

Abstract
A diastereoselective synthesis of the nucleoside adducts corresponding to a cis ring opening of the carcinogen (±)-7β,8α-dihydroxy-9α,10α-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene (BaP DE-2) by 2’-deoxyadenosine and 2’-deoxyguanosine is described. The key intermediate (±)-10α-amino-7β,8α,9α-trihydroxy-7,8,9,10-tetrahydrobenzo[a]pyrene was synthesized by a highly diastereoselective dihydroxylation wherein phenylboronic acid was a water surrogate. The resulting boronate ester was converted to a tetraol derivative in which two of the four hydroxyl groups (trans 7, 8) were protected as benzoate esters while the remaining two (cis 9, 10) were free. The cis glycol entity was then subjected to a reaction with 1-chlorocarbonyl-1-methylethylacetate to yield an intermediate chloro monoacetoxy dibenzoate. Displacement of the halide with azide, complete cleavage of the esters and catalytic reduction of the azide yielded the requisite amino triol. Fluoride displacement from appropriately protected nucleoside derivatives, 6-fluoropurine 2’-deoxyribonucleoside and 2-fluoro-2’-deoxyinosine, by the amino triol then yielded diastereomeric pairs of diol epoxide adducted 2’-deoxyadenosine (dA) and 2’-deoxyguanosine (dG) nucleosides. Small aliquots of these adducts were separated for characterization purposes. The present approach provides the first diastereoselective synthesis of the cis adducts of BaP DE-2 with 2’-deoxyguanosine as well as the first synthesis of both dA and dG adducts from a common intermediate. An informative analysis of the 1H NMR spectra of the cis adducts synthesized and comparisons to the trans adducts are reported. To gain insight into the diastereoselectivity in the key dihydroxylation step, a computational analysis, including molecular mechanics (MMFF94) and semi-empirical AM1 geometry optimizations, yielded results that are in fairly good agreement with the experimental observations.
Keywords: Benzo[a]pyrene, nucleoside adducts, diastereoselective dihydroxylation
INTRODUCTION
Polycyclic aromatic hydrocarbons, products of activities in modern society, are a class of compounds that are widely prevalent in the environment. Several members of this class that are alternant hydrocarbons, possessing a bay region, are potent carcinogens.1 In mammalian systems, the biological activity of these compounds stems from their metabolism wherein bay region diol epoxides are formed by the combined actions of cytochrome P450 and epoxide hydrolase. Thus, any given hydrocarbon is converted to four isomeric diol epoxides that are classified as series 1 (DE-1) and series 2 (DE-2) depending upon the disposition of the benzylic hydroxyl and the oxirane.2 In the series 1 compounds these are on the same side of the molecule, whereas in the series 2 isomers these are on opposite sides. Benzo[a]pyrene (BaP) is a hydrocarbon that has received significant attention due to its environmental presence and the high carcinogenicity of its diol epoxide metabolites. Figure 1 shows the structures of the series 1 and 2 diolepoxides of this compound.
Figure 1.

Structures of the series 1 and series 2 diol epoxides of BaP0.
The mechanism of action of diol epoxides is attributed to their electrophilic nature. In the presence of DNA, diol epoxides covalently bind to the exocyclic amino groups of 2’-deoxyadenosine (dA) and 2’-deoxyguanosine (dG) via a putative carbocation that is produced by protonation and ring-opening of the oxirane.3 Such a reaction therefore produces a net sixteen nucleoside adducts. Since the 4 isomeric diol epoxides have markedly different potencies, with only the (+)-BaP DE-2 being highly tumorigenic,4 it is likely that the structural features associated with each covalent adduct influence the biological outcome. Figure 2 shows the structures of the 8 nucleoside adducts formed from (+)- and (−)-BaP DE-2.
Figure 2.

Structures of the 8 nucleoside adducts (4dA + 4dG) formed from (±)-BaP DE-2.
To gain a greater insight into these structure-activity factors, research has been directed towards the synthesis of DNA fragments that contain site-specific modifications with stereochemically defined diol epoxide lesions. Such site-specifically modified DNA are valuable probes for structural studies by NMR5 and biological experimentation.6 For the synthesis of diol epoxide adducted DNA three approaches have been adopted: (a) direct reaction of DNA oligomers with diol epoxides,7 (b) reaction of DNA containing reactive nucleosides with stereochemically defined hydrocarbon amino triols,8 and (c) synthesis of individual nucleoside-diol epoxide adducts which are subsequently introduced into DNA.9,10
We have been involved with method (c), the total synthesis approach, wherein individual nucleoside adducts are synthesized and subsequently introduced into specific sites of any DNA oligomer via modified solid-phase synthesis procedures.10 Despite being somewhat labor intensive, it is more flexible and versatile in providing access to any DNA oligomer so long as synthetic routes are available to the individual diol epoxide-nucleoside adducts. Although diol epoxides are good electrophiles, the relatively low nucleophilicity of the nucleoside amino groups prompts the nucleophile-electrophile role reversal in our approach. Thus, diol epoxides are either directly converted to amino triols via amminolysis10a or they areconverted to azido triols via reaction with azide anion, that are then peracylated and reduced to yield amino triacyl derivatives.11,12 These amino hydrocarbon derivatives are then coupled with electrophilic nucleosides to produce the diol epoxide-nucleoside adducts.10 Studies by us and others have led to the understanding that fluoro nucleosides possess sufficient reactivity to couple with the sterically hindered, less reactive bay-region amines.13 We have used such an approach for the synthesis of DNA adducts arising by a trans ring opening of (±)-BaP DE-1 and DE-2.14
