

Abstract
Chk1 is a serine/threonine protein kinase which is activated by a wide range of DNA damaging agents in order to slow the cell cycle during S phase and G2/M. Abrogation of these cell cycle checkpoints using Chk1 inhibitors results in hypersensitivity to DNA damaging agents in vitro and may provide a potential therapeutic tool to sensitize tumour cells in vivo. We have generated a Cre-Lox based mouse model, where Chkl can be inducibly deleted from somatic epithelial cells in the adult mouse small intestine and liver. Loss of Chk1 in the liver is tolerated with no apparent phenotype. In contrast, loss of Chk1 within the small intestine results in immediate DNA damage, and high levels of p53-independent apoptosis leading to crypt death. However, the intestine is able to compensate for this death by undergoing complete repopulation with Chk1 proficient cells. This data therefore shows that Chk1 deficiency is cell lethal, but the intestine can tolerate such lethality at the organ level.
Keywords: Chk1, DNA damage, small intestine, apoptosis, p53
Introduction
Ionizing radiation and many chemotherapeutic agents used in the treatment of cancer induces DNA damage to kill tumour cells. These therapies not only activate apoptotic signalling pathways, but also survival mechanisms such as DNA damage-response pathways and cell cycle arrest, which allow for DNA repair to occur. DNA damage-induced cell cycle arrest depends on the activation of an evolutionarily conserved signalling network in which Chk1, a serine/threonine checkpoint kinase, plays a pivotal role (Nyberg et al., 2002; Sancar et al., 2004). Chk1 is active at the intra S-phase, G2/M, and the spindle checkpoints and recent work has indicated that abrogation of these checkpoints through inhibition of Chk1 may be a feasible cancer cell-specific therapy (Bartek & Lukas, 2003; Hapke et al., 2001; Kawabe, 2004; Tse et al., 2007). This approach is based on the fact that more than half of human cancer cells have a defective G1 checkpoint due to functional defects in the p53 and Rb pathways (Levine, 1997). Tumour cells are therefore more reliant than normal cells on the S-phase and G2 checkpoint kinases such as Chk1 to induce cell cycle arrest in response to DNA damage, to allow DNA repair and prevent cell death. There is now a significant body of in vitro data (Chen et al., 2003; Koniaras et al., 2001; Robinson et al., 2006; Wang et al., 1996; Xiao et al., 2003; Zachos et al., 2003) showing that disruption of these checkpoints through Chk1 inhibition can sensitize tumour cells to DNA-damaging agents and thus Chk1 inhibition may well be able to enhance the efficacy of current therapies. However in order to validate it as a therapeutic approach it is essential to establish the biological role of Chk1 in vivo.
Chk1 is essential for mammalian development and viability as Chk1-deficient mice are embryonic lethal due to a peri-implantation defect (Liu et al., 2000; Takai et al., 2000). Due to the lethality of the embryonic null mutation, Chk1 has also been studied in two conditional knockout mouse models using the Cre-loxP system (Lam et al., 2004; Zaugg et al., 2007). In these models the Chk1 null phenotype was restricted to either the mammary gland or the thymus and in both cases homozygous disruption of the Chk1 alleles resulted in cell death due to an increase in apoptosis (Lam et al., 2004; Zaugg et al., 2007). Previous work has also proposed Chk1 as a tumour suppressor and although this hypothesis remains unconfirmed, Lam et al. have demonstrated that the loss of a single Chk1 allele in the mouse mammary gland results in severe cell cycle mis-coordination due to checkpoint disruption (Lam et al., 2004). The observation that the loss of a single Chk1 allele results in genomic instability has also been suggested by recent work using a Chk1+/-WNT-1 transgenic model (Liu et al., 2000), whereby loss of a single Chk1 allele results in a modest increase in tumour burden, suggesting Chk1 may act as a haploinsufficient tumour suppressor. However, germline Chk1+/- mice have no detectable spontaneous tumour predisposition up to 18 months of age (Liu et al., 2000), indicating that in isolation Chk1 haploinsufficiency acts only very weakly as a tumour promoter.
Although our understanding of Chk1 in cell cycle regulation is becoming clearer it is still essential that we obtain more information regarding the in vivo consequences of Chk1 loss in somatic cells. For this purpose we have generated a mouse model in which the Chk1 allele can be conditionally deleted from the mouse small intestine and liver. In this study we demonstrate that loss of both Chk1 alleles results in a rapid induction of intestinal crypt apoptosis. However, the intestine is capable of compensating for such loss, responding with a period of intestinal cell proliferation, whereby the intestinal crypts are repopulated with wild-type cells. . In contrast loss of both Chk1 alleles in the liver appears to be tolerated with no apparent phenotype for a period of up to 9 months.
Materials and Methods
Experimental mice
Mice carrying the floxed Chk1 allele were kindly supplied by Dr Stephen J. Elledge (Lam et al., 2004). All mice were genotyped as previously described for the targeted Chk1 allele (Lam et al., 2004), the targeted p53 allele (Jonkers et al., 2001), the Rosa26R allele (Soriano, 1999) and the AhCre transgene (Ireland et al., 2004). Cre activity was induced in control and experimental mice by 3 consecutive intraperitoneal (i.p.) injections of 80mg/kg β-naphthoflavone in 24h. In addition, selected animals were injected with 100μg/kg Bromo-deoxyuridine and culled 2 hours after labelling or 10mg/kg Cisplatin and then culled 6 hours post-induction (PI). All procedures were conducted according to UK Home Office regulations.
