurs during mouse embryonic development at E12.5 and decreases thereafter (33). Thus, Sox4 and Igf2bp3 appear to be involved in embryonic development, and therefore, the postnatal declines in expression of these two genes observed in the present study may represent the vestigial remnant of expression of genes that are required during embryogenesis and are down-regulated thereafter.
Finally, we sought to determine whether the observed changes in gene expression are a function of time per se or a function of growth. In rats, PTU-induced hypothyroidism and growth retardation during the first 5 wk of life delayed the declines in expression of Igf2, Mest, and Peg3, the three genes implicated in growth regulation. These data are consistent with the hypothesis that the normal decline in expression of these genes is driven, not by age per se, but rather by the process of growth, and therefore, a prior period of growth inhibition slows this decline.
However, several issues confound this interpretation. Thyroid hormone has direct and indirect effects on the expression of many genes, and it is possible that these effects may not all reverse when the PTU is discontinued. For example, the thyroid hormone levels may not have fully recovered promptly after discontinuation of PTU. These levels were not measured in the current study, but previous studies using PTU treatment in young rats suggest that some hypothyroidism may persist for weeks after drug discontinuation (8). Persistent hypothyroidism could therefore have influenced gene expression, even at 9 wk of age, i.e. 4 wk after discontinuation of PTU. However, some of the observed differences in gene expression (i.e. Igf2, Mest, and Peg3 in heart and Mest in liver) at 7 and 9 wk had not been present at 5 wk when the hypothyroidism was presumably more severe, suggesting that the observed differences at 7 and 9 wk are not simply due to incomplete recovery but rather to the prior history of hypothyroidism. An additional complicating issue is that hypothyroidism might cause impaired lactation or suckling, and thus, the observed growth inhibition may have reflected both the direct effects of thyroid hormone deficiency and the indirect effects of malnutrition. Because of these complicating issues, to confirm our hypothesis that the changes in gene expression are driven by growth, it would be important to determine whether growth inhibition caused by conditions other than hypothyroidism also delays the changes in gene expression.
For the genes not implicated in growth regulation, Igf2bp3 and Sox4, hypothyroidism generally did not slow the decline in gene expression. Thus, the findings suggest that at least some genes that regulate growth are themselves regulated by growth, whereas the decline in expression of genes not involved in growth regulation may not driven by growth but by age per se or other developmental processes.
Based on these findings we propose the following model to explain the deceleration and eventual cessation of somatic growth in mammals. In late embryonic and early postnatal life, a network of growth-promoting genes is expressed at high levels. The resulting growth causes the expression of these growth-promoting genes to decline, which in turn slows the rate of growth. Eventually, the expression levels of these growth-promoting genes declines sufficiently to cause somatic growth to cease. If some external condition, such as hypothyroidism transiently restricts growth, then the slow growth will slow the decline in gene expression, thus preserving future growth capacity. After the period of growth inhibition, the expression levels of the growth-promoting genes will be higher than normal, and consequently, the growth rate will be greater than normal. Thus, the model provides an explanation for the phenomenon of catch-up growth, which is defined as a growth rate that is greater than normal for age after a period of growth inhibition (56,57). In this study, we focused on growth-promoting genes that show a decline in expression with age. However, the microarray analysis also identified a smaller number of genes that show an increase in expression with age in multiple organs. Thus, the model should be expanded to include the possibility of growth-inhibiting genes that are expressed at low levels in late embryonic and early postnatal life and then increase over time. Genes that increase in expression and those that decrease in expression may participate in the same networks.
The mechanism by which growth might cause these changes in gene expression is not known. Gene expression and subsequent proliferation might be regulated by biological systems that count prior cell divisions or current cell number. Such systems have been described, but little is known about the underlying cellular mechanisms (58). At least for one organ, the growth plate, the growth-limiting mechanism appears to be distinct from the mechanisms limiting growth of cultured cells (the Hayflick phenomenon) (59) and does not appear to involve telomere shortening (60).
The findings in the current study do not exclude the possibility that a systemic mechanism might be responsible for growth deceleration. However, none of the known growth-regulating hormones change in a pattern that would readily explain growth deceleration, and limited prior studies argue against systemic mechanisms (10,11,61).