In contrast to the relatively easy access to the amino triol derivatives arising by a trans ring opening of the diol epoxides, synthesis of the isomeric cis ring opened amino triols is less trivial. The latter amino triols are key entities for the synthesis of nucleoside adducts arising by a cis ring-opening of the diol epoxides. Cis epoxide ring opening of (±)-BaP DE-2 7,8-bis-O-TMS ether derivative by Ti(OiPr)4/TMSN3 in THF has been reported.15,16 However, the factors leading to this unusual stereochemical outcome are not known, and when the method was applied to 7,8-bis-O-TMS etherderivative (±)-BaP DE-1, a 1.5:1 ratio of cis/trans ring opening products was obtained.17 The synthetic challenges have therefore resulted in efforts that circumvent the need for the amino triols. For example, adducts corresponding to a cis ring-opening of (±)-BaP DE-2 by dA have been synthesized by an amino hydroxylation method involving the exocyclic amino group of dA and the olefin (±)-trans-7,8-dihydroxy-7,8-dihydro BaP.17 Whereas this method seems to work well for the synthesis of the cis dA adducts of(±)-BaP DE-2 no comparable chemistry has been reported for the dG system. On the other hand, cis dG adducts of BaP DE-2 have been synthesized via a direct ring-opening of the racemic diol epoxide with O6-allyl-3’,5’-bis-O-(tert-butyldimethylsilyl)-2’-deoxyguanosine. In one such method the diol epoxide was allowed to react with the protected dG derivative in N,N-dimethylacetamide at 90–100 °C.18 This reaction produced a mixture of trans and cis adducts in a ~1:1.4 ratio, total 47% yield after purification.18 This was followed by a report involving a “solvent-free” reaction in which (±)-BaP DE-2 was mixed with the protected dG and a few drops of CH2Cl2 in a mortar, which upon standing overnight produced a ~6:1 mixture of trans and cis adducts in a combined yield of 54%.19 More recently, a direct reaction of (±)-BaP DE-2 and protected dG in trifluoroethanol was reported.20 This reaction yielded a combined 43% of the trans and cis adducts in a 1.5:1 ratio.20 In each of these methods, the trans adducts were separated from the cis isomers by HPLC. Interestingly, when 3’,5’-bis-(O-tert-butyldimethylsilyl)-2’-deoxyadenosine was allowed to react with (±)-BaP DE-2 under the various conditions the results were as follows; under solvent-free conditions low (~5%) yield of the adducts was obtained,19 whereas in trifluoroethanol a 33% yield of only the cis ring opening product was observed.20
Cis ring opening of BaP DE-2 by the exocyclic amino group of dG can be accomplished in direct reactions with DNA. Although some control of cis versus trans ring opening can be exercised in these reactions by variation of reaction conditions, the reactions typically produce both cis and trans ring opening products that need separation.21 Since there are to date no diastereoselective total syntheses of the adducts corresponding to a cis ring opening of (±)-BaP DE-2 by dG, we have devised a novel, highly diastereoselective synthesis of a key intermediate (±)-10α-amino-7β,8α,9α-trihydroxy-7,8,9,10-tetrahydro BaP. This amino triol when coupled with C-6 and C-2 fluoro purine nucleosides afforded the corresponding dA and dG adducts of (±)-BaP DE-2. The basis of the high diastereoselection observed in a critical synthetic step has also been evaluated by molecular mechanic and semi-empirical AM1 computations and these results are in fairly good agreement with the experimental observations.
RESULTS AND DISCUSSION
Synthesis and Compound Characterization
Our planned synthesis of the key intermediate (±)-10α-amino-7β,8α,9α-trihydroxy-7,8,9,10-tetrahydro BaP dictated that the C-7 and C-8 hydroxyls remain protected, and this task would be simplest with a suitably protected dihydrodiol. Therefore, our synthesis commenced from the readily available 7,8-bis-benzoyloxy-7,8-dihydro BaP (1), that can be prepared on the multi gram scale in three steps from commercially available 7-hydroxy-7,8,9,10-tetrahydrobenzo[a]pyren-7-ol.22 Dihydrodibenzoate 1 under catalytic dihydroxylation conditions yielded a mixture of two compounds that could be separated with some difficulty (4 and 6 in Scheme 1). The 1H NMR spectra of the separated compounds after complete deprotection (NH3/MeOH, 50 °C) and reacetylation (Ac2O/pyridine/DMAP) indicated that these were two tetraol tetraacetates. Comparison of the NMR spectra of the two products to reported data for 7β,8α,9β,10β-tretraacetoxy-7,8,9,10-tetrahydro BaP (5) as well as 7β,8α,9α,10α-tetraacetoxy-7,8,9,10-tetrahydro BaP (7), that have been independently prepared by hydrolysis reactions of BaP DE-1 and DE- 2,23 showed that the major product from the dihydroxylation of 1 was 6. To ascertain the ratio of the two products, in one dihydroxylation reaction of 1, the mixture of tetraol dibenzoate products 4 and 6 were debenzoylated and reacetylated. Based upon the proton integrals of the mixture, the ratio of 5:7 was determined to be ~1:4.
Scheme 1.

This direct dihydroxylation of 1 was unfortunately not ideal due to the formation of two compounds that were not easily separable. We therefore decided to analyze the dihydroxylation of other protected dihydrodiol derivatives. For this we chose the 7,8-bis-(tert-butyldimethylsilyl)ether derivative 3. This compound was readily prepared from 1 by hydrolysis to the dihydrodiol 2 and subsequent silylation with tert-BuMe2SiOTf /Et3N (5 mol equiv each) in CH2Cl2 (92% yield). Dihydroxylation of 3 with OsO4 (1.1 mol equiv) in pyridine yielded, after one recrystallization from MeOH, a single tetraol bis-silyl ether 8 (85% yield) the structure of which was confirmed by desilylation and peracetylation to 7. These observations led us to initially investigate the factors leading to the observed diastereoselectivities in the dihydroxylation of 1 and 3. Although the addition of OsO4 in these reactions occurred predominantly from the same side as the allylic hydroxyl group, reminiscent of the allylic hydroxyl-mediated epoxidation, we believe that the facial selectivity of the dihydroxylation reactions was likely not H-bonding driven. This rationale was based on the observation that dihydrodiol 2 and its bis-(tert-butyldimethylsilyl) ether 3 undergo the same facial addition by OsO4. We also reasoned that the facial selectivity observed in the reactions of 1 and 3 was dependent on the conformation of the angular dihydro ring as determined by the C-7, C-8 substituents. For this purpose we briefly investigated the H-7 to H-8 coupling constants as a function of solvent. This data as well as other coupling constants are shown in Table 1.
Table 1.
The scalar coupling constants of the tetrahydro ring protons of compounds 1–3 in different solventsa
| JH7,H8 | JH7,H9 | JH8,H9 | JH8,H10 | JH9,H10 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| solvent | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 |
| CDCl3 | 7.5 | 11 | 9.9 | 0 | 0 | 0 | 3.6 | 1.9 | 2.4 | 1.3 | 1.9 | 2.1 | 10.2 | 10 | 10.2 |
| CD2Cl2 | 7.4 | 11.2 | 8.5 | 0.9 | 1 | 0.9 | 3.7 | 2.3 | 3.1 | 1.5 | 1.5 | 1.8 | 10.3 | 10.1 | 9.9 |
| (CD3)2CO | 7.7 | 11.2 | 8 | 0 | 0 | 0 | 3.4 | 2.3 | 3.1 | 2 | 2.4 | 1.5 | 10.1 | 10.1 | 9.4 |
| THF-d8 | 7 | 11.5 | 8.6 | 0 | 0 | 0 | 3.7 | 2 | 2.9 | 1.2 | 2.3 | 1.5 | 10.1 | 10.1 | 10.3 |
| C5H5N | 7.5 | 11.3 | 9 | 0 | 0 | 0 | 2.1 | 3.7 | 2.8 | 1.2 | 2.6 | 1.4 | 10.2 | 10.1 | 9.9 |
Data was obtained at ambient temperature, at 500 MHz.