Quantitative real-time PCR (qRT-PCR)
RNA was isolated from liver tissue or epithelial cells obtained by epithelial extraction (Bjerknes & Cheng, 1981) using a standard Trizol protocol. Reverse transcription and qRT-PCR were performed using a standard protocol. Chk1 primers were designed to the deleted exon 2 of the gene (5′-CTG GGA TTT GGT GCA AAC TT-3′ and 5′-GCC CGC TTC ATG TCT ACA AT-3′). Other primer sequences used were: GAPDH (5′-CAC TGA GCA TCT CCC TCA CA-3′ and 5′-GTG GGT GCA GCG AAC TTT AT-3′) and β-Actin (5′-ACA GCT TCT TTG CAG CTC CTT-3′ and 5′-TGG TAA CAA TGC CAT GTT CAA T-3′).
Western analysis
Protein was isolated from epithelial enriched pellets (Bjerknes & Cheng, 1981) and subsequent protein analysis, SDS-PAGE and western blotting were carried out following standard protocols. Chk1 mouse monoclonal antibody (Abgent) was used at 1:1000 and mouse monoclonal β actin (Sigma) was used at 1:12000.
Recombination analysis
Detection of the recombined allele in wild-type (AhCre+Chk1+/+) and experimental (AhCre+Chk1F/+ and AhCre+Chk1F/-) mice was carried out using a PCR-based analysis. DNA was extracted from 2mm sections of small intestine or liver and recombinant DNA was detected as previously described (Lam et al., 2004). Primers against the wild-type APC allele (Shibata et al., 1997) were used as a control.
Histology and Immunohistochemistry
Intestinal and liver tissue was fixed in ice cold 10% neutral buffered formalin for no longer than 24 hours before being processed into paraffin blocks according to standard procedures. Tissue sections (5μm) were either stained using haematoxylin and eosin for histological analysis, or were used for immunohistochemistry. The following antibodies were used for immunhistochemistry: anti-ATM (1:300; Rockland), anti-Caspase 3 (1:750; R&D systems), mouse anti-p53 (1:50; Labvision), mouse anti-H2A.X (Ser139) (1:200, Upstate), mouse anti-BrdU (1:100; Becton Dickinson), rabbit anti-p21 (1:500, Santa Cruz).
Immunofluorescence microscopy
Immunofluorescence was carried out using the above method described for immunohistochemistry using the Alexa Fluor® 488 goat anti-mouse IgG secondary antibody (1:300, Invitrogen). Stained tissue sections were scanned on a Leica TCS SP2 AOBS spectral confocal laser scanning microscope (Leica, Germany) running Leica Confocal Software. Representative regions of tissue were scanned using x40 and x 63 oil immersion objectives with appropriate excitation and emission settings for simultaneous recordings of DAPI (Ex. Max: 358; Em. Max: 461) and Alexa 488 (Ex. Max: 488; Em. Max: 519). Optical sections were sampled at approximately 0.50m intervals and maximum intensity type reconstructions then prepared from the resultant image stacks.
Results
Chk1 is rapidly but transiently lost in the mouse small intestine following β-naphthoflavone injection
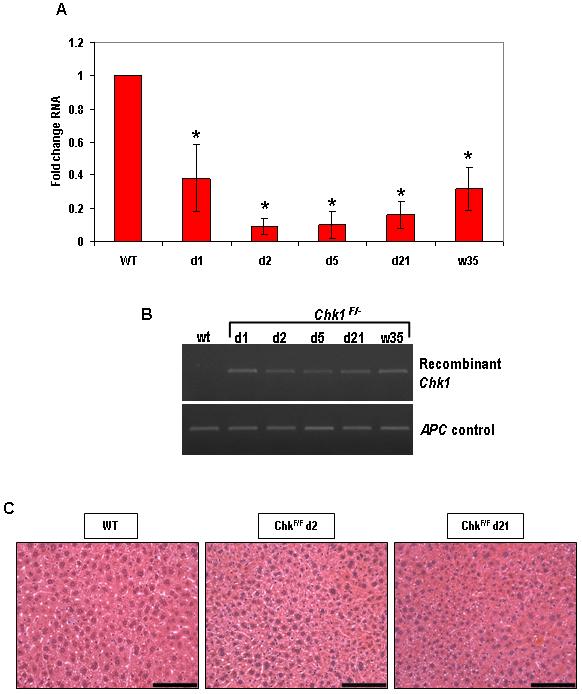
In order to delete Chk1 from the murine small intestine we crossed mice containing a Chk1 allele in which exon 2 was flanked by loxP sites (Lam et al., 2004) with mice containing a Cre transgene under the control of the CYP1A1 (AhCre) promoter (Ireland et al., 2004). We have previously shown that this construct drives high efficiency recombination within the crypt, including the stem cell compartment (Ireland et al., 2004; Sansom et al., 2005). Induction of recombination in the mouse small intestine was achieved after intraperitoneal injection of β-naphthoflavone. Cre-mediated excision of exon 2, which contains the translational initiation sequence and encodes the ATP-binding site of Chk1 (Liu et al., 2000), results in conversion to a null allele. To determine the extent of Cre-mediated recombination, qRT-PCR analysis of Chk1 expression was performed using RNA isolated from epithelial-enriched tissue samples (Figure 1A). Chk1 expression was reduced in AhCre+Chk1F/F mice compared to wild-type controls at day 1 post-induction (PI) and a significant decrease to around 10% of the wild-type level was seen at day 2 (Mann-Whitney U, n= 3, P= 0.0404), confirming that Cre-mediated loss of Chk1 had occurred. Although the level of recombination was high at day 1 and 2, by day 5 Chk1 RNA expression had returned to near wild-type levels. This high level of expression was maintained at the day 21 timepoint. The return of Chk1 expression to wild-type levels indicates that the presence of the recombined allele is transient. Western analysis was performed to support our RNA data (Figure 1B). Protein was extracted from epithelial-enriched tissue samples and the observed results were consistent with the qRT-PCR analysis. Chk1 protein levels are barely detectable at day 1 and day 2, but by day 5 and day 21 Chk1 levels had returned to wild-type levels. One likely explanation for this transient loss of Chk1 expression is that although successful recombination of the Chk1 allele occurs at day 1 PI, by day 5 cells containing the Chk1 null allele are replaced with cells containing a competent unrecombined Chk1 allele. In order to test this hypothesis we analysed genomic intestinal DNA for the presence of the recombined allele. PCR analysis was carried out using primers previously described (Lam et al., 2004) that detect a 436bp product representing the recombined Chk1 allele (Fig 1C). From our result it is clear that the recombined allele is present at day 1 PI and also day 2, but by day 5 and day 21 the AhCre+Chk1F/- mouse small intestine no longer contained the recombinant allele (Fig 1C left panel). This is consistent with the replacement of intestinal cells containing the floxed Chk1 allele with cells containing the unrecombined Chk1 allele. In contrast, PCR analysis of genomic intestinal DNA isolated from AhCre+Chk1F/+ mice demonstrated that the recombined allele remained present in the Chk1 heterozygote small intestine for up to 35 weeks (Fig 1C right panel).