We conclude that there exists an extensive genetic program occurring during the postnatal period involving up-regulation and down-regulation of thousands of genes. Of these, some appear to be organ specific, but many are regulated in a concerted fashion in multiple organs. Some of these common genes regulate cell proliferation and thus may constitute part of the mechanism that causes somatic growth to slow and eventually cease. We found evidence that several of the genes likely to participate in this growth-limiting program, Igf2, Mest, and Peg3, are themselves regulated by growth, suggesting that, in the embryo, a gene expression pattern is established that allows for rapid somatic growth of multiple tissues, but then, during postnatal life, this growth leads to negative-feedback changes in gene expression that in turn slow and eventually halt somatic growth, thus imposing a fundamental limit on adult body size.
Supplementary Material
Footnotes
This work was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health. O.N. was supported by grants from the Swedish Research Council (K2007-52X-20316-01-4), the Swedish Society of Medical Research, HKH Kronprinsessan Lovisas Förening för Barnasjukvård, Sällskapet Barnavård, and Stiftelsen Frimurare Barnhuset i Stockholm.
Disclosure Summary: The authors have nothing to declare.
First Published Online November 26, 2008
Abbreviations: CT, Threshold cycle; E, embryonic day; Mest, mesoderm-specific transcript; Peg3, paternally expressed 3; PTU, propylthiouracil; Sox4, SRY-box containing gene 4.
References
- Kobayashi T, Kronenberg H 2005 Minireview: transcriptional regulation in development of bone. Endocrinology 146:1012–1017 [DOI] [PubMed] [Google Scholar]
- Liu A, Niswander LA 2005 Bone morphogenetic protein signalling and vertebrate nervous system development. Nat Rev Neurosci 6:945–954 [DOI] [PubMed] [Google Scholar]
- Guinobert I, Viltard M, Piquemal D, Elalouf JM, Marti J, Lelievre-Pegorier M 2006 Identification of differentially expressed genes between fetal and adult mouse kidney: candidate gene in kidney development. Nephron Physiol 102:81–91 [DOI] [PubMed] [Google Scholar]
- Patterson LT, Potter SS 2004 Profiling gene expression in kidney development. Nephron Exp Nephrol 98:e109–e113 [DOI] [PubMed] [Google Scholar]
- Chang M, Parker EA, Muller TJ, Haenen C, Mistry M, Finkielstain GP, Murphy-Ryan M, Barnes KM, Sundaram R, Baron J 2008 Changes in cell-cycle kinetics responsible for limiting somatic growth in mice. Pediatr Res 64:240–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupu F, Terwilliger JD, Lee K, Segre GV, Efstratiadis A 2001 Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev Biol 229:141–162 [DOI] [PubMed] [Google Scholar]
- Wang J, Zhou J, Powell-Braxton L, Bondy C 1999 Effects of Igf1 gene deletion on postnatal growth patterns. Endocrinology 140:3391–3394 [DOI] [PubMed] [Google Scholar]
- Marino R, Hegde A, Barnes KM, Schrier L, Emons JA, Nilsson O, Baron J 2008 Catch-up growth after hypothyroidism is caused by delayed growth plate senescence. Endocrinology 149:1820–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan AY, Hartman ML 2007 IGF-I measurements in the diagnosis of adult growth hormone deficiency. Pituitary 10:151–157 [DOI] [PubMed] [Google Scholar]
- Hammerman MR 2005 Windows of opportunity for organogenesis. Transpl Immunol 15:1–8 [DOI] [PubMed] [Google Scholar]