Analysis of the of the JH7,H8 data shown in Table 1 immediately indicates that the dihydrodiol 2 exhibits a quasi diequatorial arrangement of the hydroxyls, almost to an extreme. Additionally, the JH7,H8 in 2 is relatively independent of the solvent polarity. These observations are consistent with the proposal that in the dihydrodiol an intramolecular hydrogen bond exists between the two hydroxyls.24 On the other hand, protection of the hydroxyls in 1 and 3 removes the hydrogen bond and greater flexibility is observed along with a greater diaxial orientation of the substituents. Among the two protected dihydrodiols, the bis silyl derivative 3 is more conformationally flexible and exhibits a greater diequatorial orientation of the substituents than dibenzoate 1. We believe that greater the preference for the diequatorial orientation of the substituents, greater the diastereoselection observed in the OsO4 dihydroxylation. Thus, dihydrodiol 2 yields a single detectable tetraol diastereomer, and a similar phenomenon is likely with the bis silyl compound 3. On the other hand, in the case of dibenzoate 1 where a greater preference for the diaxial substituents is seen compared to 2 and 3, two tetraol dibenzoates are produced.
The observed diastereoselectivities can be rationalized based upon the transition state structures leading to the two diastereomeric diol products in these cases. Inspection of molecular models suggests that dihydroxylation from the opposite side to the allylic C-8 OR substituent [Figure 3 pathway (a)] would lead to eclipsing interactions between the substituents on C-8 (shown in red) and those on C-9 (shown inblue). On the other hand, the substituents at C-8 and C-9 would be in a staggered conformation if the addition occurred from the same side as the allylic C-8 OR substituent [Figure 3 pathway (b)]. Therefore, such a torsional steering25 is perhaps the origin of the diastereoselectivities observed, rendering pathway (b) more favorable than (a). Such a rationale as well as the data in Table 1 suggest that a greater propensity for diequatorial orientation of the substituents in the dihydrodiol, greater the contribution to the favored route.
Figure 3.

Plausible substituent interactions leading to the product distribution in the dihydroxylation.
Although these studies formed a basis for understanding conformational influences leading to the diastereoselection, the difficulty in separating 4 from 6 as well as the fact that 8 could not be used for further reactions (explained later) raised the need for a facile route to 6. Use of PhB(OH)2 as water surrogate in OsO4-mediated dihydroxylation has been reported.26 It has also been possible to influence the facial selectivity of dihydroxylation by this modified method.27 In the current context we wondered about the course of the dihydroxylation of 1 in the presence of PhB(OH)2. We reasoned that the short B-Obonds in conjunction with the formation of fused 6- and 5-membered ring systems in the product would likely increase the contribution of path (b).
As shown in Scheme 2, boronate 9 was obtained by the reaction of 1 with catalytic OsO4/NMO/PhB(OH)2 in CH2Cl2. This reaction, which was generally high yielding, could be readily conducted on the multigram scale, with a simple precipitation from hexanes sufficient to purify 9. Within the detection limits of a 500 MHz instrument, the 1H NMR spectrum of the product obtained via this procedure showed the presence of a single boronate isomer. This boronate 9 was then oxidized with 50% H2O2 in 2:1 acetone-EtOAc to yield a tetraol dibenzoate also in generally good yield. Determination of the facial selectivity in the conversion of 1→9 came at the stage of boronate cleavage. The tetraol dibenzoate was completely hydrolyzed and peracetylated as before. The resulting tetraol tetraacetate was identical to 7 whose structure had been independently confirmed by comparison to the hydrolysis products of (±)-BaP DE-2.23 This indicated that the boronate formation had occurred from the same side as the allylic benzoate.
Scheme 2.

With the diastereoselectivity of the boronate formation established, the next stages in the synthesis centered around the selective conversion of the benzylic hydroxyl in 6 to an amino functionality with an overall retention of stereochemistry. We have reported that reaction of 1-aryl ethylene glycols with 1-chlorocarbonyl-1-methylethyl acetate (α-acetoxy-isobutyryl chloride) yields reverse, trans chlorohydrin acetates, with a benzylic halide.28 Application of this approach in the present case should provide an intermediate with a benzylic chloride that can then be manipulated to yield an amine. The overall scheme for the synthesis of (±)-10α-amino-7β,8α,9α-trihydroxy-7,8,9,10-tetrahydro BaP is shown in Scheme 3.
Scheme 3.

Reaction of 6 with 1-chlorocarbonyl-1-methylethylacetate in CH3CN yielded the putative intermediate 10. Since the benzylic halide on such an extended aryl system was expected to be highly labile, direct displacement of the chloride by azide was performed immediately upon workup. The resulting azido triacyl compound 11 was isolated, purified and characterized. Cleavage of all ester linkages yielded azido triol 12. Finally, catalytic reduction of the azide moiety provided amino triol 13 (the corresponding salt could be obtained if the reduction was performed in the presence of 6 M HCl).
Attempted reaction of the tetraol bis silyl ether 8 with 1-chlorocarbonyl-1-methylethylacetate was not very promising. This may be due either to the incompatibility of silyl groups to HCl formed in the reaction or because the benzoate groups of 6 provide electronic stabilization of the benzylic chloro intermediate 10 that the silyl groups do not offer.
In order to compare amino triol 13 with the diastereomeric (±)-10β-amino-7β,8α,9α-trihydroxy-7,8,9,10-tetrahydro BaP (product of trans ring opening of (±)-BaP DE-2) as well as other tetrahydro BaP derivatives, a small portion of 13 was converted to the tetraacetate. Table 2 is a listing of key chemical shifts and coupling constants in a series of different tetrahydro BaP derivatives. Clearly, the greatest difference lies in the H7–H8 coupling constant, with derivatives arising by a trans epoxide opening displaying a predominantly diequatorial orientation of the substituents (J7,8 = 8.3–9.5 Hz). On the other hand, in the products corresponding to a cis ring opening the substituents adopt a more diaxial orientation (J7,8 = 3.4–4.0 Hz).
Table 2.
