

Abstract
Data showing that the embryonic day 12 (E12) mouse kidney contains its own pool of endothelial progenitor cells is presented. Mechanisms that regulate metanephric endothelial recruitment and differentiation, including the hypoxia-inducible transcription factors and vascular endothelial growth factor/vascular endothelial growth factor receptor signaling system, are also discussed. Finally, evidence that glomerular endothelial cells contribute importantly to assembly of the glomerular basement membrane (GBM), especially the laminin component, is reviewed. Together, this forum offers insights on blood vessel development in general, and formation of the glomerular capillary in particular, which inarguably is among the most unique vascular structures in the body.
Key words: glomerular basement membrane, laminin, podocytes, type IV collagen, vascular endothelial growth factor
Introduction
Dr. Dale Abrahamson, University Distinguished Professor and Chair, Department of Anatomy and Cell Biology, University of Kansas Medical Center: In almost every example of primary glomerular disease (including postinfection glomerulonephritis, immunoglobulin A nephropathy, anti-GBM disease, minimal change nephrotic syndrome, focal segmental glomerulosclerosis, mesangioproliferative disease, hereditary nephritis) and metabolic or hematological diseases affecting the kidney (including diabetes mellitus, lupus, HIV), the normal architecture of the glomerular capillary wall is disrupted. These changes include loss of fenestrae and detachment of endothelial cells, focal or diffuse thickening of the GBM, and effacement and detachment of podocyte foot processes. Despite the critical importance of the glomerulus in filtration, many fundamental questions exist regarding normal glomerular development and maintenance. The GBM has long been considered to be a critical element in establishing the filtration barrier properties of the glomerular capillary. Supporting evidence has come from several decades of work using a variety of experimental physiological, pathobiological and ultrastructural tracer techniques showing that an intact GBM is essential for maintenance of normal glomerular permselective sieving properties. During the past several years, these findings have been confirmed (A) in knockout mice with targeted deficiencies in certain GBM laminin and type IV collagen chains; (B) in integrin deficient mice which lack appropriate basement membrane protein receptors in their plasma membranes; and (C) through the molecular genetic and biochemical definition of human Alport disease. More recently, as constituents of the epithelial slit diaphragm complex have been progressively revealed, a great deal of excitement and emphasis has understandably been placed on the podocyte and its role in the glomerular filtration barrier. Nevertheless, a growing realization has also developed among many investigators that no single layer of the glomerular capillary can, by itself, represent the primary barrier. Indeed, a basic tenet of cell biology is that cells and their associated extracellular matrices form an intimate microenvironment crucial for tissue differentiation and proper function. I therefore view the glomerular capillary as a continuous quilt of communicating cellular and extracellular elements, all of which are necessary for the timely formation and correct maintenance of the filtration barrier. Further, I hypothesize that errors in GBM assembly specifically injure glomerular cells, causing them to alter their differentiation programs. In turn, this induces cells to commit more errors in GBM synthesis and assembly which thereby promotes glomerular dysfunction.
Beginning with the earliest high resolution descriptions of glomerular ultrastructure in 1961,1 the microanatomy and physiology of the glomerular filtration barrier has been, and continues to be, a topic of intense interest. The flow of glomerular filtrate from the capillary lumen to the urinary space follows a strictly extracellular route, passing in sequence through the fenestrated endothelium, then the GBM, and finally across the slit diaphragms spanning the filtration slits whereupon the filtrate enters into the urinary (Bowman's) space. Recent evidence has provided a renewed appreciation for the net negative charge of the endothelial plasma membrane that may establish important barrier properties.2 Therefore, all of the current evidence indicates that the endothelium-GBM-podocyte trio constitute the filtration barrier.
Among the many achievements in nephrology research made during the past decade have been striking advances in podocyte cell biology.3–6 These have come, in part, from the availability of immortalized cell lines which have lead to a far better understanding of cell-matrix adhesion, cytoskeletal organization and transmembrane signaling properties of podocytes.5–7 In addition, a number of proteins that are either uniquely expressed or largely restricted to podocytes have been identified in the past few years that, when mutated, cause proteinuria. Some of these proteins constitute the structural elements of the slit diaphragm, a highly specialized intercellular junctional complex that, until recently, has eluded biomolecular analysis. The proteins nephrin,8–11 podocin12,13 and FAT (human homolog of the Drosophila fat tumor suppressor gene),14 have all been immunolocalized to the slit diaphragm region. Exactly how these proteins associate to constitute the zipper-like ultrastructure characteristic of the transcellular elements of the slit diaphragm is not yet clear. Both nephrin15 and podocin12 have also been shown to be embedded within lipid rafts of the foot process plasma membrane, suggesting that these proteins are involved with signaling events as well. Furthermore, compelling evidence has shown that the cytoplasmic domain of nephrin interacts with the intracellular scaffolding protein, CD2AP (CD2-associated protein),16 which, in turn, is linked to the cytoskeletal protein, α actinin-4.17 Other more widely expressed proteins have also been shown to be concentrated within the slit diaphragm region, including P-cadherin,18 which may be associated internally with ZO-1 (zonula occludens 1)19 and the catenins. The emerging picture therefore shows that the internal cytoskeletal and signaling machinery within the podocyte foot process are directly and indirectly linked to the intercellular slit diaphragm complex.3 These linkages are surely crucial for maintenance of foot process registration and the permselective barrier properties of the glomerular capillary wall. Mutations of nephrin, podocin, CD2AP, or α actinin-4 all result in foot process effacement and sometimes lethal proteinuria.
Although much progress has been made recently on the podocyte, the roles for the GBM and endothelium in glomerular homeostasis should not be overlooked. Independent of any permselective sieving properties exerted specifically by the GBM, changes in GBM composition likely affects the biosynthetic programs and function of the adherent endothelial cells and podocytes. Data supporting this comes from human Alport syndrome,20,21 canine models of Alport disease,22 and gene targeting studies in mice (laminin α5,23 laminin β2,24 and collagen α3(IV)25–27 knockouts), in which defined mutations to GBM proteins consistently result in glomerular capillary structural defects, proteinuria and renal failure. Podocyte foot process simplification occurs to some extent in all of these basement membrane mutants, and perhaps slit diaphragm complexes are misassembled as well, although there is little evidence yet that this is the case. Additionally, mutations affecting α3β1 integrin, which is the principal integrin in the peripheral glomerular wall and located specifically at the foot process-GBM interface, results in failure of foot process development and perinatal death.28 Importantly, the GBM is also disorganized in these integrin mutants, which again reflects reciprocal interactions occurring between adherent cells and the GBM. I believe that the term “dynamic reciprocity,” which was applied independently by Drs. Mina Bissell and Helene Sage 27 years ago29,30 to reflect the relationship between cells and their extracellular matrices, aptly describes the situation in the glomerular capillary wall.
Several comprehensive reviews on development of the kidney glomerulus and on the composition and pathology of the glomerular basement membrane have appeared recently.31–34 Here, I focus on glomerular endothelial cells and discuss their embryonic origins, factors that stimulate their differentiation, and their roles in assembly of the GBM during glomerular development.
Nephrogenesis
The permanent, metanephric kidney begins its formation at embryonic day 11 in mice, day 12 in rats, and during the 4th–5th week of gestation in humans.35 As the ureteric bud emerges from the mesonephric duct and enters the metanephric anlage, mesenchymal cells begin condensing around the bud's advancing tip. The condensed mesenchyme then converts to epithelial cells that sequentially develop through a series of nephric structures, which are termed vesicle, comma- and S-shaped, developing capillary loop, and maturing glomerulus stages.35,36 The growing ureteric bud branches repeatedly (and ultimately comprises the renal collecting tubules), and induces the aggregation of additional clusters of metanephric mesenchymal cells around each branch tip. This process of ureteric bud growth and branching, mesenchymal cell induction and condensation, epithelial transition and glomerular and tubule development occurs reiteratively until the full endowment of nephrons has developed. The induction of new nephrons concludes ∼1 week after birth in rodents,35 and during the 34th gestational week in humans.37 The mechanisms causing cessation of nephrogenesis at these times are unknown, but they may involve inhibition of ureteric bud growth and branching, and/or consumption of the pool of metanephric mesenchyme. Although glomerular hypertrophy and tubule elongation can occur to compensate for kidney loss later in life, there apparently is no inherent ability to regenerate new nephrons once the full complement has formed during renal organogenesis. Nevertheless, the promise of stem cell biology offers hope that regenerative procedures can eventually be devised to replace irretrievably damaged nephrons with new ones.
Glomerulogenesis
During the earliest stages of nephron develpment, mesenchymal cells are induced to aggregate near ureteric bud tips and this aggregate then converts to an epithelial cluster (vesicle). These cells undergo polarization, and begin assembling a basement membrane at their basal surfaces that contains laminin, type IV collagen and proteoglycans.38–40 Soon thereafter, an invagination of the vesicle occurs at its lower aspect, giving rise to a vascular cleft, and producing the comma- and then S-shaped stages of nephron development (Fig. 1). Endothelial precursors (angioblasts), and probably mesangioblasts as well, migrate into the vascular cleft to eventually form the glomerular capillaries. These cells assemble a thin basement membrane matrix that separates them from the visceral epithelial cells of Bowman's capsule, which are immediately posterior to the vascular cleft. In comma- and S-shaped nephrons these cells are seen as a simple row of columnar or cuboidal epithelium, and they will eventually differentiate into epithelial podocytes (Figs. 1 and 2). These cells, too, are attached to a basement membrane matrix, so that the basement membrane within the vascular cleft actually consists as a dual structure; one beneath the developing endothelium, and a second beneath developing podocytes (Fig. 3). This dual basement membrane will soon fuse to produce a common glomerular basement membrane shared on its inner surface by endothelial cells and on its outer surface by podocytes (Fig. 3). Overlying the visceral (podocyte) cell layer are the parietal epithelial cells, which eventually will comprise the flat epithlelial cells that line Bowman's capsule.
Figure 1.

Light micrograph of section of developing rat kidney showing comma-shaped nephric figure. A vascular cleft (large arrow) has formed and is the site of angioblast ingress. Visceral epithelial cells (VE) will develop into podocyes, and parietal epithelial cells (PE) will line Bowman's capsule. A small Bowman's space (BS) can be observed. Note mitotic figures (double arrows) in developing tubular segment of the forming nephron. UD: Ureteric duct. Reproduced with permission (ref. 36).
Figure 2.