- Stevens DG, Boyer MI, Bowen CV 1999 Transplantation of epiphyseal plate allografts between animals of different ages. J Pediatr Orthop 19:398–403 [PubMed] [Google Scholar]
- Varrault A, Gueydan C, Delalbre A, Bellmann A, Houssami S, Aknin C, Severac D, Chotard L, Kahli M, Le DA, Pavlidis P, Journot L 2006 Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev Cell 11:711–722 [DOI] [PubMed] [Google Scholar]
- Lui JC, Finkielstain GP, Barnes KM, Baron J 2008 An imprinted gene network that controls mammalian somatic growth is down-regulated during postnatal growth deceleration in multiple organs. Am J Physiol Regul Integr Comp Physiol 295:R189–R196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean-Faucher C, Berger M, de TM, Veyssiere G, Jean C 1978 Developmental patterns of plasma and testicular testosterone in mice from birth to adulthood. Acta Endocrinol (Copenh) 89:780–788 [DOI] [PubMed] [Google Scholar]
- Meehan TP, Harmon BG, Overcast ME, Yu KK, Camper SA, Puett D, Narayan P 2005 Gonadal defects and hormonal alterations in transgenic mice expressing a single chain human chorionic gonadotropin-lutropin receptor complex. J Mol Endocrinol 34:489–503 [DOI] [PubMed] [Google Scholar]
- Matsushita K, McCray Jr PB, Sigmund RD, Welsh MJ, Stokes JB 1996 Localization of epithelial sodium channel subunit mRNAs in adult rat lung by in situ hybridization. Am J Physiol 271:L332–L339 [DOI] [PubMed] [Google Scholar]
- Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ 1996 Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol 271:H2183–H2189 [DOI] [PubMed] [Google Scholar]
- Ogawa Y, Okada H, Heya T, Shimamoto T 1989 Controlled release of LHRH agonist, leuprolide acetate, from microcapsules: serum drug level profiles and pharmacological effects in animals. J Pharm Pharmacol 41:439–444 [DOI] [PubMed] [Google Scholar]
- Schilham MW, van EM, van de WM, Clevers HC 1993 The murine Sox-4 protein is encoded on a single exon. Nucleic Acids Res 21:2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker EA, Hegde A, Buckley M, Barnes KM, Baron J, Nilsson O 2007 Spatial and temporal regulation of GH-IGF-related gene expression in growth plate cartilage. J Endocrinol 194:31–40 [DOI] [PubMed] [Google Scholar]
- Divjak M, Glare EM, Walters EH 2002 Improvement of non-radioactive in situ hybridization in human airway tissues: use of PCR-generated templates for synthesis of probes and an antibody sandwich technique for detection of hybridization. J Histochem Cytochem 50:541–548 [DOI] [PubMed] [Google Scholar]
- Zhou J, Chin E, Bondy C 1991 Cellular pattern of insulin-like growth factor-I (IGF-I) and IGF-I receptor gene expression in the developing and mature ovarian follicle. Endocrinology 129:3281–3288 [DOI] [PubMed] [Google Scholar]
- Meisami E 1984 Complete recovery of growth deficits after reversal of PTU-induced postnatal hypothyroidism in the female rat: a model for catch-up growth. Life Sci 34:1487–1496 [DOI] [PubMed] [Google Scholar]
- Fleeman TL, Cappon GD, Hurtt ME 2004 Postnatal closure of membranous ventricular septal defects in Sprague-Dawley rat pups after maternal exposure with trimethadione. Birth Defects Res B Dev Reprod Toxicol 71:185–190 [DOI] [PubMed] [Google Scholar]
- Holinka CF, Tseng YC, Finch CE 1978 Prolonged gestation, elevated preparturitional plasma progesterone and reproductive aging in C57BL/6J mice. Biol Reprod 19:807–816 [DOI] [PubMed] [Google Scholar]
- Marty MS, Crissman JW, Carney EW 2001 Evaluation of the male pubertal onset assay to detect testosterone and steroid biosynthesis inhibitors in CD rats. Toxicol Sci 60:285–295 [DOI] [PubMed] [Google Scholar]