Chemical shifts and coupling constants of the tetrahydro ring protons in a series of BaP derivatives
| compound | H-7 | H-8 | H-9 | H-10 |
|---|---|---|---|---|
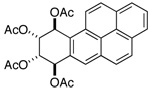 |
d 6.96 | dd 5.89 | dd 5.95 | d 7.12 |
| From ref. 23(CDCl3): J7,8 = 8.8; J8,9 = 2.5; J9,10 = 3.6 | ||||
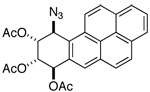 |
d 6.79 | dd 5.83 | dd 5.86 | d 5.64 |
| 300 MHz (CDCl3): J7,8 = 8.3; J8,9 = 2.4; J9,10 = 4.0 | ||||
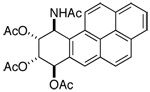 |
d 6.76 | dd 5.72 | App t 5.82 | dd 6.10 |
| From ref. 10a (Acetone-d6): J7,8 = 9.5; J8,9 = 2.3; J9,10 = 3.4; J10,NH = 8.0 | ||||
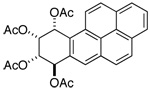 |
d 6.67 | app t 5.68 | dd 5.99 | d 7.36 |
| 300 MHz (CDCl3): J7,8 = 3.5; J8,9 = 2.5; J9,10 = 4.7 | ||||
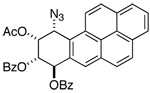 |
d 7.08 | dd 6.05 | dd 6.22 | d 6.00 |
| 500 MHz (CDCl3): J7,8 = 4.0; J8,9 = 2.4; J9,10 = 5.0 | ||||
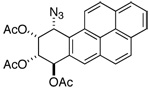 |
d 6.64 | dd 5.71 | dd 5.90 | d 5.74 |
| 300 MHz (CDCl3): J7,8= 3.6; J8,9 = 2.4; J9,10 = 5.4 | ||||
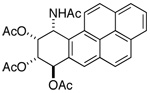 |
d 6.57 | br m 5.64 | dd 5.80 | dd 6.53 |
| 500 MHz (Acetone-d6): J7,8 = 3.4; J8,9 = 2.4; J9,10 = 5.4; J10,NH= 9.8 | ||||
With the synthesis of the amino triol and the relative stereochemical assignment completed, the final stage in the synthesis was the preparation of the dA and dG adducts. Although we and others have developed metal-mediated reactions for the assembly of carcinogen-nucleoside lesions,29 the presence of free hydroxyl groups was a consideration. In order to avoid unwanted problems, displacement of fluoride from fluorinated nucleosides10,14,30 was the method chosen for the present work. Reactions of 6-fluoro-9-(2-deoxy-3,5-bis-O-(tert-butyldimethylsilyl)-β-D-erythropentofuranosyl)purine and 3’,5’-bis-O-(tert-butyldimethylsilyl)-2-fluoro-2’-deoxyinosine with 13 were conducted in DMSO in the presence of N,N-diisopropylethylamine and hexamethyldisiloxane at 85–90 °C to yield the corresponding 2’-deoxyadenosine and 2’-deoxyguanosine adducts, respectively (Scheme 4). Reactions leading to the dA adduct pair 14a, 15a proceeded in good yield (65–70%) whereas those leading to the dG adduct pair 16a, 17a were lower yielding (ca 30–40%).
Scheme 4.

Small amounts of pure diastereomers from each pair were separated and characterized as their triacetates (14b–17b) via NMR spectroscopy. Since the chemical shift differences between the individual cis and trans ring opened adducts could be important in the analysis of DNA structures containing them, a comparison of key chemical shift data is provided in Table 3. The chemical shifts of the sugar and the tetrahydrobenzo ring protons of the adducted mononucleotides were unequivocally assigned from an analysis of corresponding 2-D NOESY and COSY spectra. Of the two benzylic resonances (H-10 and H-7), H-10 is easily identified from its coupling to both H-9 and the amino NH. Assignment of H-10 is also confirmed from NOE interaction with H-11. On the other hand, H-7 shows NOE with H-6 singlet.
Table 3.
Chemical shift data for the cis and trans ring opened (±)-BaP DE-2 nucleoside adducts
| adducta | H-1’ | H-2’β | H-2’α | H-7 | H-8 | H-9 | H-10 |
|---|---|---|---|---|---|---|---|
| 2’-Deoxyadenosine adducts via cis ring opening of BaP DE-2b,c | |||||||
| 10-S | 6.46 | 2.67 | 2.45 | 6.65 | 5.75 | 6.01 | 7.15 |
| 10-R | 6.48 | 2.58 | 2.43 | 6.66 | 5.75 | 6.00 | 7.16 |
| 2’-Deoxyadenosine adducts via trans ring opening of BaP DE-2b,d,e | |||||||
| 10-S | 6.47 | 2.61 | 2.45 | 6.78 | 5.90 | 6.15 | 6.58 |
| 10-R | 6.43 | 2.70 | 2.43 | 6.76 | 5.91 | 6.14 | 6.59 |
| 2’-Deoxyguanosine adducts via cis ring opening of BaP DE-2 d,f | |||||||
| 10-S | 6.37 | 2.75 | 2.39 | 6.52 | 5.60 | 5.80 | 6.58 |
| 10-R | 6.28 | 3.20 | 2.25 | 6.53 | 5.61 | 5.84 | 6.59 |
| 2’-Deoxyguanosine adducts via trans ring opening of BaP DE-2 d,e,f | |||||||
| 10-S | 6.37 | 2.66 | 2.37 | 6.68 | 5.75 | 5.96 | 6.01 |
| 10-R | 6.26 | 3.32 | 2.17 | 6.67 | 5.76 | 5.90 | 5.98 |
R and S refer to the configuration at the point of attachment (C-10) of the BaP moiety to the amino group of the nucleoside.
Obtained in CDCl3.
Acquired at 600 MHz.
Acquired at 500 MHz.
Independently synthesized and analyzed.
Obtained in DMSO-d6.
Good line shape and resolution for the dA adducts were observed in CDCl3 (deacidified) or acetone-d6 and the NMR spectra can be obtained conveniently in either. The dG adducts on the other hand, provided good line shape and resolution only in DMSO-d6, and this appears to be the case with such hydrocarbon adducts generally. As can be seen from Table 3, among the pairs of dA adducts derived from a cis or trans opening the differences in chemical shifts of the protons shown are relatively small (|δΔ| = 0.01–0.09 ppm for both the cis and trans ring-opened pairs). In contrast, among the pairs of dG adducts these chemical shift differences become larger ((|δΔ| = 0.01–0.45 ppm for the cis ring-opened pair and 0.01–0.66 ppm for the trans ring-opened pair). In terms of the magnitude of the difference, the protons most affected are theH-2’β in each case, followed by H-2’α and then H-1’. Among the dG adducts, the 10R isomers show significantly downfield shifted H-2’β and this is more pronounced for the trans adduct. The stereochemical disposition of the 2’-protons in the adducted mononucleosides was obtained from the NOESY spectra of one adduct from each pair. In this analysis the more upfield H-2’ displayed a stronger nOe with H-1’ and was hence labeled as α. Correspondingly, the more downfield H-2’ was labeled β.