Figure 1. Induction with β-naphthoflavone causes recombination in the small intestine, leading to conditional loss of Chk1.
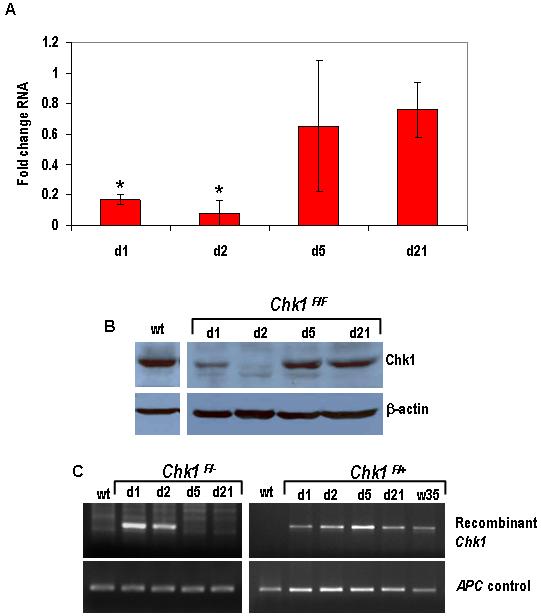
Cre activity was induced in wild-type (AhCre+Chk1+/+) and experimental (AhCre+Chk1F/+ and AhCre+Chk1F/F) by 3 consecutive i.p. injections of 80mg/kg β-naphthoflavone in 24h. Animals were culled at various time-points (day 1 to day 21, and week 35) after induction and tissue was isolated from both control and experimental animals.
(A) Loss of Chk1 RNA from the small intestine was confirmed by qRT-PCR using RNA derived from samples of tissue enriched for intestinal epithelium. The PCR was carried out using primers designed to detect transcript from the deleted exon 2 of Chk1 (see Materials and Methods) and the data shown is in terms of fold-change of the Chk1/β-actin ratio compared to AhCre+Chk1+/+. *, P < 0.05
(B) Loss of Chk1 protein was demonstrated by western analysis. Protein was isolated from tissue samples enriched for intestinal epithelium cells and subjected to SDS-PAGE. Blotted membranes were incubated with a Chk1 mouse monoclonal antibody overnight and the membrane was re-probed with a β-actin mouse monoclonal antibody to confirm equal protein loading.
(C) Recombination in the small intestine was confirmed by semi-quantitative PCR using primers designed to detect the recombined Chk1 allele. DNA was isolated from tissue harvested from Cre-induced wild-type and experimental animals (AhCre+Chk1F/- left panel, AhCre+Chk1F/+ right panel) and detection of the recombined allele yielded a 436 bp product. Primers against the wild-type APC allele were used as a control.
Chk1 loss results in apoptosis and crypt death
In order to assess the phenotypic consequence of Chk1 loss in the mouse small intestine and to gain a further understanding of the kinetics involved in the loss of the recombined allele, we examined histological sections from AhCre+Chk1F/F mice at various timepoints (Fig 2A). Histological analysis demonstrated a clear change in the AhCre+Chk1F/F mouse small intestine at day 2 PI, with the cells of the intestinal crypts undergoing apoptosis (Fig 2A, Chk1F/F d2). This increase in cell death at day 2 was independently confirmed by immunohistochemical staining with an antibody against active caspase 3 (Fig 2B). Day 5 histology showed virtually no apoptosis and instead showed the characteristic appearance of intestinal crypt repopulation, including rapid cell proliferation and overgrowth of the crypts (Fig 2A, Chk1F/F d5). By day 21 crypt histology had reverted back to that of wild-type (Fig 2A, Chk1F/F d21).
Figure 2. Loss of Chk1 results in crypt death due to an increase in apoptosis.