Electron micrograph of vascular cleft region of an S-shaped stage nephron from a newborn rat that had received an injection of anti-laminin IgG directly conjugated to horseradish peroxidase (HRP). An erythrocyte (E) is densely stained due to the peroxidatic activity of hemoglobin. HRP is present throughout the full thickness of the developing GBM separating the endothelium (En) and primitive glomerular epithelium (Ep). Some strands of basement membrane-like material (arrows) lie within the cleft near putative mesangioblasts (M). HRP is also present in developing tubular basement membranes (TBM) beneath developing tubular epithelium (TEp). Reproduced with permission (ref. 43).
Figure 3.
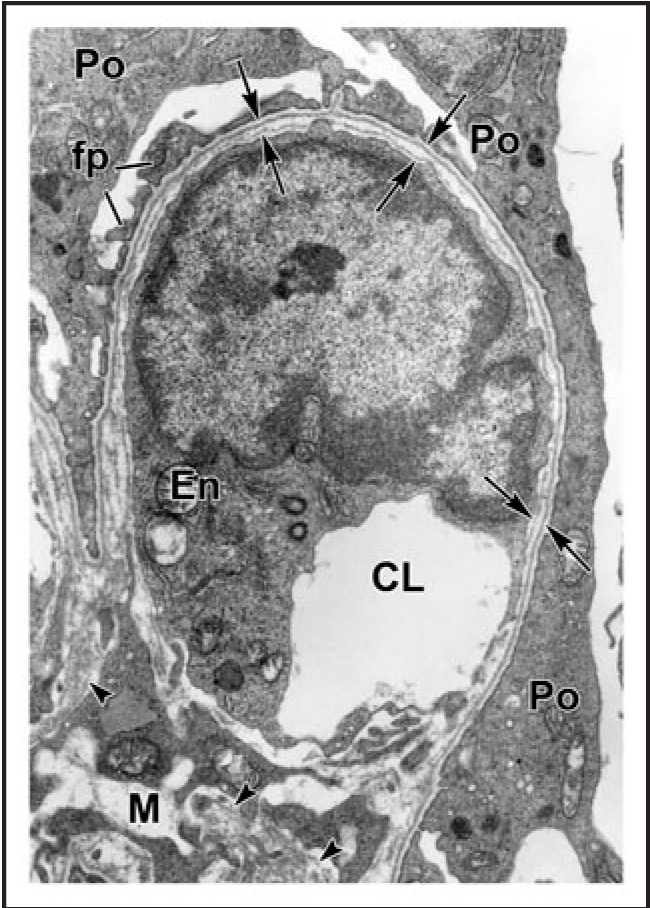
Electron micrograph of developing glomerular capillary loop. The endothelial cell (En) is large and contains only a few fenestrations at this stage. Only a few, relatively broad foot processes (fp) are present in the podocyte (Po) cell layer. A double basement membrane between the endothelium and podocytes can be seen clearly (arrows). Note the loose mesangial matrix (arrowheads) in the mesangium (M). Reproduced with permission (ref. 36).
Anterior to the vascular cleft are epithelial cells that ultimately develop into the tubular portion of the nephron, including the proximal, Henle's loop and distal tubule (Figs. 1 and 2). During the early S-shaped phase of nephron development, the distal segment fuses with the same ureteric bud branch tip that initially induced the nephric fuigure. The lumen of the developing nephron is from then on connected to what will become the collecting system.
As indicated previously, endothelial cells within the vascular clefts of early nephrons will go on to form glomerular capillary loops. At first, the endothelium appears as squat, cube-shaped cells (Fig. 3) but they soon flatten considerably and become extensively fenestrated. The initial fenestrae that form are spanned by diaphragms (like fenestrated capillaries elsewhere in the body) but, unique to the glomerulus, these fenestral diaphragms completely disappear.41 During the past several years, the apical endothelial cell surface layer (ESL),42 which is comprised of the membrane-bound glycocalyx and membrane-associated endothelial cell coat, has been shown to be an important part of the glomerular filtration barrier. Specifically, the ESL contains negatively charged glycoproteins, glycosaminoglycans and membrane-bound and secreted proteoglycans, together with serum glycoproteins.42 Whether the composition of the ESL evolves during glomerular development has not yet been investigated thoroughly, however.
Many changes also occur within the podocyte cell layer in capillary loop stage glomuleruli. The visceral epithelial cells begin as tall cuboidal/low columnar epithelium with typical apical junctional complexes. As the glomerular capillaries begin to form, the developing podocytes project basolateral cytoplasmic extensions that interdigitate with similar extensions from neighboring cells to ultimately form foot processes (pedicels). The apical junctional complexes migrate basolaterally alongside the forming foot processes and somehow convert to the slit diaphragm complex spanning the filtration slits. A number of different studies have shown that the glomerular endothelium and podocytes are both actively secreting basement membrane proteins throughout glomerular development,38,40,41,43–45 but many questions regarding assembly of the GBM remain. Likewise, and similar to the situation with the ESL, the maturation of the podocyte surface glycocalyx also needs further attention.
In maturing stage glomeruli, capillary loops inflate and endothelial and podocyte cell layers begin to resemble their fully mature counterparts. Though lengths of dual, unfused basement membranes are uncommonly seen in maturing glomeruli, subepithelial outpockets or complex scrolls of basement membrane are often seen beneath podocytes at this stage, especially in regions where foot process formation appears to still be active (Fig. 4). These subepithelial basement membrane segments seem to be spliced into the existing, largely fused GBM, possibly to provide the basement membrane matrix necessary for the expanding capillaries.46 Throughout glomerular development, different isoforms of the GBM proteins, laminin and type IV collagen, appear and disappear (discussed later), but little is known about what regulates these isoform substitutions. Regardless, once full glomerular maturation has been achieved, laminin and type IV collagen synthesis slow considerably.
Figure 4.

In maturing stage glomeruli, a well developed, thin and extensively fenestrated endothelium (En) is evident. Double basement membranes are generally absent, but several examples of subepithelial projections of basement membrane can be seen (*). In the podocyte layer (Po) foot process (fp) interdigitation is well underway, and the filtration slits are spanned by slit diaphragms (arrows). CL: capillary lumen; US: urinary space. Reproduced with permission (ref. 38).
Embryonic Origin of Glomerular Endothelial Cells
Until relatively recently, most experimental evidence showed that kidney vascular endothelium, including that lining the glomerular capillaries, originated from outside the kidney.47 Much of this evidence came from experiments in which fetal kidney rudiments from rodents or chick were grafted onto quail chorioallantoic membranes, and grafts were then cultured in ovo.48 Species specific immunolabeling or discrimination of quail nuclear markers showed in most instances that glomerular endothelial and mesangial cells within grafts originated from the host chorioallantoic membrane, indicating that they were derived from an extrarenal source.47,48
More recently, my laboratory attempted to create hybrid glomeruli by transplanting mouse metanephroi into the anterior eye chamber of host rats, and vice versa. Approximately 1 week after transplantation, grafts were harvested, sectioned and immunolabeled with species-specific monoclonal anti-basement membrane antibodies. In every case, however, we found that the GBMs within grafts, as well as most microvascular basement membranes throughout grafts, were of graft, not host, origin.49 These unexpected findings therefore indicated that the developing kidney generated its own endothelium, at least when grafted into this ectopic, anterior chamber site. We then extended these experiments and grafted E12 mouse kidneys under the renal capsules of adult ROSA26 mouse hosts. These animals ubiquitously express the bacterial transgene LacZ, encoding β-galactosidase, which is an enzyme that provides a convenient cell lineage marker. Again, none of the microvessels and glomeruli within the engrafted kidney bore cells of host lineage.50 However, when we grafted E12 kidneys under the renal capsules of newborn ROSA26 hosts, (a time when nephrogenesis was still taking place), host derived endothelial cells expressing β-galactosidase could now be identified within glomeruli and microvessels developing within the graft (Fig. 5). Similarly, when we grafted E12 ROSA26 metanephroi into wildtype newborn host mouse kidney, hybrid microvessels in the host near the margins of engrafted tissue were identified that contained endothelial cells of both graft and host origin.50
Figure 5.
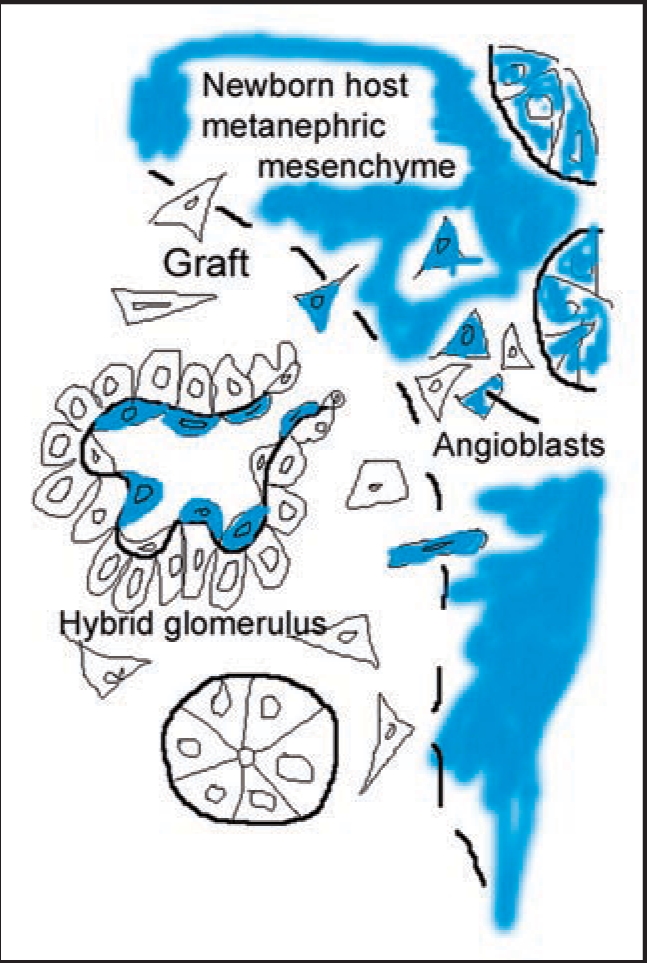
Diagram illustrating the formation of a hybrid glomerulus containing host- and graft-derived angioblasts, and graft-derived podocytes. Reproduced with permission (ref. 101).