In a study of a large database of DNA chemical shifts,31 the center of the shift distributions of the H-2’ protons in pyrimidines is 2.0 ppm and that of the purines is 2.6 ppm, with very little overlap between them. The chemical shifts of the deoxyribose ring protons, especially those at C-2’, are predicted to be affected by the magnitude of glycosidic torsion angle that results from ring current and the local magnetic anisotropy of their bases.32 In the case of the BaP adducts, the presence of the pyrene ring can greatly affect the chemical shifts of the sugar protons due to the ring current of the fairly large aromatic residue. A preliminary examination of the spatial proximity of the BaP ring to the sugar moieties in the dA and dG adducts has been conducted by molecular modeling (MM2). As may be anticipated, in the case of the dG adducts the pyrene ring is closer to the deoxyribose moiety, whereas in the case of the dA adducts the pyrene ring is farther away. Consistent with this, the chemical shifts of the 2’ protons of the dA adducts are influenced to a lesser extent by the BaP residue than those of the dG.
For purposes of comparison Figure 4 shows the CD spectra of BaP dA and dG adducts resulting from cis and trans ring opening. Although the literature amply documents CD spectra of such PAH diol epoxide-nucleoside adducts, Figure 4 highlights some differences. Whereas the cis and trans ring opened dA adduct pair bear a nearly mirror image relationship, the cis and trans ring opening dG adduct pair show subtly different CD spectra. Thus, these minor differences are related to the stereochemistry of the epoxide ring opening. However, the CD spectra are consistent with previously published CD data of BaP-nucleoside adducts.33
Figure 4.

Circular dichroism spectra (in MeOH) of 4 out of the 8 (±)-BaP DE-2 dA and dG adducts. Panel A shows a pair of dA adducts: blue = 10S trans, red = 10R cis. Panel B shows a pair of dG adducts: blue = 10S trans, red = 10R cis.
Computational Analysis
The potential energies of the lowest energy conformations obtained after the MC conformational searches for the compounds in Figure 5 show that for the BaP tetraol dibenzoate pair (BaP1 and BaP2),BaP1 dominates, and for the pair of boronate esters (BaPB1 and BaPB2), BaPB1 dominates (Table A in the Supporting Information). Of the flexible torsion angles in the tetraol dibenzoates and their boronate esters (Table 4), there are two torsion angles for each of the tetraol dibenzoate isomers and five for each of their boroate esters. Only those torsion angles at the point of reaction and where the isomers differ in absolute configuration are considered.
Figure 5.

Structures of diastereomeric tetraol dibenzoates and their boronate esters with torsion angles (BaP1 is the major isomer 6 in Scheme 1 and BaPB1 is isomer 9 isolated in Scheme 2).
Table 4.
Monte Carlo torsion angles for the tetraol dibenzoates and their boronate esters (degrees)
| Angle | Atoms | BaP1a | BaP2 | BaPB1b | BaPB2 |
|---|---|---|---|---|---|
| χ’α | H-O-C-H | −73.2 | 73.8 | -- | -- |
| χ’β | H-O-C-H | −71.1 | 96.8 | -- | -- |
| χ’1 | C-C-B-O | -- | -- | −37.8 | −24.7 |
| χ’2 | C-B-O-C | -- | -- | −107.2 | 119.1 |
| χ’3 | B-O-C-H | -- | -- | −145.8 | 137.2 |
| χ’4 | B-O-C-H | -- | -- | 80.5 | −75.9 |
| χ’5 | C-B-O-C | -- | -- | 132.1 | −144.6 |
The BaP1 isomer is favored by 86%.
The BaPB1 isomer is favored by 99%.
The geometries resulting for the AM1 calculations (Table 5) yield heats of formation (Table B in the Supporting Information) that also favor the BaP1 and BaPB1 isomers. The standard thermodynamic quantities computed at 298.15 K and 1.00 atm also favor the BaP1 and the BaPB1 in both the enthalpic contribution and entropic contributions, but the enthalpic contributions dominate (Table C in the Supporting Information). The energy difference and Boltzmann weighting factors calculated using the solvation potential energies (Table D in the Supporting Information) also favor the BaP1 isomer.
Table 5.
AM1 torsion angles for the tetraol dibenzoates and their boronate esters (degrees)
| Angle | Atoms | BaP1 a | BaP2 | BaPB1 b | BaPB2 |
|---|---|---|---|---|---|
| χ’α | H-O-C-H | −69.3 | 164.8 | -- | -- |
| Χ’β | H-O-C-H | −71.8 | 78.5 | -- | -- |
| Χ’1 | C-C-B-O | -- | -- | 0.9 | −5.6 |
| Χ’2 | C-B-O-C | -- | -- | −178.7 | 179.2 |
| χ’3 | B-O-C-H | -- | -- | −118.9 | 121.0 |
| χ’4 | B-O-C-H | -- | -- | 114.8 | −112.4 |
| χ’5 | C-B-O-C | -- | -- | −178.6 | 176.4 |
The BaP1 isomer is favored by 95–96%.
The BaPB1 isomer is favored by 88%.
For the tetraol dibenzoate and their boronate isomers, compared to the torsion angles of the Monte Carlo (MC) geometries, the torsion angles of the AM1 geometries are more symmetrical for the various isomers in which the angles are almost the same in magnitude but differ in direction (Tables 4 and 5). Although there are small variations in the energy differences (see the Computational Analysis section in the Supporting Information), all methods of determining equilibrium molecular geometries consistently agree on which isomer is dominant. Indeed, the predictions are borne out experimentally as well.
Structures resulting from the calculation of electron density surfaces (Figure 1 A–D in the Supporting Information) and calculation of electron density surfaces onto which the value of the electrostatic potential is mapped (Figure 6 A–D) depict the conformational arrangement of the major and minor isomers. The electrostatic potential is the hypothetical energy of a positive test charge at some location around a molecule. The electrostatic potential is positive in a region of excess positive charge and charge-molecule interaction is repulsive. Similarly, in a region of excess negative charge, the electrostatic potential is negative and the charge-molecule interaction is attractive. The electrostatic potential is shown by mapping it onto a surface of electron density, which depicts the size of the molecule, obtained from quantum mechanical calculation. An electrostatic potential map is a good indication of the size and shape of a molecule, the charge distribution, and the site of chemical reactivity.
Figure 6.