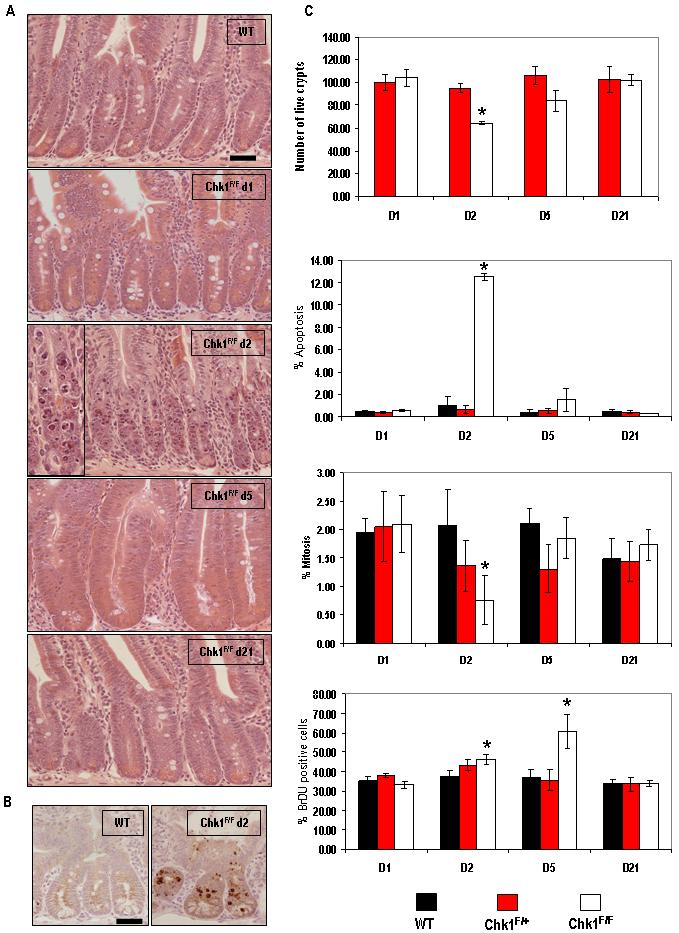
(A) H&E-stained histological sections of mouse small intestine from a wild-type mouse (AhCre+Chk1+/+) and experimental (AhCre+Chk1F/F) mice at various timepoints PI. A significant increase in apoptosis in Chk1-deficient tissue can be seen at day 2 PI (A, ChkF/F d2, see inset for magnified view) when compared to wild-type. This increase in apoptosis was independently confirmed by IHC using an antibody specific for cleaved caspase 3 (B). (C) Tissue sections were used to quantify live crypts (data shown is in terms of number of crypts scored per section and normalised to wild type controls), apoptosis and mitosis (both shown as percentage of total cells per crypt). Data shown are means ± S.D. of three independent experiments. (black bars, AhCre+Chk1+/+; red bars, AhCre+Chk1F/+ and white bars, AhCre+Chk1F/F). *, P < 0.05. To examine S-phase labelling in vivo, control (AhCre+Chk1+/+) and experimental (AhCre+Chk1F/+, AhCre+Chk1F/F) animals were injected with 100μg BrdU and culled 2hrs after labelling. BrdU incorporation was determined by IHC and quantified by scoring positively stained cells (C, bottom graph). Bar, 25μm.
In order to quantify the increase in apoptosis and characterise any other phenotypic changes indicated by histological analysis, crypt death, mitotic index and levels of apoptosis was scored from the hematoxylin-and-eosin-stained sections as previously described (Merritt et al., 1997). These results confirmed a 12-fold increase in apoptosis at the day 2 timepoint in AhCre+Chk1F/F mice (Fig 2C), which was accompanied by a reduction in mitotic figures (2.7-fold, P=0.0404) and an overall decrease in the number of live crypts (30%, P=0.0404). No other timepoint demonstrated a significant variation to wild-type, other than an increase in BrdU uptake at day 2 and day 5 in AhCre+Chk1F/F mice (Fig 2C). This increase in BrdU uptake at day 2 PI (Mann-Whitney U, n= 3, P= 0.0404) without an increase in mitosis may reflect the ability of Chk1 null cells to synthesise DNA without being able to successfully complete mitosis. In contrast, the increase in mitosis and BrdU uptake at day 5 PI (Mann-Whitney U, n= 3, P= 0.0404) reflects the rapid proliferation occurring during the repopulation phase of the crypt.
Histological sections from AhCre+Chk1F/+ small intestine were examined and scored for crypt death, mitotic index and apoptosis (Fig 2C). No variation from wild-type was observed over any of the timepoints, indicating that within the mouse small intestine loss of a single Chk1 allele does not result in a significant phenotype (see also Supplementary Fig 1). This lack of phenotype, in addition to the persistent presence of the recombined allele up to 9 months (see Fig 1C), indicates that heterozygous loss of Chk1 is tolerated in the mouse small intestine.
Chk1 deficiency results in an increase in DNA damage and p53 induction
The above observations demonstrate that the conditional homozygous loss of Chk1 in the mouse small intestine results in cell death. Previous work has shown that reduction in Chk1 levels results in genomic instability and DNA damage, which may provide a likely explanation for the increase in apoptosis (Lam et al., 2004). In order to define these molecular events in the small intestine, sections were stained for phospho-H2A.X and phospho-ATM, both of which become phosphorylated and activated in the presence of DNA damage (Bakkenist & Kastan, 2003; Rogakou et al., 1998). Our results show an increase in phospho-H2A.X and phospho-ATM staining at day 2 PI (Fig 3A and 3B), indicating that AhCre+Chk1F/F intestinal cells are accumulating DNA damage. It is important to note that previous reports have shown H2A.X phosphorylation to occur following apoptotic DNA fragmentation (Rogakou et al., 2000). However, we observe activation of H2A.X at day 1 PI (Fig 3A), prior to the activation of caspase-3 or the appearance of apoptotic bodies, suggesting that loss of Chk1 directly causes DNA damage and that apoptosis is a secondary event, which is consistent with previous studies (Lam et al., 2004; Syljuasen et al., 2005). In addition immunofluorescence was carried out to further characterise the increase in phospho-H2A.X expression observed at day 1 PI. Although the increase in H2A.X phosphorylation was mainly associated with a diffuse pattern, some focal H2A.X staining was observed (see Supplementary Fig 2) which is consistent with DNA damage being a consequence of Chk1 loss rather than initiated apoptosis.