In additional experiments, we grew embryonic kidneys from Flk1 (VEGF receptor-2)—LacZ heterozygous mice in organ culture. Under routine culture conditions, Flk1-LacZ positive microvessels do not develop in vitro, despite the extensive tubulogenesis that occurs as well as differentiation of podocytes in avascular glomerular epithelial tufts.51 When we transplanted these cultured kidneys into anterior eye chambers of wild type host mice, the grafts developed well vascularized glomeruli lined by Flk1-LacZ-expressing cells, demonstrating once again that the endothelium originates from the engrafted kidney itself and not from the host.51 Other researchers have reached similar conclusions. For example, immunolocalization studies have shown that angioblasts are present in metanephric mesenchyme of prevascular embryonic rat kidney.52 Also, just as we observed with grafts of Flk1-LacZ transgenics, when avascular metanephroi from E11 Tie1-LacZ transgenic mice are transplanted into newborn wildtype hosts, there is glomerular expression of Tie1-LacZ within glomeruli that form within grafts.53 Similarly, other results have shown that juxtaglomerular cells in developing kidney also develop from metanephric mesenchyme, although probably not from the endothelial lineage.54
Taken together, we believe that the initial discrepancy between our anterior chamber and intrarenal graft results and those obtained from the earlier avian graft experiments can now be resolved. If embryonic kidneys are implanted into “embryonic” sites that contain angioblasts, such as on chorioallantoic membranes of eggs or beneath developing kidney capsules of infant rodents, hybrid glomeruli and microvessels within grafts can result that contain endothelial cells of both graft and host origin (Fig. 5). In contrast, if embryonic kidneys are grafted into fully differentiated sites that no longer contain angioblasts, such as into mature kidney or anterior eye chamber, glomerular and vascular hybrids do not occur and all of the microvascular endothelium stems from the graft. Our results do not rule out the possibility that kidney angioblasts might migrate into metanephroi from some external source prior to E12. On the other hand, angioblasts might also derive directly from the metanephric mesenchyme, as do all of the other epithelial cells of the nephron. There must also be some coordinated mechanism for connecting the developing renal microvasculature with the larger systemic blood supply.
Regulation of Glomerular Endothelial Cell Development
Many mechanisms are involved with development of the vasculature in general, including receptor-ligand signaling networks, cell-cell and cell-matrix interactions, transcription factors,55,56 and probably microRNA as well as complex epigenetic influences. Among all of these, perhaps the vascular endothelial growth factor (VEGF) and VEGF receptor (VEGFR1 and VEGFR2) signaling system is best understood. Vegf null mice die by E9.5 with severe vascular deficits and even Vegf heterozygous animals die by E12 with vascular phenotypes. These results indicate that the loss of just one Vegf allele dysregulates normal vascular development.57,58 Mice that are homozygous (but not heterozygous) for the human VEGFR2 and VEGFR1 homologs die somewhat later in gestation with milder but nevertheless lethal vascular phenotypes encompassing failure of endothelial differentiation59 and vessel integrity,60 respectively.
Much of the VEGF important for capillary formation is believed to come from developing podocytes.61 Angioblasts bearing VEGF receptors may bind the ligand which then stimulates their migration into vascular clefts of comma-and S-shaped nephrons,62 from which the glomerular capillaries subsequently blossom. Continued expression of VEGF by podocytes and binding to endothelial cells may promote formation of fenestrae.63 Indeed, the glomerulus is one of the very few capillaries where VEGF and VEGFR expression are both maintained in adulthood. VEGF-VEGFR signaling in glomeruli therefore probably exerts functions extending well beyond those needed for mobilization of angioblasts and initial formation of the capillary tuft, and may include maintenance of the highly differentiated phenotype seen in the endothelium However, the exact role that this ligand/receptor pair plays in mediating glomerular development has been hard to understand completely because VEGF and VEGFR2 knockout mice die before glomerular vascular formation begins. On the other hand, injection of anti-VEGF IgGs into newborn mouse kidney cortices blocks glomerular development in vivo.64 More recently, conditional deletion of VEGF specifically in podocytes (in animals carrying nephrin-cre recombinase and floxed VEGF-A alleles) results in perinatal death and abnormal vascularized glomeruli.65 When the VEGF164 isoform is overexpressed specifically in podocytes, a collapsing glomerulopathy occurs and death results at postnatal day 5.66 Clearly, VEGF needs to be maintained within an optimal range for normal development of the glomerulus.
One of the key ways in which VEGF and VEGFR genes are regulated is through the stabilization of hypoxia-inducible transcription factors (HIFs), which consist of heterodimers of HIFα and β subunits.67 At normoxia, the HIFa subunit is hydroxylated at a proline residue, which leads to binding of von Hippel-Lindau protein (VHL), polyubiquitination, and ultimately, degradation of the complex in the proteasome. In contrast, hypoxic conditions inhibit the prolyl hydroxylase enzyme, there is no binding by VHL, and HIFα thereby escapes proteasomal degradation. In these cases, HIFα translocates to the nucleus, where it heterodimerizes with HIFβ. These hypoxia-stabilized HIFα/β heterodimers then bind to hypoxia-responsive elements (HREs) located in promoter/enhancer regions of inducible genes,68 many of which encode proteins expressed in response to hypoxic stress. Among the more than 70 different genes known to be transcriptionally activated by HIFs are erythropoietin, transferrin, VEGF, VEGFR1 and VEGFR2.66
At least three distinct HIFα and two β subunits are known at present, making various HIF isoforms possible. HIF stabilization is demonstrably enhanced in cells experiencing hypoxia, such as in rapidly growing tumors but also during normal organogenesis, and high expression levels for VEGF and VEGFR occur in these tissues as well. Increased VEGF/VEGFR signaling has a number of downstream effects, including the stimulation of mitosis in endothelial progenitor cells, phosphorylation of the anti-apoptotic kinases Akt/PKB69 and focal adhesion kinase,70 and upregulation of the survival factors Bcl2 and A1.71 Many of these events lead to the production of new vascular networks, which can then provide normal oxygen tensions to sites that were formerly hypoxic.
A number of studies have examined expression of the different HIFα and β subunits in developing human, rat and mouse kidney using in situ hybridization and immunohistochemistry. Both HIF-1α and -2α have been localized in glomeruli, with specific immunolocalization of -2α protein to some immature podocytes (which are rich sources of VEGF).72,73 HIF-2α is also expressed robustly by developing vascular endothelial cells in the kidney (most of which express VEGFR2) whereas HIF-1α is found in cortical and medullary collecting duct epitheilium. Despite the continued expression of VEGF and VEGFR in fully mature glomeruli, HIF-1α and -2α protein are not detectable. HIF-1β appears to be expressed ubiquitously expressed by all cells in kidney. HIF-2β distribution is also fairly widespread during early stages of nephrogenesis but later becomes significantly restricted to nuclei of epithelial cells in the thick ascending limb of Henle's loop.74
Previously, we speculated that the selective expression of certain HIF isoforms in different tissue compartments of developing kidney might reflect coordinated regulation of different sets of HIF target genes.75 For example, expression of one HIF isoform in epithelial cells may selectively promote expression of VEGF, whereas expression of a different HIF isoform in endothelial cells may promote expression of its cognate receptor, VEGFR.75 To address this hypothesis, we examined kidney development in Hif2a null mice that express the transcriptional reporter, LacZ, under control of the Hif2a locus.76 The first report on these mice showed that homozygous mutants were embryonic lethals with a catecholamine defect and heart failure, but possible errors in kidney development or renal vascular patterning had not been investigated.77 In our studies, Hif2a/LacZ was intensely expressed by all vascular endothelial cells and smooth muscle cells, with occasional expression by podocytes and tubular epithelium (Fig. 6).76 Surprisingly, we observed no defects in glomerular development or vascular formation in Hif2a knockout mice. Additionally, homozygous metanephroi responded no differently from wildtypes in an organ culture assay under hypoxia, and synthesized similar amounts of VEGF. We concluded that HIF2-α was not essential for kidney glomerular and microvascular development, though we could not rule out a role for it later in development under normal or hypoxic stress.76
Figure 6.

HIF-2α expression in kidney. (A) E13.5 metanephros from a HIF2α+/− stained for β-galactosidase histochemistry shows a branching, vessel-like pattern. (B) E14 HIF2a−/− kidney showing β-galactosidase reaction product in glomerular endothelial cells migrating into the vascular cleft (VC), and in capillary loop stage glomeruli (G). Also, small arrows denote individual cells in the metanephric mesenchyme also containing β-galactosidase. (C) The same slide in B was also labeled with the lectin, BsLB4, an endothelial marker. (D) Co-localization of β-galactosidase and BsLB4 shows complete overlap. (E) Comma-shaped nephric figure from newborn mouse showing HIF2α/LacZ expression by developing podocytes (*). (F) View of newborn mouse showing widespread expression of HIF2α/LacZ in vascular endothelial cells and glomeruli. (G) Boxed region from (F) is shown at higher power. Note HIF2α/LacZ expression in some (arrows) but not all podocytes (arrowheads). (H) Frozen section of a 4 week HIF2α+/− mouse showing intense β-galactosidase product in an endothelial pattern, and in smooth muscle cells of arteries (arrowhead). (I) Frozen section of a 4 week HIF2α+/− mouse showing a small artery. Reaction product is seen in both endothelial cells (arrow) and smooth muscle cells (arrowheads). (J) Vascular endothelial cells (arrow) are positive for HIF2α/LacZ and a few tubular epithelial cells (arrowhead) also express HIF2α/LacZ. Reproduced with permission (ref. 76).
Importantly, individuals with mutations in the tumor suppressor protein VHL, a key mediator of HIFα chain degradation which reduces expression of HIF target genes, are prone to developing hemangioblastomas and clear cell-renal cell carcinomas.78–80 Aiming to understand more about the possible roles of VHL and HIFs in kidney vascular development, three different groups have selectively deleted VHL in mouse podocytes using Cre excision. Adding more confusion, however, three different phenotypes have been reported and none of them include errors in vascular patterning during glomerulogenesis. First, my own group observed completely normal glomerular development and kidney growth but, beginning several weeks after birth, we noted that many, but not all, mutants became proteinuric, some severely and fatally so.81 In general, this appeared to be a slowly progressive disorder, and all animals also showed focal podocyte foot process effacement and GBM thickening, but with no evidence of an inflammatory component. We are still in the process of characterizing this mutant, but believe that the glomerular disease we observe is linked to basement membrane assembly abnormalities and a resulting matrix-cell signaling defect. In stark contrast to our results, a second study reported a rapidly progressive crescentic glomerulonephritis, proteinuria, fibrin deposition and upregulation of the HIF target gene, Cxcr4.82 A third study reported only occasional proteinuria, glomerulomegaly and glomerulosclerosis.83 Although the phenotypes in mature mice from these three studies differ, the fact that none of the investigators observed errors in glomerular or renal vascular development indicate that release of HIFα from VHL-mediated degradation in podocytes fails to deregulate initial endothelial differentiation.