Structures of the tetraol dibenzoates BaP1 (A), BaP2 (B) as well as the corresponding boronate analogues BaPB1 (C) and BaPB2 (D), resulting from calculation of the electron density surface onto which the value of the electrostatic potential has been mapped. The different values of the electrostatic potential at the surface are represented by different colors: red represents regions of most negative electrostatic potential, blue represents regions of most positive electrostatic potential and green represents regions of zero potential. Potential increases in the order red < orange < yellow < green < blue.
EXPERIMENTAL SECTION
Synthesis and Compound Characterization
For space considerations all synthetic procedures and compound characterization details are provided in the Supporting Information.
Computational Analysis
A computational search of the isomeric tetraol derivatives of BaP (Figure 5) was performed with the molecular modeling package PC SPARTAN Pro (Wavefunction, Inc.) using a Monte Carlo (MC) method plus the MMF94 force field. The minimum energy structures were next subject to a semi-empirical quantum mechanical geometric optimization using AM1. Following AM1 calculation, the energy due to solvation for the tetraol dibenzoate isomers was calculated using the SM5.4 model of Chambers et al. (Table C in the Supporting Information).34 Details of the computational procedures can be found in the Supporting Information.
CONCLUSIONS
In conclusion, we have developed an efficient, highly diastereoselective synthesis of (±)-10α-amino-7β,8α,9α-trihydroxy-7,8,9,10-tetrahydrobenzo[a]pyrene, a key component utilized for preparing nucleoside adducts of the carcinogen BaP DE-2 corresponding to a cis ring-opening of the oxirane in the metabolite. This amino triol was utilized for the synthesis of the nucleoside conjugates and in this process good yields of the dA adducts were realized. Although the yields leading to the dG adducts were modest, we are currently considering methods wherein the described chemistry can be modified and coupled with Pd-catalysis, methodology that has proven to be highly effective for the synthesis of these types ofadducts.29 The chemical syntheses of carcinogen-nucleoside adducts becomes more significant when considering the minor DNA adducts formed. For instance in a direct reaction of (7R,8S,9S,10R)-BaP DE-2 with DNA, of the total nucleoside adducts formed, the cis ring opened dA and dG adducts are only ~1%each.35 With (7S,8R,9R,10S) enantiomer, within detection limits, no cis ring opened dA adducts were observed.35 Since minor or undetected adducts could play a disproportionate role in carcinogenesis, the present synthesis allows access to the unstudied, cis ring opened BaP DE-2 adduct.5c In the course of this synthesis we have also uncovered factors that control facial selectivity of addition to BaP dihydrodiol and its derivatives. To understand these computational techniques have been utilized. The results from the computation parallel quite closely the observed facial selectivities in the key OsO4 dihydroxylation leading to either the tetraol dibenzoates or the boronate esters. Although computational approaches have been utilized to understand conformational aspects of diol epoxide-nucleoside adducts and their impact on biochemical processes,36 a computation-based understanding of the chemistry of PAH derivatives has been lacking. The combination of molecular mechanics with quantum mechanics offers a useful approach in the computational evaluation of flexible organic molecules, and could offer insight into other reactions of PAH derivatives that may be useful in developing chemical syntheses such as those described here.
Supplementary Material
Complete experimental details including those for the computational analysis. 1H NMR spectra for the BaP derivatives 3, 6, 8, 9, 11–13 and nucleoside adducts 14b–17b. This information is available free of charge via the Internet at http://www.pubs.acs.org.
ACKNOWLEDGMENT
This paper is dedicated to Prof. Roland E. Lehr (1942–2003), a mentor and colleague. Support of this work by NIH grants 1R15 CA094224-01 (NCI) and S06 GM008168-24S1 (NIGMS) to MKL, ND EPSCoR grant to KAT, ND EPSCoR Doctoral Dissertation Fellowship to FNN and support for BZ via NIH RCMI grant 5G 12 RR03060-20 is gratefully acknowledged. Dr. M. Cosman is thanked for useful discussions and S. Patil for performing one adduct-forming reaction.
Contributor Information
Mahesh K. Lakshman, Email: lakshman@sci.ccny.cuny.edu.
Kathryn A. Thomasson, Email: kthomasson@chem.und.edu.
REFERENCES
- 1.Reviewed in: Yang SK, Silverman BD, editors. Polycyclic Aromatic Hydrocarbon Carcinogenesis: Structure-Activity Relationships. Volumes I and II. Boca Raton, FL: CRC Press; 1988. Harvey RG, editor. Polycyclic Aromatic Hydrocarbons: Chemistry and Carcinogenicity. Cambridge, U.K.: Cambridge Unviersity Press; 1991. Harvey RG, editor. Polycyclic Hydrocarbons and Carcinogenesis. Washington, DC: ACS Symposium Series 283; 1985.
- 2.Thakker DR, Levin W, Wood AW, Conney AH, Yagi H, Jerina DM. In: Drug Stereochemistry-Analytical Methods and Pharmacology. Wainer IW, Drayer DE, editors. New York: Marcel Dekker, Inc.; 1988. pp. 271–296. [Google Scholar]
- 3.Jerina DM, Chadha A, Cheh AM, Schurdak ME, Wood AW, Sayer JM. In: Biological Reactive Intermediates IV. Witmer CM, Snyder R, Jollow DJ, Kalf GF, Kocsis JJ, Sipes IG, editors. New York: Plenum Press; 1991. pp. 533–553. [Google Scholar]
- 4.Jerina DM, Sayer JM, Agarwal SK, Yagi H, Levin W, Wood AW, Conney AH, Pruess-Schwartz D, Baird WM, Pigott MA, Dipple A. In: Biological Reactive Intermediates III. Kocsis JJ, Jollow DJ, Witmer CM, Nelson JO, Snyder R, editors. New York: Plenum Press; 1986. pp. 11–30. [DOI] [PubMed] [Google Scholar]
- 5.For a review see: Geacintov NE, Cosman M, Hingerty BE, Amin S, Broyde S, Patel DJ. Chem. Res. Toxicol. 1997;10:111–146. doi: 10.1021/tx9601418. For other recent representative examples see: Sayer JM, Shah JH, Liang C, Xie G, Kroth H, Yagi H, Liu X, Yeh HJC, Jerina DM. Polycyclic Aromatic Compounds. 1999;17:95–104. Suri AK, Mao B, Amin S, Geacintov NE, Patel DJ. J. Mol. Biol. 1999;292:289–307. doi: 10.1006/jmbi.1999.2974. Mao B, Gu Z, Gorin A, Chen J, Hingerty BE, Amin S, Broyde S, Geacintov NE, Patel DJ. Biochemistry. 1999;38:10831–10842. doi: 10.1021/bi991212f. Li ZI, Kim H-Y, Tamura PJ, Harris CM, Harris TM, Stone MP. Biochemistry. 1999;38:2969–2981. doi: 10.1021/bi982072x. Li Z, Kim H-Y, Tamura PJ, Harris CM, Harris TM, Stone MP. Biochemistry. 1999;38:14820–14832. doi: 10.1021/bi991607z. Li Z, Kim H-Y, Tamura PJ, Harris CM, Harris TM, Stone MP. Biochemistry. 1999;38:16045–16057. doi: 10.1021/bi9903650. Volk DE, Rice JS, Luxon BA, Yeh HJC, Liang C, Xie G, Sayer JM, Jerina DM, Gorenstein DG. Biochemistry. 2000;39:14040–14053. doi: 10.1021/bi001669l. Pradhan P, Tirumala S, Liu X, Sayer JM, Jerina DM, Yeh HJC. Biochemistry. 2001;40:5870–5881. doi: 10.1021/bi002896q. Lin CH, Huang X, Kolbanovskii A, Hingerty BE, Amin S, Broyde S, Geacintov NE, Patel DJ. J. Mol. Biol. 2001;306:1059–1080. doi: 10.1006/jmbi.2001.4425. Li Z, Tamura PJ, Wilkinson AS, Harris CM, Harris TM, Stone MP. Biochemistry. 2001;40:6743–6755. doi: 10.1021/bi002785r. Kim H-YH, Wilkinson AS, Harris CM, Harris TM, Stone MP. Biochemistry. 2003;42:2328–2338. doi: 10.1021/bi020462k.