Figure 3. Chk1 deficiency results in DNA damage and activation of apoptotic markers.
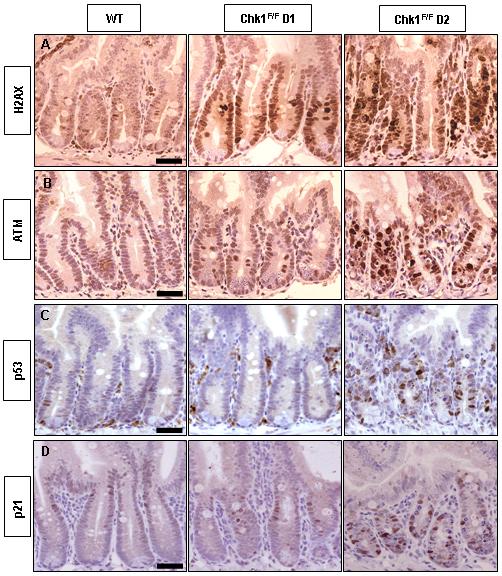
(A-D) IHC analysis of intestinal tissue sections from wild-type (AhCre+Chk1+/+) and experimental (AhCre+Chk1F/F) mice with antibodies detecting phosphorylated-H2A.X (A), phosphorylated-ATM (B), p53 (C) and p21 (D). Each row of panels represents analysis using a single antibody on a wild-type mouse (left), and an AhCre+Chk1F/F mouse at day 1 (middle) and day 2 PI (right). Bar, 25μm.
IHC staining against p53 and p21, was carried out to address any potential involvement of these apoptotic mediators. Both p21 and p53 levels were increased at day 2, coinciding with the observed increase in apoptosis (Fig 3C and 3D). These results suggests that loss of Chk1 in the mouse small intestine results in DNA damage and ultimately leads to apoptosis through a p53-dependent pathway. In contrast, IHC analysis of AhCre+Chk1F/+ small intestine at all timepoints did not reveal increases in phospho-H2A.X (see Supplementary Fig 1), phospho-ATM, p53, or p21 (data not shown). In addition deletion of a single Chk1 allele did not sensitise cells to apoptosis following exposure to the cytotoxic drug Cisplatin (see Supplementary figure 6). These results, in combination with the BrdU data in Fig 2, indicate that loss of a single Chk1 allele has no detectable functional consequences in the mouse small intestine. This contrasts with the findings of Lam et al. where Chk1 heterozygosity in LMECs leads to an increase in the proportion of BrdU and phospho-H2A.X staining, reflecting cell cycle defects and genomic instability (Lam et al., 2004).
p53 deletion fails to rescue Chk1 null cells from apoptosis
The involvement of p53 in mediating apoptosis following loss of Chk1 is unclear. Our data indicate that intestinal cell apoptosis involves activation of both p53 and p21. In addition, avian DT40 cells lacking functional p53 can successfully replicate in the absence of Chk1 (Zachos et al., 2003). However, both embryonic lethality and apoptosis during T cell development following Chk1 deletion have both been reported to be independent of p53 status (Liu et al., 2000; Zaugg et al., 2007). In order to define the role of p53 in the apoptosis seen in the mouse intestine, we generated AhCre+p53F/FChk1F/F mice using a previously described p53 allele (Jonkers et al., 2001). Our results show that loss of p53 did not alter the phenotype associated with Chk1 deletion in the mouse small intestine (Fig 4). Levels of apoptosis between and AhCre+p53F/FChk1F/F and AhCre+p53+/+Chk1F/F were equivalent at day 2 PI, as was crypt viability, mitosis (Fig 4B) and DNA damage (see Supplementary Fig 4). However in the AhCre+p53F/FChk1F/F mice apoptosis occurred without the induction of p53 or p21 (Fig 4A). Overall it is clear that although p53 is induced upon deletion of Chk1, the apoptotic phenotype of Chk1 deficient crypts is p53-independent.
Figure 4. p53 deficiency does not affect the apoptotic phenotype of Chk1 deficient crypts.
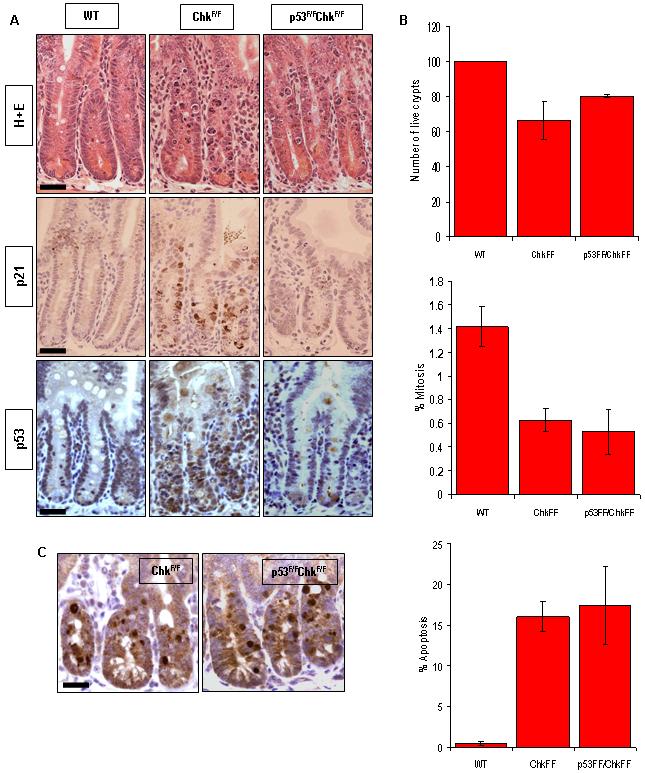
Cre activity was induced in wild-type (AhCre+Chk1+/+) and experimental (AhCre+p53F/FChk1F/F and AhCre+p53+/+Chk1F/F) mice. Animals were culled at day 2 after induction and tissue was isolated from both control and experimental animals. (A) Histological sections of mouse small intestine from mice at day 2 PI were stained with H&E (top) or subjected to IHC analysis using antibodies specific for p21 (middle) and p53 (bottom). (B) H&E-stained tissue sections were used to quantify live crypts, apoptosis and mitosis, as described in Figure 2. (C) Apoptosis levels were independently confirmed by IHC using a caspase 3 antibody. Bar, 25μm.