- Nathan BM, Hodges CA, Supelak PJ, Burrage LC, Nadeau JH, Palmert MR 2006 A quantitative trait locus on chromosome 6 regulates the onset of puberty in mice. Endocrinology 147:5132–5138 [DOI] [PubMed] [Google Scholar]
- Evans AM 1986 Age at puberty and first litter size in early and late paired rats. Biol Reprod 34:322–326 [DOI] [PubMed] [Google Scholar]
- Omoto Y, Lathe R, Warner M, Gustafsson JA 2005 Early onset of puberty and early ovarian failure in CYP7B1 knockout mice. Proc Natl Acad Sci USA 102:2814–2819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curley JP, Pinnock SB, Dickson SL, Thresher R, Miyoshi N, Surani MA, Keverne EB 2005 Increased body fat in mice with a targeted mutation of the paternally expressed imprinted gene Peg3. FASEB J 19:1302–1304 [DOI] [PubMed] [Google Scholar]
- DeChiara TM, Efstratiadis A, Robertson EJ 1990 A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature 345:78–80 [DOI] [PubMed] [Google Scholar]
- Schilham MW, Oosterwegel MA, Moerer P, Ya J, de Boer PA, van de WM, Verbeek S, Lamers WH, Kruisbeek AM, Cumano A, Clevers H 1996 Defects in cardiac outflow tract formation and pro-B-lymphocyte expansion in mice lacking Sox-4. Nature 380:711–714 [DOI] [PubMed] [Google Scholar]
- Liao B, Hu Y, Herrick DJ, Brewer G 2005 The RNA-binding protein IMP-3 is a translational activator of insulin-like growth factor II leader-3 mRNA during proliferation of human K562 leukemia cells. J Biol Chem 280:18517–18524 [DOI] [PubMed] [Google Scholar]
- Shi W, Lefebvre L, Yu Y, Otto S, Krella A, Orth A, Fundele R 2004 Loss-of-imprinting of Peg1 in mouse interspecies hybrids is correlated with altered growth. Genesis 39:65–72 [DOI] [PubMed] [Google Scholar]
- Johnson MD, Wu X, Aithmitti N, Morrison RS 2002 Peg3/Pw1 is a mediator between p53 and Bax in DNA damage-induced neuronal death. J Biol Chem 277:23000–23007 [DOI] [PubMed] [Google Scholar]
- DeChiara TM, Robertson EJ, Efstratiadis A 1991 Parental imprinting of the mouse insulin-like growth factor II gene. Cell 64:849–859 [DOI] [PubMed] [Google Scholar]
- Stylianopoulou F, Efstratiadis A, Herbert J, Pintar J 1988 Pattern of the insulin-like growth factor II gene expression during rat embryogenesis. Development 103:497–506 [DOI] [PubMed] [Google Scholar]
- Brown AL, Graham DE, Nissley SP, Hill DJ, Strain AJ, Rechler MM 1986 Developmental regulation of insulin-like growth factor II mRNA in different rat tissues. J Biol Chem 261:13144–13150 [PubMed] [Google Scholar]
- Kanwar YS, Kumar A, Ota K, Lin S, Wada J, Chugh S, Wallner EI 2002 Identification of developmentally regulated mesodermal-specific transcript in mouse embryonic metanephros. Am J Physiol Renal Physiol 282:F953–F965 [DOI] [PubMed] [Google Scholar]
- Li F, Wang X, Capasso JM, Gerdes AM 1996 Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol 28:1737–1746 [DOI] [PubMed] [Google Scholar]
- Winick M, Noble A 1965 Quantitative changes in DNA, RNA, and protein during prenatal and postnatal growth in the rat. Dev Biol 12:451–466 [DOI] [PubMed] [Google Scholar]
- Thurlbeck WM 1975 Postnatal growth and development of the lung. Am Rev Respir Dis 111:803–844 [DOI] [PubMed] [Google Scholar]
- Rechler MM, Nissley SP 1990 Insulin-like growth factors. In: Sporn MB, Roberts AB, eds. Peptide growth factors and their receptors I. New York: Springer-Verlag; 263–362 [Google Scholar]