Other growth factor-receptor signaling systems important for vessel development systemically are also crucial for glomerular capillary formation, including Tie/angiopoietin, and PDGFR/PDGF.55,56 Immature glomerular endothelial cells express Tie-2, and one of its ligands, angiopoietin-1, has been shown to be critical for blood vessel organization. Another Tie-2 ligand, angiopoietin-2, may regulate vascular permeability and stability. Therefore, the coordinated expression of angiopoietins-1 and -2 may modulate the later, maturation and stabilization phases of glomerular development.84 Importantly, Tie-2 and at least some members of the angiopoietins also contain defined HREs in their promoter/enhancer elements, making their transcriptional control by HIFs seem possible.67 An HRE is also present in the PDGFB gene, although this may not necessarily be responsive to hypoxia.85 PDGFB protein is secreted by podocytes during early glomerular development, which may be an important signal for recruitment of mesangioblasts, which express the PDGFB receptor, PDGFRβ.86 Both PDGF and PDGFRβ become confined to the mesangium in later developmental stages, which may provide autocrine signals required for maturation of the mesangium.85
Axonal guidance receptors and ligands are also found in developing glomeruli, as they are in developing vessels elsewhere in the embryo.87–89 For example, neuropilin-1 (Np1), a co-receptor with VEGFR2 for VEGF164 (but lacking a cytoplasmic signaling motif), immunolocalizes to glomerular endothelial cells. The Np1 ligands, semaphorins-3A and -3F, have been found in podocytes, raising the possibility that semaphorin-Np1 signaling between podocytes and endothelium may help pattern glomerular vascularization.90 However, one report showed that Np1 itself is expressed by podocytes in vivo, suggesting an autocrine role.91 Other studies have shown that podocyte-derived VEGF may similarly act as an autocrine survival factor for cultured podocytes.92 These same experiments documented an upregulation of VEGFR2 in cultured podocytes. This suggests that VEGF/VEGFR2 signaling may be important not only for glomerular capillary formation and maintenance but for podocyte differentiation as well.92
Other receptor-ligand signaling systems that may help regulate glomerular formation include members of the Eph/ephrin receptor/counter receptor families.55,93 For example, the receptor tyrosine kinase EphB1 and its ligand, ephrin-B1, which itself is also a transmembrane protein receptor, are both distributed in similar patterns in developing kidney microvessels.93 Although the exact functions for Eph/ephrin signaling in the developing glomerulus are still unknown, but knockout mice display lethal vascular systemic phenotypes including defects in vessel patterning, sprouting and remodeling.55 Reciprocal gradients of Eph and ephrin protein concentrations have been identified in the developing brain, where they appear to provide temporal and spatial cues for neuronal patterning in the visual system. Conceivably, analogous events may also be occurring in the developing glomerulus.55
Assembly of the GBM Laminin and Type IV Collagen Networks
Basement membranes, including the GBM, are comprised of laminin, type IV collagen, entactin/nidogen and proteoglycans. I will restrict the balance of the discussion to formation of the laminin and type IV collagen networks in the GBM.
Most laminins are composed of three evolutionarily related α, β and γ chains.94 There are five different α, four β and three γ chains that associate nonrandomly and become disulfide bonded to each other to form at least 15 distinct heterotrimers, most of which are ∼800 kDa.95 Heterotrimer assembly occurs within the endoplasmic reticulum before secretion. Once secreted, laminin interacts with its cellular receptors, such as integrins and dystroglycan, and copolymerizes to form laminin networks.96
Interestingly, only a select few of the known laminin heterotrimers are expressed in glomeruli, and only a single isoform, laminin α5β2γ1 (designated LM-521 in a recently adopted nomenclature),94 is present in mature GBM.97 On the other hand, several different laminin isoforms are found in the mesangial matrix, including LM-111, LM-211, LM-411 and LM-511.97 Why the GBM and mesangial matrix are so different with respect to their laminin composition is unclear, but this may reflect diverse functions with respect to adhesive and other cellular behaviors in these different glomerular compartments.
The laminin composition of the GBM also changes considerably during glomerulogenesis.98 From the comma-shape through the S-shape stages, LM-111 and LM-411 are both present, but they are rapidly removed from the GBM at the inception of the capillary loop stage. Beginning with the late S-shape stage and continuing thereafter, LM-521 is deposited into the GBM, where it persists as the only GBM laminin isoform through adulthood. Post-fixation immunoelectron microscopy experiments have shown that both glomerular endothelial cells and podocytes jointly produce laminin (Fig. 7),44 and the different laminin isoforms are also synthesized by both cell types at the appropriate developmental stage.99
Figure 7.
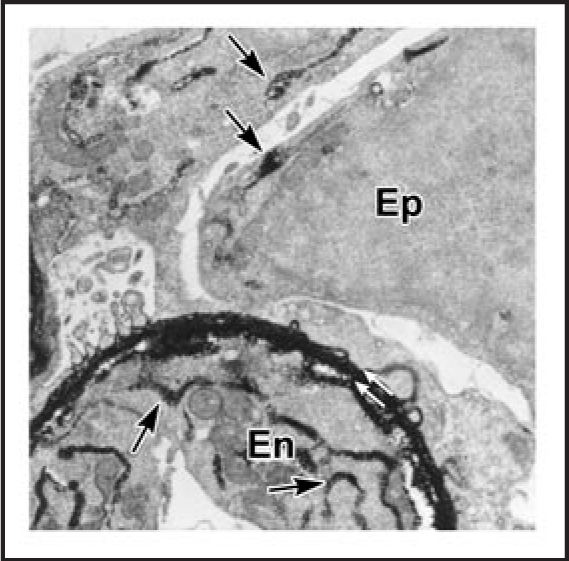
Electron microscopy of lightly fixed sections from newborn rat kidney incubated in vitro with anti-laminin IgG-HRP. Note HRP is present within double basement membrane (white arrows) and within biosynthetic apparatus (arrows) of the endothelium (En) and epithelium (Ep). Reproduced with permission (ref. 43).
Why these laminin transitions occur in developing GBM is uncertain, but they may be necessary for acquisition and maintenance of the intricate cellular form displayed by the glomerular endothelium and/or podocytes. Clues that this may be true have come from experiments in which certain laminin chains have been deleted in knockout mice. Along these lines, targeted mutations of the Lama5 gene, which encodes laminin α5, results in a failure of glomeruli to vascularize, and the primitive basement membrane that does assemble soon completely disappears in the glomerular epithelial tufts found in this mutant.100
Knowing that both endothelial cells and podocytes undertake the laminin transition program, and taking advantage of the ability to create glomerular hybrids by grafting, we sought to rescue the Lama5 null phenotype.101 To do so, Lama5 knockout metanephroi from E12 embryos were transplanted into kidneys of newborn, ROSA26 hosts, which undergo laminin transitioning normally. Five-7 days after grafting, kidneys were removed, and hybrid glomeruli within grafts were located by β-galactosidase histochemisty (Fig. 8). The identification of vascularized glomeruli within engrafted Lama5 knockout kidney therefore confirmed at least partial rescue of the Lama5 null phenotype.101 We then undertook confocal microscopy and immunolabeled croyostat sections with anti-laminin α1 and anti-laminin α5 antibodies. As shown in Figure 9, laminin α5 was localized immediately beneath the host-derived, wild type endothelium in hybrid glomeruli. Surprisingly, we also observed that there was persistence of laminin α1 on the podocyte surface of GBMs in glomerular hybrids, subjacent to the Lama5 mutant podocytes. The GBM was therefore distinctly stratified, with a subendothelial layer of laminin α5, and a subepithelial layer of laminin α1 (Fig. 9). Further, electron microscopy of these glomerular hybrids, showed a poorly condensed GBM between the normal endothelial cells and mutant podocytes, and a complete absence of foot process formation in the podocyte layer (Fig. 10). Our chief conclusions from these experiments were that (A) laminin derived from the endothelium remains closely adherent to that cell layer and does not project across the full width of the GBM, and (B) that podocytes fail to form foot processes in the absence of laminin α5 (or in the presence of laminin α1).100 This later point is also consistent with the morphology of the incipient podcytes that normally underlie the vascular clefts of comma- and S-shaped nephrons; there are no foot processes found in these regions where the unfused basement membrane contains laminin α1 (but not laminin α5).
Figure 8.

Sections showing hybrid glomeruli. Top panels are from separate samples processed for LacZ, lower panels show corresponding serial sections immunolabeled for laminin. (A and C) Host tissue is intensely blue and can easily be distinguished from graft (dashed black line demarcates margin between host and graft tissue). Note ingress of a number of host-derived cells into graft, and the formation of hybrid glomeruli (arrows) containing host (blue) endothelial cells. (B and D) Immunofluorescence images of serial sections doubly labeled for laminin α1 (green) and α5 (red) chains. Laminin α5 protein is present in GBMs of hybrid glomeruli. (* marks same tubule in serial sections). Reproduced with permission (ref. 100).
Figure 9.

Separate confocal images of the same hybrid glomerulus, dually labeled for laminin α1 (green) and laminin α5 (red). Note laminin α5 presence in vascular stalk (VS) as well as GBM. A higher power view of merged image (C) is shown in (D). GBM in glomerular hybrid is stratified; laminin α5 is on endothelial surface, whereas laminin α1 (which ordinarily is not present in capillary loop stage glomeruli) occupies podocyte surface of GBM. Central areas of signal overlap appear yellow. Reproduced with permission (ref. 100).
Figure 10.