In addition to the day 2 time point we examined histological sections from AhCre+p53F/FChk1F/F mice at day 5 and day 7 PI (see Supplementary Fig 5A). Histological analysis demonstrated that although p53 deficiency has no effect on the immediate apoptotic phenotype of Chk1 loss, p53 status does impact on the repopulation which follows Chk1-dependent crypt death. At day 5 there is a marked difference in the histology of the AhCre+p53F/FChk1F/F and the AhCre+p53+/+Chk1F/F mice, as the small intestine of the AhCre+p53F/FChk1F/F mice shows a high level of crypt death and no sign of re-population. Re-population appears to be a later event in these mice occurring at day 7 (see Supplementary Fig 5A). These phenotypic changes indicated by histological analysis were quantified by scoring for crypt death, mitotic index and apoptosis as previously described (Merritt et al., 1997). These results (see Supplementary Fig 5B, C and D) confirmed a significant decrease in the number of live crypts at day 5 (44.83, P= 0.0113) and a moderate increase in the level of apoptosis. At day 7 it appears that the small intestine of the AhCre+p53F/FChk1F/F mice has returned to its normal state with an increase in the number of live crypts, and a decrease in apoptosis to resemble that seen in wild-type animals. From our results it appears that the concurrent loss of p53 with Chk1 does not protect the intestine from the early wave of apoptosis but instead generates a delay in repopulation of approximately 2 days.
Previous data has indicated a potentially important interaction of p53 and Chk1 in the cellular response to cytotoxic drugs, whereby the sensitisation of cells by Chk1 inhibitors may itself be optimal in a p53 null environment (Koniaras et al., 2001; Tse et al., 2007). In order to investigate this interaction in vivo we compared the levels of apoptosis in intestinal epithelial cells following a dose of Cisplatin. We found that although apoptosis was heavily dependent on a functional p53 allele (approximately 6 fold greater in p53 wildtype animals, P=0.0012), there was no difference between the Chk1+/+ and Chk1f/+ genotypes (see Supplementary Fig 6).
β-naphthoflavone injection results in Chk1 loss in the liver
In addition to the mouse small intestine, previous work has shown that the AhCre promoter is able to drive extensive recombination in the mouse liver (Ireland et al., 2004). Induction of recombination was achieved through IP injection of β-naphthoflavone and qRT-PCR analysis demonstrated a significant decrease in Chk1 expression in AhCre+Chk1F/F mice compared to wild-type controls at day 1 PI (Mann-Whitney U, n= 3, P= 0.0404) which was decreased to around 10% of the wild-type at day 2, similar to that seen in the intestine (Mann-Whitney U, n= 3, P= 0.0404). However unlike the intestine, a significant reduction in Chk1 expression was maintained at day 21 and even out to a week 35 timepoint (Fig 5A). In addition, PCR analysis demonstrated the persistent presence of the recombined Chk1 allele throughout the timecourse of the experiment, indicating that the liver is able to tolerate the loss of Chk1 over a long period of time (Fig 5B).
Figure 5. Induction with β-naphthoflavone causes recombination in the liver, leading to conditional loss of Chk1 expression.
Cre activity was induced in wild-type (AhCre+Chk1+/+) and experimental (AhCre+Chk1F/F) by 3 consecutive i.p. injections of 80mg/kg β-naphthoflavone in 24h. Animals were culled at various time-points (day 1 to day 21) after induction and tissue was isolated from both control and experimental animals. (A) Loss of Chk1 RNA expression from the liver was determined by qRT-PCR. The PCR was carried out using primers designed to detect transcript from the deleted exon 2 of Chk1 (see Materials and Methods) and the data shown is in terms of fold-change of the Chk1/β-actin ratio compared to AhCre+Chk1+/+. *, P < 0.05. (B) Recombination in the liver was confirmed by semi-quantitative PCR using primers designed to detect the recombined Chk1 allele. DNA was isolated from tissue harvested from Cre-induced wild-type and experimental animals and detection of the recombined allele yielded a 436 bp product. Primers against the wild-type APC allele were used as a control. (C) H&E-stained histological sections of mouse liver from AhCre+Chk1F/F mice at various timepoints (day 2 and day 21) PI demonstrates no change in histology when compared to wild-type (AhCre+Chk1+/+).
In order to assess the phenotypic consequence of Chk1 loss in the liver, we examined histological sections from AhCre+Chk1F/F mice at various time points. Our histological analysis showed no phenotypic consequence to loss of Chk1 expression in the liver (see Fig 5C) again indicating that homozygous loss of the Chk1 allele is tolerated in the liver and does not result in the same dramatic apoptosis observed in the intestine.