- 6.For some representative examples see: Kramata P, Zajc B, Sayer JM, Jerina DM, Wei CS-J. J. Biol. Chem. 2003;278:14940–14948. doi: 10.1074/jbc.M211557200. Huang X, Kolbanovskiy A, Wu X, Zhang Y, Wang Z, Zhuang P, Amin S, Geacintov NE. Biochemistry. 2003;42:2456–2466. doi: 10.1021/bi026912q. Xie Z, Braithwaite E, Guo D, Zhao B, Geacintov NE, Wang Z. Biochemistry. 2003;42:11253–11262. doi: 10.1021/bi0346704. Zou Y, Shell SM, Utzat CD, Luo C, Yang Z, Geacintov NE, Basu AK. Biochemistry. 2003;42:12654–12661. doi: 10.1021/bi034446e. Khan QA, Kohlhagen G, Marshall R, Austin CA, Kalena GP, Kroth H, Sayer JM, Jerina DM, Pommier Y. Proc. Natl. Acad. Sci. USA. 2003;100:12498–12503. doi: 10.1073/pnas.2032456100. Zou Y, Ma H, Minko IG, Shell SM, Yang Z, Qu Y, Xu Y, Geacintov NE, Lloyd RS. Biochemistry. 2004;43:4196–4205. doi: 10.1021/bi035992a. Zang H, Harris TM, Guengerich FP. Chem. Res. Toxicol. 2005;18:389–400. doi: 10.1021/tx049683c. Baskunov VB, Subach FV, Kolbanovskiy A, Kolbanovskiy M, Eremin SA, Johnson F, Bonala R, Geacintov NE, Gromova ES. Biochemistry. 2005;44:1054–1066. doi: 10.1021/bi048130y.
- 7.(a) Cosman M, Ibanez V, Geacintov NE, Harvey RG. Carcinogenesis. 1990;11:1667–1672. doi: 10.1093/carcin/11.9.1667. [DOI] [PubMed] [Google Scholar]; (b) Mao B, Xu J, Li B, Margulis LA, Smirnov S, Ya NQ, Courtney SH, Geacintov NE. Carcinogenesis. 1995;16:357–365. doi: 10.1093/carcin/16.2.357. [DOI] [PubMed] [Google Scholar]
- 8.(a) Kim SJ, Stone MP, Harris CM, Harris TM. J. Am. Chem. Soc. 1992;114:5480–5481. [Google Scholar]; (b) Harris TM, Harris CM, Kim SJ, Han S, Kim H-Y, Zhou L. Polycyclic Aromatic Compounds. 1994;6:9–16. [Google Scholar]; (c) DeCorte BL, Tsarouhtsis D, Kuchimanchi S, Cooper MD, Horton P, Harris CM, Harris TM. Chem. Res. Toxicol. 1996;9:630–637. doi: 10.1021/tx9501795. [DOI] [PubMed] [Google Scholar]; (d) Cooper MD, Hodge RP, Tamura PJ, Wilkinson AS, Harris CM, Harris TM. Tetrahedron Lett. 2000;41:3555–3558. [Google Scholar]
- 9.(a) Steinbrecher T, Becker A, Stezowski JJ, Oesch F, Seidel A. Tetrahedron Lett. 1993;34:1773–1774. [Google Scholar]; (b) Lee H, Luna E, Hinz M, Stezowski JJ, Kiselyov AS, Harvey RG. J. Org. Chem. 1995;60:5604–5613. [Google Scholar]
- 10.(a) Lakshman MK, Sayer JM, Jerina DM. J. Am. Chem. Soc. 1991;113:6589–6594. [Google Scholar]; (b) Lakshman MK, Sayer JM, Yagi H, Jerina DM. J. Org. Chem. 1992;57:4585–4590. [Google Scholar]; (c) Lakshman MK, Sayer JM, Jerina DM. J. Org. Chem. 1992;57:3438–3443. [Google Scholar]
- 11.(a) Lakshman M, Nadkarni DV, Lehr RE. J. Org. Chem. 1990;55:4892–4897. [Google Scholar]; (b) Lakshman MK, Chaturvedi S, Lehr RE. Synth. Commun. 1994;24:2983–2988. [Google Scholar]
- 12.Steinbrecher T, Wameling C, Oesch F, Seidel A. Angew. Chem. Int. Ed. Engl. 1993;32:404–406. [Google Scholar]
- 13.(a) Lakshman M, Lehr RE. Tetrahedron Lett. 1990;31:1547–1550. [Google Scholar]; (b) Kim SJ, Harris CM, Jung K-Y, Koreeda M, Harris TM. Tetrahedron Lett. 1991;32:6073–6076. [Google Scholar]
- 14.Chaturvedi S, Lakshman MK. Carcinogenesis. 1996;17:2747–2752. doi: 10.1093/carcin/17.12.2747. [DOI] [PubMed] [Google Scholar]
- 15.Jhingan AK, Meehan T. J. Chem. Res. (S) 1991:122–123. [Google Scholar]
- 16.In our hands, the ring opening of (±)-BaP DE-2 7,8-bis-O-TMS ether with Ti(OiPr)4/TMSN3 in THF (ref. 15) led to the formation of two compounds as observed by TLC, each containing TMS groups. We have also found that cis ring opening of (±)-BaP DE-2 7,8-bis-O-TMS can be attained with BF3.Et2O/TMSN3 to yield the corresponding azido triol in about 30% yield.