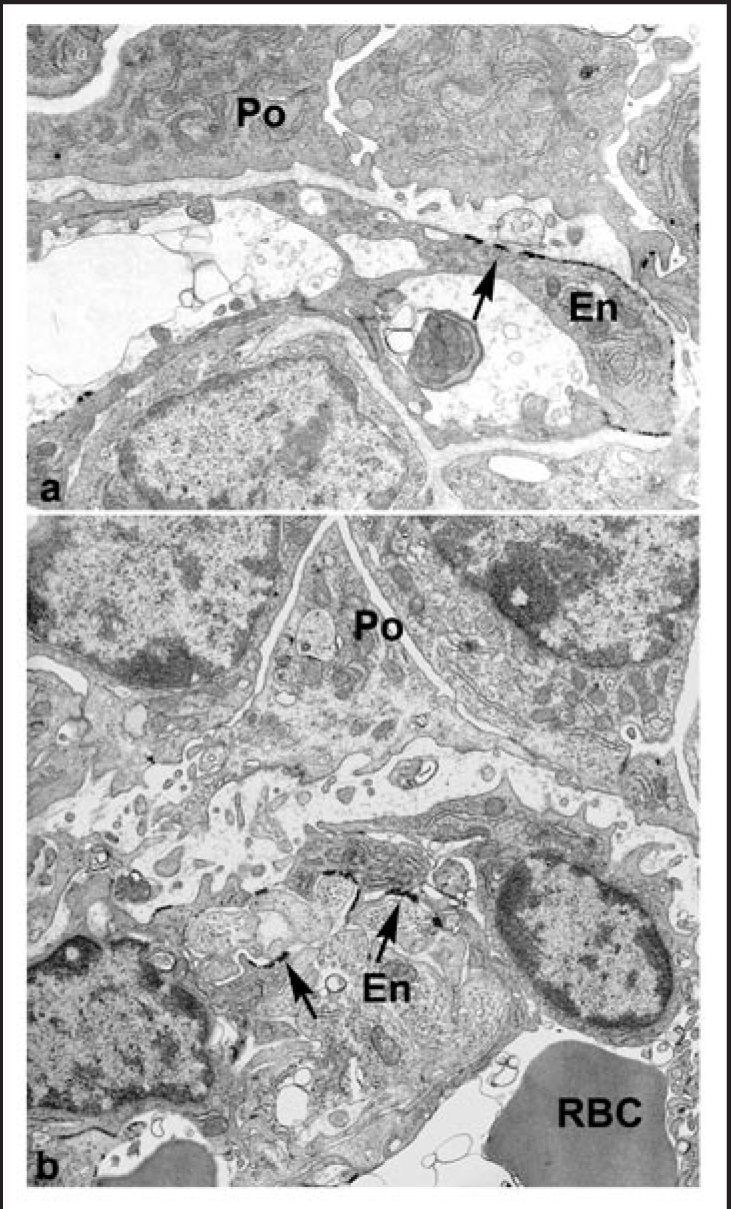
Ultrastructural examination of hybrid glomeruli. Tissue was developed with Bluo-gal, an alternative substrate for β-galactosidase, to mark endothelial cells of host (ROSA26) origin. Host-derived, Bluo-gal positive endothelial cells (En; arrows) and Bluo-gal negative podocytes (Po). In general, GBMs are poorly organized than and podocyte foot processes are absent. RBC: erythrocyte, signifying perfusion. Reproduced with permission (ref. 100).
Finally, we used the anti-laminin α5 immunolabel and confocal microscopy to quantify the relative abundance of this chain in normal and hybrid GBMs. Our results showed that up to ∼1/2 of the full complement of laminin α5 could be found in glomerular hybrids containing wildtype endothelial cells, as compared with completely normal, wildtype host glomeruli (Fig. 11).101 We interpreted this to signify that glomerular endothelial cells can contribute up to ∼1/2 of the total GBM laminin, at least under the grafting conditions we used in this experiment.101
Figure 11.

To evaluate the relative contribution of endothelial cells to developing GBMs, immunofluorescence signal strengths across GBMs were quantified from host glomeruli (A) and compared with those from hybrid glomeruli (B) in adjacent areas containing metanephric grafts. Histogram plots show peak intensity values for laminin α5 chain (red) at bisected regions of the GBM. Only background levels for laminin α1 chain (green) are observed at these same points in normal glomeruli (A), whereas hybrids show abnormally high levels of α1 in outer layer of GBMs (B). Reproduced with permission (ref. 100).
Unlike the need for successful laminin α1-to-laminin α5 transition for normal glomerulogenesis, laminin β1-to-laminin β2 transitioning is not absolutely necessary. Normal glomerulogenesis proceeds in Lamb2 knockout mice,24 probably because LM-511 can partially substitute functionally for LM-521. On the other hand, the glomeruli that do form in Lamb2 knockouts become progressively abnormal; the mice develop foot process effacement, proteinuria, and eventually progress to renal failure.24 Humans with mutations in LAMB2 (Pierson syndrome) often suffer congenital nephrotic syndrome and severe neuromuscular junction abnormalities (another site where laminin β2 is expressed).101
Six genetically distinct type IV collagen α chains self assemble to form three different triple helical protomers; α1α2α1, α3α4α5 and α5α6α5.103,104 These protomers interact with each other through covalent and non-covalent bonds and crosslinks to form a three-dimensional meshwork within basement membranes. As is true for the laminins, there is also a striking substitution of type IV collagen within the GBM as it develops.
The immature GBM of the comma-and S-shaped nephrons contain collagen α1α2α1(IV) and α3α4α5(IV) can be detected for the first time in GBMs of capillary loop stage glomeruli.98 As the GBM matures, there is the elimination of collagen α1α2α1(IV), and collagen α3α4α5(IV) is by far the most prevalent isoform in the GBM.98,105 By contrast, the mesangial matrix contains collagen α1α2α1(IV) and collagen α3α4α5(IV) is absent. Bowman's capsule basement membrane, on the other hand, contains both α1α2α1(IV) and α5α6α5(IV).105
The importance of the collagen α3,α4,α5 (IV) chain network to the proper function of the GBM is evident by the effects of mutations in genes that encode these chains. The α3(IV) chain contains the major autoantigen in Goodpasture glomerulonephritis,107 and the α3, α4 or α5 (IV) chain genes are mutated in Alport syndrome.108–110 The failure of formation of a collagen α3α4α5(IV) network leads to protracted presence of the fetal α1α2α1(IV) network, which is more susceptible to proteolytic injury.111,112 Homozygous mutant mice with deletion of the α3(IV) collagen chain (Alport mice) die of renal failure 2–4 months after birth.113–115 GBMs in type IV collagen α3 mutants at the time of death are multilaminated and closely resemble those of humans with Alport syndrome. There are also defects in podocyte foot process architecture, with widespread effacement occurring during late stages of the disease. Interestingly, numerous changes in the molecular composition of GBM are also accompanied with the onset of renal dysfunction in this mutant, which include loss of collagens α4 and α5(IV), retention of collagen α1(IV) and 2(IV) chains, appearance of fibronectin and collagen VI, increased levels of perlecan,113 and reappearance of laminin α1 and β1.116 Post-fixation immunoelectron microscopy has shown that glomerular endothelial cells and podocytes both participate in the ectopic deposition of LM-111 in Alport mouse GBM.116 When double knockout mice containing null mutations for both α3(IV) collagen and integrin α1 are generated, less severe defects are seen during early life, but the animals eventually progress to renal failure.117
In experiments in mice, we have immunolocalized collagen α1α2α1(IV) to developing endothelial cells and to podocytes of immature glomeruli.118 On the other hand, collagen α3α4α5(IV) could be immunolocalized only to developing podocytes; endothelial cells were consistently negative. Additionally, attempts to correct the GBM phenotype by grafting E12 Alport metanephroi into wildtype newborn mouse hosts failed. Unlike the situation with the laminin α5 mutant rescues described earlier, GBMs of hybrid glomeruli containing host-derived, wild type endothelial cells and collagen α3(IV) null podocytes did not immunolabel with antibodies specific for collagen α3α4α5(IV), which again signifies that this protomer is of podocyte, not endothelial, origin.118 These findings indicate that Alport disease is a genetic disorder specifically of the podocyte in kidney.
How laminin and collagen IV isoform substitutions are regulated at either the gene or protein level are not at all clear. Patients with nail patella syndrome, however, have mutations encoding a LIM homeodomain transcription factor, LMX1B, and some of these patients have GBM abnormalities as well. Mice with null mutations for this protein show diminished levels of α3(IV) and α4(IV) collagen, and LMX1B has been shown to bind to putative enhancer sequences located in intron 1 of both mouse and human COL4A4 genes.119 This suggests that LMX1B may regulate expression of α3(IV) and α4(IV) collagen but how this is coordinated with other basement membrane proteins is not known.
Acknowledgements
I thank Eileen Roach for help with the illustrations. Funds came from NIH grants R01DK052483, P01DK065123, P20RR024124.
Abbreviations
- E
embryonic day
- CD2AP
CD2-associated protein
- ESL
endothelial cell surface layer
- FAT
human homology of drosophila fat tumor suppressor gene
- GBM
glomerular basement membrane
- HIF
hypoxia-inducible transcription factor
- HRE
hypoxia-responsive elements
- LM
laminin
- PDGF
platelet derived growth factor
- PDGFR
platelet derived growth factor receptor
- ROSA26
reverse orientation splice acceptor, strain 26
- VEGF
vascular endothelial growth factor
- VEGFR
vascular endothelial growth factor receptor
- VHL
von Hippel-Lindau protein
- ZO-1
zonula occludens-1 protein
Note
Edited transcripts of research conferences sponsored by Organogenesis and the Washington University George M. O'Brien Center for Kidney Disease Research (P30 DK079333) are published in Organogenesis. These conferences cover organogenesis in all multicellular organisms including research into tissue engineering, artificial organs and organ substitutes and are participated in by faculty at Washington University School of Medicine, St. Louis Missouri.
Footnotes
Previously published online as an Organogenesis E-publication: http://www.landesbioscience.com/journals/organogenesis/article/7577
Questions and Answers
Dr. Feng Chen, Assistant Professor of Medicine, Washington University School of Medicine: Just a quick question about the similarity between the Alport mutant GBM and the normal embryonic GBM, because structurally they have similarities and the Alport GBM is leaky. How does leakiness of the embryonic GBM impact on embryonic physiology and development?
Dr. Abrahamson: I think that it is pretty well established that the immature glomerulus, certainly at that relatively early stage of development, the filtration barrier is incomplete and is leaky. However, apparently it is not so leaky that it is harmful to the rest of the nephron or somehow the proximal tubules are able to deal with and handle whatever protein does pass across the immature glomerular capillary wall. That is a good question, but once the glomerulus gets to be about, in the mouse, probably about five or six days or seven days old, then the filtration barrier in that particular glomerulus becomes more or less impermeable to albumin.