Discussion
Chk1 is currently regarded as a promising drug target to enhance the sensitivity of cytotoxic drugs in cancer therapy. This is based on considerable literature generated from cell culture experiments, and there are a number of compounds currently entering clinical trials (Tse et al., 2007). In spite of this, there is a relative paucity of information regarding the inhibition of Chk1 in vivo. The data presented in this paper adds to two previous reports analysing the functional consequences of Chk1 loss in the developing mammary gland and thymus (Lam et al., 2004; Zaugg et al., 2007). However unlike these reports, the mouse intestine allows functional analysis of Chk1 loss in the somatic cells of a fully established adult organ system. The mouse intestine is continually turning over with replication of stem cells in the crypt and subsequent differentiation and migration to the villus tip where they are consequently shed. It represents a relevant system to analyse Chk1 as it is biologically well-defined, it represents a major site for cancer formation, and is also responsible for some of the most significant drug-related side effects in chemotherapy.
The work presented in this paper clearly demonstrates that deletion of Chk1 in the mouse small intestine is highly traumatic. Transcript and protein are both lost within 24 hours of recombination and this loss of Chk1 results in DNA damage and apoptosis, both of which peak at 48 hours. However, this Cre-mediated deletion of Chk1 is short-lived. The small intestine is able to recover by recruiting epithelial stem cells in which recombination has not occurred to replace the dying Chk1 deficient cells, this leads to a phase of repopulation whereby rapid proliferation of intestinal epithelial cells occurs. By day 5, all cells with the recombined Chk1 allele have been eliminated, indicating that there is a strong selective disadvantage for them. This is similar to previous reports, although in both the thymus and mammary gland there was no repopulation following the loss of Chk1 but simply an inability to form functional adult tissue structures (Lam et al., 2004; Zaugg et al., 2007). Therefore, our results, in accordance with previous studies indicate that Chk1 is essential for the viability of proliferating cells. In contrast, the consequence of Chk1 loss in non-proliferating cells is much less clear. Indeed, we have shown that loss of Chk1 in the liver, a largely post-mitotic organ, is tolerated with very little effect, indicating that Chk1 function may only be required in actively cycling cells, an observation previously seen in other post-mitotic cells (Lam et al., 2004; Zaugg et al., 2007). However, this apparent functional redundancy in the liver is not reflected by low protein expression or increased turnover as Western analysis indicates that Chk1 levels are higher than that seen in rapidly dividing intestinal epithelial cells (see Supplementary Fig 7).
Although we have shown that the loss of Chk1 results in p53 upregulation and apoptosis, and previous work has demonstrated that only the p53-deficient DT-40 tumor cell line is able to tolerate Chk1 deletion (Zachos et al., 2003), our results clearly show that the coincident deletion of p53 did not rescue the Chk1 null phenotype in the mouse small intestine, indicating that perhaps an alternative apoptotic pathway is activated by Chk1 loss. Indeed, previous work has shown that Chk1 has a protective function in the regulation of centrosome-associated Cdk1/cyclin B and is essential for the critical timing of cell division (Kramer et al., 2004). Therefore loss of Chk1 would result in unscheduled mitotic entry which would possibly explain the p53-independent mode of cell death that we observed. Whilst the apoptotic phenotype remained unchanged by the loss of p53 and Chk1, our results at day 5 and day 7, indicate that the additional loss of p53 does result in a significantly more severe phenotype whereby the crypts of small intestine crypts are unable to recover as quickly, due to delayed repopulation.
We have also investigated the loss of a single Chk1 allele in the mouse small intestine. In contrast to previous reports, whereby Chk1 haploinsufficiency results in significant cell cycle defects and genomic instability (Lam et al., 2004; Liu et al., 2000) our model demonstrated no variation from wild-type phenotype in AhCre+Chk1F/+ mice relating to BrdU uptake or DNA damage. In addition, Chk1 haploinsufficiency in our model did not potentiate the cytotoxicity of a DNA-damaging agent in a p53-deficient environment, also suggesting that loss of a single allele does not lead to a significant phenotype. Although we cannot fully explain this observation, it is possible that the requirement of Chk1 differs between tissues, or that expression levels vary, so that loss of a single allele may be more crucial in the mammary gland as opposed to the intestine.
Chk1 inhibitors continue to be tested in clinical trials. The findings in this and other reports are likely to have therapeutic implications. Complete ablation of Chk1 function leads to the death of replicating somatic cells. This would appear to undermine the feasibility of using inhibitors as a targeted therapy in clinical practice. However the AhCre expression system is inducible and transient, and thus we have been able to show that providing that loss of Chk1 activity is only temporary, the functional integrity of the intestine remains intact due to its capacity to repopulate. In addition, the in vivo consequences of partial Chk1 inhibition remain to be clarified. Although we have not observed an intestinal phenotype in Chk1 heterozygotes, the findings of Lam et al. (Lam et al., 2004), are consistent with a role as a haploinsufficient tumour suppressor. This would clearly have implications for any drug that partially inhibits Chk1 function, especially if given over a sustained period and in combination with DNA damaging agents. Inhibitors of Chk1 remain a viable therapeutic tool but it is clear a comprehensive understanding of its in vivo biology is essential to ensure a safe and effective integration with current treatment modalities.
Supplementary Material
Acknowledgements
This work was supported by Cancer Research UK. Particular thanks go to Mark Bishop, Lucie Pietzka and Derek Scarborough for technical assistance. The authors declare no conflicts of interest.