- Scott J, Cowell J, Robertson ME, Priestley LM, Wadey R, Hopkins B, Pritchard J, Bell GI, Rall LB, Graham CF 1985 Insulin-like growth factor-II gene expression in Wilms' tumour and embryonic tissues. Nature 317:260–262 [DOI] [PubMed] [Google Scholar]
- Grant ES, Ross MB, Ballard S, Naylor A, Habib FK 1998 The insulin-like growth factor type I receptor stimulates growth and suppresses apoptosis in prostatic stromal cells. J Clin Endocrinol Metab 83:3252–3257 [DOI] [PubMed] [Google Scholar]
- Stewart CE, Rotwein P 1996 Growth, differentiation, and survival: multiple physiological functions for insulin-like growth factors. Physiol Rev 76:1005–1026 [DOI] [PubMed] [Google Scholar]
- Sun FL, Dean WL, Kelsey G, Allen ND, Reik W 1997 Transactivation of Igf2 in a mouse model of Beckwith-Wiedemann syndrome. Nature 389:809–815 [DOI] [PubMed] [Google Scholar]
- Lefebvre L, Viville S, Barton SC, Ishino F, Keverne EB, Surani MA 1998 Abnormal maternal behaviour and growth retardation associated with loss of the imprinted gene Mest. Nat Genet 20:163–169 [DOI] [PubMed] [Google Scholar]
- Lefebvre L, Viville S, Barton SC, Ishino F, Surani MA 1997 Genomic structure and parent-of-origin-specific methylation of Peg1. Hum Mol Genet 6:1907–1915 [DOI] [PubMed] [Google Scholar]
- Li L, Keverne EB, Aparicio SA, Ishino F, Barton SC, Surani MA 1999 Regulation of maternal behavior and offspring growth by paternally expressed Peg3. Science 284:330–333 [DOI] [PubMed] [Google Scholar]
- Schilham MW, Clevers H 1998 HMG box containing transcription factors in lymphocyte differentiation. Semin Immunol 10:127–132 [DOI] [PubMed] [Google Scholar]
- Ya J, Schilham MW, de Boer PA, Moorman AF, Clevers H, Lamers WH 1998 Sox4-deficiency syndrome in mice is an animal model for common trunk. Circ Res 83:986–994 [DOI] [PubMed] [Google Scholar]
- Cheung M, bu-Elmagd M, Clevers H, Scotting PJ 2000 Roles of Sox4 in central nervous system development. Brain Res Mol Brain Res 79:180–191 [DOI] [PubMed] [Google Scholar]
- Reppe S, Rian E, Jemtland R, Olstad OK, Gautvik VT, Gautvik KM 2000 Sox-4 messenger RNA is expressed in the embryonic growth plate and regulated via the parathyroid hormone/parathyroid hormone-related protein receptor in osteoblast-like cells. J Bone Miner Res 15:2402–2412 [DOI] [PubMed] [Google Scholar]
- Sekiya I, Vuoristo JT, Larson BL, Prockop DJ 2002 In vitro cartilage formation by human adult stem cells from bone marrow stroma defines the sequence of cellular and molecular events during chondrogenesis. Proc Natl Acad Sci USA 99:4397–4402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersma B, Otten BJ, Stoelinga GB, Wit JM 1996 Catch-up growth after prolonged hypothyroidism. Eur J Pediatr 155:362–367 [DOI] [PubMed] [Google Scholar]
- Boersma B, Wit JM 1997 Catch-up growth. Endocr Rev 18:646–661 [DOI] [PubMed] [Google Scholar]
- Gomer RH 2001 Not being the wrong size. Nat Rev Mol Cell Biol 2:48–54 [DOI] [PubMed] [Google Scholar]
- Nilsson O, Mitchum Jr RD, Schrier L, Ferns SP, Barnes KM, Troendle JF, Baron J 2005 Growth plate senescence is associated with loss of DNA methylation. J Endocrinol 186:241–249 [DOI] [PubMed] [Google Scholar]
- Nwosu BU, Nilsson O, Mitchum Jr RD, Coco M, Barnes KM, Baron J 2005 Lack of telomere shortening with age in mouse resting zone chondrocytes. Horm Res 63:125–128 [DOI] [PubMed] [Google Scholar]
- Nilsson O, Baron J 2004 Fundamental limits on longitudinal bone growth: growth plate senescence and epiphyseal fusion. Trends Endocrinol Metab 15:370–374 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.