Dr. Maggie Chen, Postdoctoral fellow in nephrology, Washington University School of Medicine: You showed a high amount of laminin alpha1 and alpha5 in Alport mice. Laminin alpha1 is the infantile form. When in the Alport mice age does it disappear?
Dr. Abrahamson: Yes, it is and in fact, that is a good question, In the Alport condition both the endothelial cells and the podocytes restart laminin alpha1 synthesis. It does normally downregulate on schedule but as the Alport disease progresses, in the alpha3 type 4 collagen null mouse, the biosynthesis of the laminin alpha1 restarts again in the loops and, together with the overexpression of laminin alpha5, is deposited in those sub epithelial projections that are so characteristic of Alport. Did that answer the question?
Dr. Robert Mecham, Alumni Endowed Professor of Cell Biology and Physiology, Washington University School of Medicine: Dale, very nice work. In your Rosa mouse transplant models you are seeing new blood vessel arise through angiogenesis from blood vessels in the host tissue. Do you think that is the same process that happens when blood vessels are forming in the embryonic kidney, and if not then could you be looking at apples and oranges in terms of laminin expression?
Dr. Abrahamson: That is a good question as well. Again, I think it is because we have this pool of responsive angioblasts, both in the kidney as well as in host tissue, so you are absolutely right there is ingress of host derived endothelial cells through a vasculogenic and/or angiogenic process. But I could show you also that there is bidirectional exchange so that if you graft a fetal embryonic day 12 Rosa kidney into a wild type newborn host, you also have egress of graft derived endothelial cells out of the graft, resulting in hybrid microvessels in neighboring host tissue. Although we can't rule entirely out that this grafting procedure would create artifactual vascular development processes, but because we have angioblasts both coming out of and going into grafted kidney, I think it is reasonable to assume that what we are looking at probably reflects what is happening in the native tissue.
Dr. Mecham: As you know, lung has two vascular beds, the systemic circulation that invades developing lung through an angiogenic process and the pulmonary circulation which arises de novo in the embryonic lung. I am curious. Does the kidney also show those two processes?
Dr. Abrahamson: I don't know for sure. However, I would say that in these experimental grafting approaches that we have used, and think others have used too using other knockouts or other transgenics, that we have clearly been able to show that under those experimental conditions there is principally a vasculogenic, endogenous origin of the endothelial primordia in the kidney by E12. Now a very important problem or question is how does this extensive microvascular network that the kidney apparently is capable of developing independently on its own, then connect with the much larger systemic vascular supply and when does that happen and how is that regulated. As far as I am aware, we don't have a clue.
Dr. Nguyet Nguyen, Assistant Professor of Medicine, Washington University School of Medicine: I have a question regarding your laminin alpha1 antibody. I don't recall exactly what epitope it recognizes. However, does the epitope get processed during maturation of the glomerulus, such that it is no longer seen within the glomerular cleft?
Dr. Abrahamson: The antibody is a monoclonal rat anti-mouse laminin antibody designated 8B3. The epitope it recognizes is at the end of the long arm of the laminin alpha1 chain. The precise amino acid sequence though has not been determined. The issue of masking or proteolysis is always something that one has to be careful about. It could be happening, but on the other hand this antibody does label other basement membranes in the kidney including the mesangial matrix at different times of development. The possibility exists that protein may be processed differently in the GBM, but on the other hand this antibody definitely is recognizing laminin epitopes elsewhere in the vicinity.
Dr. Nguyen: In EMs of your hybrid implants, I recall there was no lamina densa and there was no podocyte extension. Do you think that podocyte extension requires lamina densa formation? Certainly by immunofluoresence you can see the presence of laminin alpha5 and alpha1. However the EMs show no true lamina densa.
Dr. Abrahamson: That is an excellent point and you know, we really don't know how this basement membrane, particularly in this stratified hybrid condition, is put together. It definitely does not look like a normal intact basement membrane despite the immuno-signal that we see by immunofluoresence. Whether the lamina densa formation is important to induce foot process formation, that is certainly possible. But I favor the idea instead that either the prolonged presence of laminin alpha1 and/or the absence of laminin alpha5 in the matrix that the podocyte sees results in a failure of foot processes to form. But it certainly could be that the micro architecture of the stratified basement membrane in these glomerular hybrids also does not support foot process formation.
Dr. Nguyen: Does the normal podocyte foot process physically attach to the lamina densa?
Dr. Abrahamson: Probably not, there is a lamina rara externa on that surface immediately beneath the podocytes. Certainly in EM preparations you can see these fibular, vertical extensions of fibers that project from the laminin densa onto the membrane of the cell. But there is some debate about the true structure of basement membranes, and some of what we see might possibly be due to artifacts introduced during preparation of the tissue. Those are good questions.
Dr. George Jarad, Instructor in Medicine, Washington University School of Medicine: For how long do you track the transplanted embryonic kidney?
Dr. Abrahamson: Usually when we do them in the sub capsule in the kidney about 5–7 days. We have gone maybe as long as 8–10 days but typically we keep them for 5–7 days.
Dr. Jarad: If you were to track transplanted embryonic kidney development for a longer period of time, would you observe alpha5 laminin, or the laminin which is made by endothelial cells, in the other parts of the GBM?
Dr. Abrahamson: It could be, it could be worth trying I suppose. We haven't done that. The only thing I would say is that if you look at the normal glomerulus as it develops in situ, the laminin alpha1 as I showed you in the last couple of slides is very rapidly cleared out of that vascular cleft before the capillary loops form. So we are definitely getting a very lengthy persistence of laminin alpha1 in these hybrid GBMs—well beyond when it would normally be present. It is worth doing I suppose to try and set up some grafts and run them out for longer periods. They tend not to do so well, these intrarenal grafts, for whatever reason, when we keep them for lengthy periods, but it would be worth taking a look at.
Dr. Feng Chen: What do you think is the signal for the laminin isoform switch?
Dr. Abrahamson: I don't know what the switch is. It could be a clock.
Dr. Feng Chen: The cells are not a homogenous population. They have different cell cycles. If the switch were related to the cell cycle one might expect to see expression of the immature type, the alpha1 beta1, in some locations, and expression of the more mature type, the alpha5 beta2 in others and so on.
Dr. Abrahamson: We are trying to look at that, but it is not obvious, what the control elements might be, I don't know.
Dr. Feng Chen: Is that kind of switch common during development?
Dr. Abrahamson: Yes, the globin genes are all lined up on the same chromosome. They sequentially turn on and off as development progresses.
Dr. Feng Chen: Is it observed elsewhere for matrix components such as the alpha1, beta1?
Dr. Abrahamson: There is some of that in the lung I believe.
Dr. Feng Chen: Maybe there is some kind of structural requirement for such an abrupt switch. In other parts of the glomerulus we don't see anything like it.
Dr. Abrahamson: One of the many mysteries of life, I guess. It would be a good career goal to understand this laminin isoform switching—it may provide clues on the pathogenesis of fibrosis in general.
References
- 1.Farquhar MG, Wissig SL, Palade GE. Glomerular permeability I. ferritin transfer across the normal glomerular capillary wall. J Exp Med. 1961;113:47–87. doi: 10.1084/jem.113.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jeansson M, Haraldsson B. Morphological and functional evidence for an important role of the endothelial cell glycocalyx in the glomerular barrier. Am J Physiol Renal Physiol. 2006;290:111–116. doi: 10.1152/ajprenal.00173.2005. [DOI] [PubMed] [Google Scholar]
- 3.Kerjaschki D. Caught flat-footed: Podocyte damage and the molecular bases of focal glomerulosclerosis. J Clin Invest. 2001;108:1583–1587. doi: 10.1172/JCI14629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miner JH. Focusing on the glomerular slit diaphragm. Podocin enters the picture. Am J Pathol. 2002;160:3–5. doi: 10.1016/S0002-9440(10)64341-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Somlo S, Mundel P. Getting a foothold in nephrotic syndrome. Nat Genet. 2000;24:333–335. doi: 10.1038/74139. [DOI] [PubMed] [Google Scholar]
- 6.Mundel P, Reiser J, Zuniga Mejia Borja A, Pavenstadt H, et al. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp Cell Res. 1997;236:248–258. doi: 10.1006/excr.1997.3739. [DOI] [PubMed] [Google Scholar]