- 17.Pilcher AS, Yagi H, Jerina DM. J. Am. Chem. Soc. 1998;120:3520–3521. [Google Scholar]
- 18.Kroth H, Yagi H, Seidel A, Jerina DM. J. Org. Chem. 2000;65:5558–5564. doi: 10.1021/jo000522x. [DOI] [PubMed] [Google Scholar]
- 19.Ramesha AR, Kroth H, Jerina DM. Org. Lett. 2001;3:531–533. doi: 10.1021/ol0003580. [DOI] [PubMed] [Google Scholar]
- 20.Ramesha AR, Kroth H, Jerina DM. Tetrahedron Lett. 2001;42:1003–1005. [Google Scholar]
- 21. Cosman M, de los Santos C, Fiala R, Hingerty BE, Ibanez V, Luna E, Harvey R, Geacintov NE, Broyde S, Patel DJ. Biochemistry. 1993;32:4145–4155. doi: 10.1021/bi00067a001. Cosman M, de los Santos C, Fiala R, Hingerty BE, Singh SB, Ibanez V, Margulis LA, Live D, Geacintov NE, Broyde S, Patel DJ. Proc. Natl. Acad. Sci. USA. 1992;89:1914–1918. doi: 10.1073/pnas.89.5.1914. Cosman M, Hingerty BE, Luneva N, Amin S, Geacintov NE, Broyde S, Patel DJ. Biochemistry. 1996;35:9850–9863. doi: 10.1021/bi9605346. Funk M, Pontén I, Seidel A, Jernström B. Bioconjugate Chem. 1997;8:310–317. doi: 10.1021/bc9700188. (e) Personal communication Dr. Monique Cosman (Lawrence Livermore National Laboratories).
- 22.McCaustland DJ, Engel JF. Tetrahedron Lett. 1975;16:2549–2552. [Google Scholar]
- 23.Yagi H, Thakker DR, Hernandez O, Koreeda M, Jerina DM. J. Am. Chem. Soc. 1977;99:1604–1611. doi: 10.1021/ja00447a053. [DOI] [PubMed] [Google Scholar]
- 24.(a) Tierney B, Burden P, Hewer A, Ribeiro O, Walsh C, Rattle H, Grover PL, Sims P. J. Chromatogr. A. 1979;176:329–335. [Google Scholar]; (b) Zajc B, Grahek R, Kocijan A, Lakshman MK, Košmrlj J, Lah J. J. Org. Chem. 2003;68:3291–3294. doi: 10.1021/jo020607t. [DOI] [PubMed] [Google Scholar]
- 25.Martinelli MJ, Peterson BC, Khau VV, Hutchison DR, Leanna MR, Audia JE, Droste JJ, Wu Y-D, Houk KN. J. Org. Chem. 1994;59:2204–2210. [Google Scholar]
- 26.Iwasawa N, Kato T, Narasaka K. Chem. Lett. 1988:1721–1724. [Google Scholar]; (b) Sakurai H, Iwasawa N, Narasaka K. Bull. Chem. Soc. Jpn. 1996;69:2585–2594. [Google Scholar]
- 27.Gypser A, Michel D, Nirschl DS, Sharpless KB. J. Org. Chem. 1998;63:7322–7327. doi: 10.1021/jo980850l. [DOI] [PubMed] [Google Scholar]
- 28.(a) Lakshman MK, Zajc B. Tetrahedron Lett. 1996;37:2529–2532. [Google Scholar]; (b) Lakshman MK, Chaturvedi S, Zajc B, Gibson DT, Resnick SM. Synthesis. 1998:1352–1356. [Google Scholar]
- 29.(a) Reviewed in Lakshman MK. Curr. Org. Synth. 2005;2:83–112. Johnson F, Bonala R, Tawde D, Torres MC, Iden CR. Chem. Res. Toxicol. 2002;15:1489–1494. doi: 10.1021/tx0256174. Lakshman MK, Gunda P. Org. Lett. 2003;5:39–42. doi: 10.1021/ol027084w.
- 30.Zajc B, Lakshman MK, Sayer JM, Jerina DM. Tetrahedron Lett. 1992;33:3409–3412. [Google Scholar]
- 31.van de Ven FJM, Hilbers CW. Nucleic Acids Res. 1988;16:5713–5726. doi: 10.1093/nar/16.13.5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wijmenga SS, Kruithof M, Hilbers CW. J. Biomol. NMR. 1997;10:337–350. doi: 10.1023/A:1018348123074. [DOI] [PubMed] [Google Scholar]
- 33.Cheng SC, Hilton BD, Roman JM, Dipple A. Chem. Res. Toxicol. 1989;2:334–340. doi: 10.1021/tx00011a011. [DOI] [PubMed] [Google Scholar]
- 34.Chambers CC, Hawkins GD, Cramer CJ, Truhlar DG. J. Phys. Chem. 1996;100:16385–16398. [Google Scholar]
- 35.Szeliga J, Dipple A. Chem. Res. Toxicol. 1998;11:1–11. doi: 10.1021/tx970142f. [DOI] [PubMed] [Google Scholar]
- 36.For examples see: Xie X-M, Geacintov NE, Broyde S. Chem. Res. Toxicol. 1999;12:597–609. doi: 10.1021/tx990021a. Xie X-M, Geacintov NE, Broyde S. Biochemistry. 1999;38:2956–2968. doi: 10.1021/bi9825605. Tan J, Geacintov NE, Broyde S. Chem. Res. Toxicol. 2000;13:811–822. doi: 10.1021/tx000094q. Kozack R, Seo K-Y, Jelinsky SA, Loechler EL. Mutat. Res. 2000;450:41–59. doi: 10.1016/s0027-5107(00)00015-4. Seo K-Y, Jelinsky SA, Loecher EL. Mutat. Res. 2000;463:215–246. doi: 10.1016/s1383-5742(00)00047-8. Ruan Q, Zhuang P, Li S, Perlow R, Srinivasan AR, Lu X-J, Broyde S, Olson WK, Geacintov NE. Biochemistry. 2001;40:10458–10472. doi: 10.1021/bi002643x. Yan SF, Wu M, Geacintov NE, Broyde S. Biochemistry. 2004;43:7750–7765. doi: 10.1021/bi0499516.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Complete experimental details including those for the computational analysis. 1H NMR spectra for the BaP derivatives 3, 6, 8, 9, 11–13 and nucleoside adducts 14b–17b. This information is available free of charge via the Internet at http://www.pubs.acs.org.