References
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–9. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- Bjerknes M, Cheng H. Methods for the isolation of intact epithelium from the mouse intestine. Anat Rec. 1981;199:565–74. doi: 10.1002/ar.1091990412. [DOI] [PubMed] [Google Scholar]
- Chen Z, Xiao Z, Chen J, Ng SC, Sowin T, Sham H, et al. Human Chk1 expression is dispensable for somatic cell death and critical for sustaining G2 DNA damage checkpoint. Mol Cancer Ther. 2003;2:543–8. [PubMed] [Google Scholar]
- Hapke G, Yin MB, Rustum YM. Targeting molecular signals in chk1 pathways as a new approach for overcoming drug resistance. Cancer Metastasis Rev. 2001;20:109–15. doi: 10.1023/a:1013116826788. [DOI] [PubMed] [Google Scholar]
- Ireland H, Kemp R, Houghton C, Howard L, Clarke AR, Sansom OJ, et al. Inducible Cre-mediated control of gene expression in the murine gastrointestinal tract: effect of loss of beta-catenin. Gastroenterology. 2004;126:1236–46. doi: 10.1053/j.gastro.2004.03.020. [DOI] [PubMed] [Google Scholar]
- Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29:418–25. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- Kawabe T. G2 checkpoint abrogators as anticancer drugs. Mol Cancer Ther. 2004;3:513–9. [PubMed] [Google Scholar]
- Koniaras K, Cuddihy AR, Christopoulos H, Hogg A, O’Connell MJ. Inhibition of Chk1-dependent G2 DNA damage checkpoint radiosensitizes p53 mutant human cells. Oncogene. 2001;20:7453–63. doi: 10.1038/sj.onc.1204942. [DOI] [PubMed] [Google Scholar]
- Kramer A, Mailand N, Lukas C, Syljuasen RG, Wilkinson CJ, Nigg EA, et al. Centrosome-associated Chk1 prevents premature activation of cyclin-B-Cdk1 kinase. Nat Cell Biol. 2004;6:884–91. doi: 10.1038/ncb1165. [DOI] [PubMed] [Google Scholar]
- Lam MH, Liu Q, Elledge SJ, Rosen JM. Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell. 2004;6:45–59. doi: 10.1016/j.ccr.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–31. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–59. [PMC free article] [PubMed] [Google Scholar]
- Merritt AJ, Allen TD, Potten CS, Hickman JA. Apoptosis in small intestinal epithelial from p53-null mice: evidence for a delayed, p53-independent G2/M-associated cell death after gamma-irradiation. Oncogene. 1997;14:2759–66. doi: 10.1038/sj.onc.1201126. [DOI] [PubMed] [Google Scholar]
- Nyberg KA, Michelson RJ, Putnam CW, Weinert TA. Toward maintaining the genome: DNA damage and replication checkpoints. Annu Rev Genet. 2002;36:617–56. doi: 10.1146/annurev.genet.36.060402.113540. [DOI] [PubMed] [Google Scholar]
- Robinson HM, Jones R, Walker M, Zachos G, Brown R, Cassidy J, et al. Chk1-dependent slowing of S-phase progression protects DT40 B-lymphoma cells against killing by the nucleoside analogue 5-fluorouracil. Oncogene. 2006;25:5359–69. doi: 10.1038/sj.onc.1209532. [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J Biol Chem. 2000;275:9390–5. doi: 10.1074/jbc.275.13.9390. [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- Sansom OJ, Griffiths DF, Reed KR, Winton DJ, Clarke AR. Apc deficiency predisposes to renal carcinoma in the mouse. Oncogene. 2005;24:8205–10. doi: 10.1038/sj.onc.1208956. [DOI] [PubMed] [Google Scholar]
- Shibata H, Toyama K, Shioya H, Ito M, Hirota M, Hasegawa S, et al. Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science. 1997;278:120–3. doi: 10.1126/science.278.5335.120. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–1. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Syljuasen RG, Sorensen CS, Hansen LT, Fugger K, Lundin C, Johansson F, et al. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol. 2005;25:3553–62. doi: 10.1128/MCB.25.9.3553-3562.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai H, Tominaga K, Motoyama N, Minamishima YA, Nagahama H, Tsukiyama T, et al. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(-/-) mice. Genes Dev. 2000;14:1439–47. [PMC free article] [PubMed] [Google Scholar]
- Tse AN, Carvajal R, Schwartz GK. Targeting checkpoint kinase 1 in cancer therapeutics. Clin Cancer Res. 2007;13:1955–60. doi: 10.1158/1078-0432.CCR-06-2793. [DOI] [PubMed] [Google Scholar]
- Wang Q, Fan S, Eastman A, Worland PJ, Sausville EA, O’Connor PM. UCN-01: a potent abrogator of G2 checkpoint function in cancer cells with disrupted p53. J Natl Cancer Inst. 1996;88:956–65. doi: 10.1093/jnci/88.14.956. [DOI] [PubMed] [Google Scholar]
- Xiao Z, Chen Z, Gunasekera AH, Sowin TJ, Rosenberg SH, Fesik S, et al. Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J Biol Chem. 2003;278:21767–73. doi: 10.1074/jbc.M300229200. [DOI] [PubMed] [Google Scholar]
- Zachos G, Rainey MD, Gillespie DA. Chk1-deficient tumour cells are viable but exhibit multiple checkpoint and survival defects. Embo J. 2003;22:713–23. doi: 10.1093/emboj/cdg060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaugg K, Su YW, Reilly PT, Moolani Y, Cheung CC, Hakem R, et al. Cross-talk between Chk1 and Chk2 in double-mutant thymocytes. Proc Natl Acad Sci U S A. 2007;104:3805–10. doi: 10.1073/pnas.0611584104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.