- 7.Shankland SJ, Pippin JW, Reiser J, Mundel P. Podocytes in culture: past, present and future. Kidney Int. 2007;72:26–36. doi: 10.1038/sj.ki.5002291. [DOI] [PubMed] [Google Scholar]
- 8.Ruotsalainen V, Ljungberg P, Wartiovaara J, Lenkkeri U, Kestila M, Jalanko H, et al. Nephrin is specifically located at the slit diaphragm of glomerular podocytes. Proc Natl Acad Sci USA. 1999;96:7962–7967. doi: 10.1073/pnas.96.14.7962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holzman LB, St John PL, Kovarila A, Verma R, Holthofer H, Abrahamson DR. Nephrin localizes to the slit pore of the glomerular epithelial cell. Kidney Int. 1999;56:1481–1491. doi: 10.1046/j.1523-1755.1999.00719.x. [DOI] [PubMed] [Google Scholar]
- 10.Holthofer H, Ahola H, Solin ML, Wang S, Palmern T, Luimula P, et al. Nephrin localizes at the podocyte filtration slit area and is characteristically spliced in the human kidney. Am J Pathol. 1999;155:1681–1687. doi: 10.1016/S0002-9440(10)65483-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawachi H, Koike H, Kurihara H, Yaoita E, Orikasa M, Shia MA, Sakai T, Yamamoto T, Salant DJ. Cloning of rat nephrin: Expression in developing glomeruli and in proteinuric states. Kidney Int. 2000;57:1949–1961. doi: 10.1046/j.1523-1755.2000.00044.x. [DOI] [PubMed] [Google Scholar]
- 12.Schwarz K, Simons K, Reiser J, Kriz W, Holzman LB, Shaw AS, Mundel P. Podocin is a raft-associated component of the glomerular slit diaphragm that interacts with CD2AP and nephrin. J Am Soc Nephrol. 2001;12:60. doi: 10.1172/JCI12849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roselli S, Gribouval O, Boute N, Sich M, Benessy F, Attie T, et al. Podocin localizes in the kidney to the slit diaphragm area. Am J Pathol. 2002;160:131–139. doi: 10.1016/S0002-9440(10)64357-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inoue T, Yaoita E, Kurihara H, Shimuzu F, Sakai T, Koboyashi T, et al. FAT is a component of glomerular slit diaphragms. Kidney Int. 2001;59:1003–1012. doi: 10.1046/j.1523-1755.2001.0590031003.x. [DOI] [PubMed] [Google Scholar]
- 15.Simons M, Schwarz K, Kriz W, Miettinen A, Reiser J, Mundel P, Holthofer H. Involvement of lipid rafts in nephrin phosphorylation and organization of the glomerular slit diaphragm. Am J Pathol. 2001;159:1069–1077. doi: 10.1016/S0002-9440(10)61782-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shih NY, Li J, Karpitskii V, Nguyen A, Dustin ML, Kanagawa O, et al. Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science. 1999;286:312–315. doi: 10.1126/science.286.5438.312. [DOI] [PubMed] [Google Scholar]
- 17.Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet. 2000;24:251–256. doi: 10.1038/73456. [DOI] [PubMed] [Google Scholar]
- 18.Reiser J, Kriz W, Kretzler M, Mundel P. The glomerular slit diaphragm is a modified adherens junction. J Am Soc Nephrol. 2000;11:1–8. doi: 10.1681/ASN.V1111. [DOI] [PubMed] [Google Scholar]
- 19.Schnabel E, Anderson JM, Farquhar MG. The tight junction protein ZO-1 is concentrated along slit diaphragms of the glomerular epithelium. J Cell Biol. 1990;111:1255–1263. doi: 10.1083/jcb.111.3.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kashtan CE. Familial hematuria due to type IV collagen mutations: Alport syndrome and thin basement membrane nephropathy. Curr Opin Pediatr. 2004;16:177–181. doi: 10.1097/00008480-200404000-00011. [DOI] [PubMed] [Google Scholar]
- 21.Kashtan CE. Familial hematurias: What we know and what we don't. Pediatr Nephrol. 2005;20:1027–1035. doi: 10.1007/s00467-005-1859-z. [DOI] [PubMed] [Google Scholar]
- 22.Heikkila P, Tryggvason K, Thorner P. Animal models of Alport syndrome: Advancing the prospects for effective gene therapy. Exp Nephrol. 2000;8:1–7. doi: 10.1159/000020641. [DOI] [PubMed] [Google Scholar]
- 23.Miner JH, Li C. Defective glomerulogenesis in the absence of laminin alpha5 demonstrates a developmental role for the kidney glomerular basement membrane. Dev Biol. 2000;217:278–289. doi: 10.1006/dbio.1999.9546. [DOI] [PubMed] [Google Scholar]
- 24.Noakes PG, Miner JH, Gautam M, Cunningham JM, Sanes JR, Merlie JP. The renal glomerulus of mice lacking s-laminin/laminin β2: Nephrosis despite molecular compensation by laminin β1. Nat Genet. 1995;10:400–406. doi: 10.1038/ng0895-400. [DOI] [PubMed] [Google Scholar]
- 25.Cosgrove D, Meehan DT, Grunkemeyer JA, Kornak JM, Sayers R, Hunter WJ, et al. Collagen COL4A3 knockout: A mouse model for autosomal Alport syndrome. Genes Dev. 1996;10:2981–2992. doi: 10.1101/gad.10.23.2981. [DOI] [PubMed] [Google Scholar]
- 26.Miner JH, Sanes JR. Molecular and functional defects in kidneys of mice lacking collagen alpha3(IV): Implications for Alport syndrome. J Cell Biol. 1996;135:1403–1413. doi: 10.1083/jcb.135.5.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu W, Phillips CL, Killen PD, Hlaing T, Harrison WR, Elder FF, et al. Insertional mutation of the collagen genes Col4a3 and Col4a4 in a mouse model of Alport syndrome. Genomics. 1999;61:113–124. doi: 10.1006/geno.1999.5943. [DOI] [PubMed] [Google Scholar]
- 28.Kreidberg JA, Donovan MJ, Goldstein SL, Rennke H, Shepherd K, Jones RC, Jaenisch R. Alpha3 beta1 integrin has a crucial role in kidney and lung organogenesis. Development. 1996;122:3537–3547. doi: 10.1242/dev.122.11.3537. [DOI] [PubMed] [Google Scholar]
- 29.Bissell MJ, Hall HG, Parry G. How does the extracellular matrix direct gene expression? J Theor Biol. 1982;99:31–68. doi: 10.1016/0022-5193(82)90388-5. [DOI] [PubMed] [Google Scholar]
- 30.Sage H. Collagens of basement membranes. J Invest Dermatol. 1982;79:51–59. doi: 10.1111/1523-1747.ep12545773. [DOI] [PubMed] [Google Scholar]
- 31.Miner JH, Abrahamson DR. Molecular and cellular mechanisms of glomerular capillary development. In: Alpern RJ, Hebert SC, editors. Seldin and Giebisch's The Kidney. 4th edition. Burlington, MA: Academic Press; 1988. pp. 691–706. [Google Scholar]
- 32.Quaggin SE, Kreidberg JA. Development of the renal glomerulus: good neighbors and good fences. Development. 2008;135:609–620. doi: 10.1242/dev.001081. [DOI] [PubMed] [Google Scholar]
- 33.Khoshnoodi J, Pedchenko V, Hudson BG. Mammalian collagen IV. Microsc Res Tech. 2008;71:357–370. doi: 10.1002/jemt.20564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gubler MC. Inherited diseases of the glomerular basement membrane. Nat Clin Pract Nephrol. 2008;4:24–37. doi: 10.1038/ncpneph0671. [DOI] [PubMed] [Google Scholar]
- 35.Saxen L. Organogenesis of Kidney. Cambridge: Cambridge University Press; 1987. pp. 1–173. [Google Scholar]
- 36.Abrahamson DR. Glomerulogenesis in the developing kidney. Semin Nephrol. 1991;11:375–389. [PubMed] [Google Scholar]
- 37.Woolf AS. The life of the human kidney before birth: Its secrets unfold. Pediatr Res. 2001;49:8–10. doi: 10.1203/00006450-200101000-00006. [DOI] [PubMed] [Google Scholar]
- 38.Abrahamson DR. Structure and development of the glomerular capillary wall and basement membrane. Am J Physiol. 1987;253:783–794. doi: 10.1152/ajprenal.1987.253.5.F783. [DOI] [PubMed] [Google Scholar]
- 39.Ekblom P. Formation of basement membranes in the embryonic kidney: an immunohistological study. J Cell Biol. 1981;91:1–10. doi: 10.1083/jcb.91.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lelongt B, Makino H, Kanwar YS. Maturation of the developing renal glomerulus with respect to basement membrane proteoglycans. Kidney Int. 1987;32:498–506. doi: 10.1038/ki.1987.238. [DOI] [PubMed] [Google Scholar]
- 41.Reeves WH, Kanwar YS, Farquhar MG. Assembly of the glomerular filtration surface. Differentiation of anionic sites in glomerular capillaries of newborn rat kidney. J Cell Biol. 1980;85:735–753. doi: 10.1083/jcb.85.3.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haroldsson B, Nystrom J, Deen WM. Properties of the glomerular basement barrier and mechanisms of proteinuria. Physiol Rev. 2008;88:451–487. doi: 10.1152/physrev.00055.2006. [DOI] [PubMed] [Google Scholar]
- 43.Abrahamson DR. Origin of the glomerular basement membrane visualized after in vivo labeling of laminin in newborn rat kidneys. J Cell Biol. 1985;100:1988–2000. doi: 10.1083/jcb.100.6.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kanwar YS, Jakubowski ML, Rosenzweig LJ, Gibbons JT. De novo cellular synthesis of sulfated proteoglycans of the developing renal glomerulus in vivo. Proc Natl Acad Sci USA. 1984;81:7108–7111. doi: 10.1073/pnas.81.22.7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sariola H, Timpl R, von der Mark K, Mayne R, Fitch JM, Linsenmayer TF, et al. Dual origin of the glomerular basement membrane. Dev Biol. 1984;101:86–96. doi: 10.1016/0012-1606(84)90119-2. [DOI] [PubMed] [Google Scholar]
- 46.Abrahamson DR, Perry EW. Evidence for splicing new basement membrane into old during glomerular development in newborn rat kidneys. J Cell Biol. 1986;103:2489–2498. doi: 10.1083/jcb.103.6.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hyink DP, Abrahamson DR. Origin of the glomerular vasculature in the developing kidney. Semin Nephrol. 1995;15:300–314. [PubMed] [Google Scholar]
- 48.Sariola H, Ekblom P, Lehtonen E, Saxen L. Differentiation and vascularization of the metanephric kidney grafted on the chorioallantoic membrane. Dev Biol. 1983;96:427–435. doi: 10.1016/0012-1606(83)90180-x. [DOI] [PubMed] [Google Scholar]
- 49.Hyink DP, Tucker DC, St John PL, et al. Endogenous origin of glomerular endothelial and mesangial cells in grafts of embryonic kidneys. Am J Physiol. 1996;270:886–899. doi: 10.1152/ajprenal.1996.270.5.F886. [DOI] [PubMed] [Google Scholar]
- 50.Robert B, St John PL, Hyink DP, Abrahamson DR. Evidence that embryonic kidney cells expressing flk-1 are intrinsic, vasculogenic angioblasts. Am J Physiol Renal Physiol. 1996;271:744–753. doi: 10.1152/ajprenal.1996.271.3.F744. [DOI] [PubMed] [Google Scholar]
- 51.Robert B, St John PL, Abrahamson DR. Direct visualization of renal vascular morphogenesis in Flk1 heterozygous mutant mice. Am J Physiol Renal Physiol. 1998;275:164–172. doi: 10.1152/ajprenal.1998.275.1.F164. [DOI] [PubMed] [Google Scholar]
- 52.Tufro A, Norwood VF, Carey RM, Gomez RA. Vascular endothelial growth factor induces nephrogenesis and vasculogenesis. J Am Soc Nephrol. 1999;10:2125–2134. doi: 10.1681/ASN.V10102125. [DOI] [PubMed] [Google Scholar]
- 53.Loughna S, Hardman P, Landels E, et al. A molecular and genetic analysis of renal glomerular capillary development. Angiogenesis. 1997;1:84–101. doi: 10.1023/A:1018357116559. [DOI] [PubMed] [Google Scholar]
- 54.Sequeira Lopez ML, Pentz ES, Robert B, et al. Embryonic origin and lineage of juxtaglomerular cells. Am J Physiol Renal Physiol. 2001;281:345–356. doi: 10.1152/ajprenal.2001.281.2.F345. [DOI] [PubMed] [Google Scholar]
- 55.Daniel TO, Abrahamson DR. Endothelial signal integration in vascular assembly. Annu Rev Physiol. 2000;62:649–671. doi: 10.1146/annurev.physiol.62.1.649. [DOI] [PubMed] [Google Scholar]
- 56.Robert B, Abrahamson DR. Control of glomerular capillary development by growth factor/receptor kinases. Pediatr Nephrol. 2001;16:294–301. doi: 10.1007/s004670000534. [DOI] [PubMed] [Google Scholar]
- 57.Carmeliet P, Ferreira V, Breier G, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 58.Ferrara N, Carver-Moore K, Chen H, et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 59.Shalaby F, Rossant J, Yamaguchi TP, Gertsenestein M, Wu XF, Breitman ML, et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 60.Fong G-H, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- 61.Simon M, Grone HJ, Johren O, et al. Expression of vascular endothelial growth factor and its receptors in human renal ontogenesis and in adult kidney. Am J Physiol. 1995;268:240–250. doi: 10.1152/ajprenal.1995.268.2.F240. [DOI] [PubMed] [Google Scholar]
- 62.Tufro A. VEGF spatially directs angiogenesis during metanephric development in vitro. Dev Biol. 2000;227:558–566. doi: 10.1006/dbio.2000.9845. [DOI] [PubMed] [Google Scholar]
- 63.Esser S, Wolburg K, Wolburg H, Breier G, Kurzchalia T, Risau W. Vascular endothelial growth factor induces endothelial fenestrations in vitro. J Cell Biol. 1998;140:947–959. doi: 10.1083/jcb.140.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kitamoto Y, Tokunaga H, Tomita K. Vascular endothelial growth factor is an essential molecule for mouse kidney development: glomerulogenesis and nephrogenesis. J Clin Invest. 1997;99:2351–2357. doi: 10.1172/JCI119416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eremina V, Cui S, Gerber H, et al. Vascular endothelial growth factor-A signaling in the podocyte-endothelial compartment is required for mesangial cell migration and survival. J Am Soc Nephrol. 2006;17:724–735. doi: 10.1681/ASN.2005080810. [DOI] [PubMed] [Google Scholar]
- 66.Eremina V, Sood M, Haigh J, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111:701–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE. 2005;306:12. doi: 10.1126/stke.3062005re12. [DOI] [PubMed] [Google Scholar]
- 68.Maxwell PH, Ratcliffe PJ. Oxygen sensors and angiogenesis. Semin Cell Dev Biol. 2002;13:29–37. doi: 10.1006/scdb.2001.0287. [DOI] [PubMed] [Google Scholar]
- 69.Gerber HP, Hillan KJ, Ryan AM, Kowalski J, Keller GA, Rangell L, et al. VEGF is required for growth and survival in neonatal mice. Development. 1999;126:149–159. doi: 10.1242/dev.126.6.1149. [DOI] [PubMed] [Google Scholar]
- 70.Abedi H, Zachary I. Vascular endothelial growth factor stimulates tyrosine phosphorylation and recruitment to new focal adhesions of focal adhesion kinase and paxillin in endothelial cells. J Biol Chem. 1997;272:15442–15451. doi: 10.1074/jbc.272.24.15442. [DOI] [PubMed] [Google Scholar]
- 71.Gerber HP, Dixit V, Ferrara N. Vascular endothelial growth factor induces expression of the anti-apoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J Biol Chem. 1998;273:13313–13316. doi: 10.1074/jbc.273.21.13313. [DOI] [PubMed] [Google Scholar]
- 72.Bernhardt WM, Schmitt R, Rosenberger C, et al. Expression of hypoxia-inducible transcription factors in developing human and rat kidneys. Kidney Int. 2006;69:114–122. doi: 10.1038/sj.ki.5000062. [DOI] [PubMed] [Google Scholar]
- 73.Freeburg PB, Robert B, St John PL, Abrahamson DR. Podocyte expression of hypoxia-inducible factor (HIF)-1 and HIF-2 during glomerular development. J Am Soc Nephrol. 2003;14:927–938. doi: 10.1097/01.asn.0000059308.82322.4f. [DOI] [PubMed] [Google Scholar]
- 74.Freeburg PB, Abrahamson DR. Divergent expression patterns for hypoxia-inducible factor1beta and aryl hydrocarbon receptor nuclear transporter-2 in developing kidney. J Am Soc Nephrol. 2004;15:2569–2578. doi: 10.1097/01.ASN.0000141464.02967.29. [DOI] [PubMed] [Google Scholar]
- 75.Freeburg PB, Abrahamson DR. Hypoxia-inducible factors and kidney vascular development. J Am Soc Nephrol. 2003;14:2723–2730. doi: 10.1097/01.asn.0000092794.37534.01. [DOI] [PubMed] [Google Scholar]
- 76.Steenhard BM, Freeburg PB, Isom K, Stroganova L, Borza DB, Hudson BG, et al. Kidney development and gene expression in the HIF2a knockout mouse. Dev Dyn. 2007;236:1115–1125. doi: 10.1002/dvdy.21106. [DOI] [PubMed] [Google Scholar]
- 77.Tian H, Hammer RE, Matsumoto AM, Russell DW, McKnight SL. The hypoxia-responsive transcription factor EPAS1 is essential for catecholamine homeostasis and protection against heart failure during embryonic development. Genes Dev. 1998;12:3320–3324. doi: 10.1101/gad.12.21.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Haase VH. The VHL/HIF oxygen sensing pathway and its rele3vance to kidney disease. Kidney Int. 2006;69:1302–1307. doi: 10.1038/sj.ki.5000221. [DOI] [PubMed] [Google Scholar]
- 79.Haase VH. Hypoxia-inducible factors in the kidney. Am J Physiol Renal Physiol. 2006;291:271–281. doi: 10.1152/ajprenal.00071.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kaelin WG. The von Hippel-Lindau tumor suppressor gene and kidney cancer. Clin Can Res. 2004;10:6290–6295. doi: 10.1158/1078-0432.CCR-sup-040025. [DOI] [PubMed] [Google Scholar]
- 81.Steenhard B, Isom K, Stroganova L, St John P, Freeburg P, Holzman L, et al. Podocyteselective deletion of von Hippel-Lindau (VHL) protein causes albuminuria. J Am Soc Nephrol. 2005;16:667. [Google Scholar]
- 82.Ding M, Cui S, Li C, Jothy S, Haase V, Steer BM, et al. Loss of the tumor suppressor Vhlh leads to upregulation of Cxcr4 and rapidly progressive glomerulonephritis in mice. Nat Med. 2006;12:1081–1087. doi: 10.1038/nm1460. [DOI] [PubMed] [Google Scholar]
- 83.Brukamp K, Jim B, Moeller MJ, Haase VH. Hypoxia and podocyte specific Vhlh deletion confer risk of glomerular disease. Am J Physiol Renal Physiol. 2007;293:1397–1407. doi: 10.1152/ajprenal.00133.2007. [DOI] [PubMed] [Google Scholar]
- 84.Woolf AS, Yuan HT. Angiopoietin growth factors and Tie receptor tyrosine kinases in renal vascular development. Pediatr Nephrol. 2001;16:177–184. doi: 10.1007/s004670000509. [DOI] [PubMed] [Google Scholar]
- 85.Ulleras E, Wilcock A, Miller SJ, Franklin GC. The sequential activation and repression of the human PDGF-B gene during chronic hypoxia reveals antagonistic roles for the depletion of oxygen and glucose. Growth Factors. 2001;19:233–245. doi: 10.3109/08977190109001089. [DOI] [PubMed] [Google Scholar]
- 86.Alpers CE, Seifert RA, Hedkins KL, Johnson RJ, Bowen-Pope DR. Developmental patterns of PDGF B-chain, PDGF-receptor and β-actin expression in human glomerulogenesis. Kidney Int. 1992;42:390–399. doi: 10.1038/ki.1992.300. [DOI] [PubMed] [Google Scholar]
- 87.Eichmann A, Makinen T, Alitalo K. Neural guidance molecules regulate vascular remodeling and vessel navigation. Genes Dev. 2005;19:1013–1021. doi: 10.1101/gad.1305405. [DOI] [PubMed] [Google Scholar]
- 88.Klagsbrun M, Eichmann A. A role for axon guidance receptors and ligands in blood vessel development and tumor angiogenesis. Cytokine Growth Fact Rev. 2005;16:535–548. doi: 10.1016/j.cytogfr.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 89.Robert B, Zhao X, Abrahamson DR. Coexpression of neuropilin-1, Flk1 and VEGF(164) in developing and mature mouse kidney glomeruli. Am J Physiol Renal Physiol. 2000;279:275–282. doi: 10.1152/ajprenal.2000.279.2.F275. [DOI] [PubMed] [Google Scholar]
- 90.Villegas G, Tufro A. Ontogeny of semaphorins 3A and 3F and their receptors neuropilins 1 and 2 in the kidney. Mech Dev. 2002;119:149–153. doi: 10.1016/s0925-4773(03)00108-4. [DOI] [PubMed] [Google Scholar]
- 91.Harper SJ, Xing CY, Whittle C, et al. Expression of neuropilin-1 by human glomerular epithelial cells in vitro and in vivo. Clin Sci. 2001;101:439–446. [PubMed] [Google Scholar]
- 92.Guan F, Villegas G, Teichman J, et al. Autocrine VEGF system in podocytes regulates podocin and its interaction with CD2AP. Am J Physiol Renal Physiol. 2006;291:422–428. doi: 10.1152/ajprenal.00448.2005. [DOI] [PubMed] [Google Scholar]
- 93.Daniel TO, Stein E, Cerretti DP, St John PL, Robert B, Abrahamson DR. ELK and LERK-2 in developing kidney and microvascular endothelial assembly. Kidney Int. 1996;50:73–81. [PubMed] [Google Scholar]
- 94.Aumailley M, Bruckner-Tuderman L, Carter WG, et al. A simplified laminin nomenclature. Matrix Biol. 2005;24:326–332. doi: 10.1016/j.matbio.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 95.Miner JH, Yurchenco PD. Laminin functions in tissue morphogenesis. Annu Rev Cell Dev Biol. 2004;20:255–284. doi: 10.1146/annurev.cellbio.20.010403.094555. [DOI] [PubMed] [Google Scholar]
- 96.Colognato H, Yurchenco PD. Form and function: the laminin family of heterotrimers. Dev Dyn. 2000;218:213–234. doi: 10.1002/(SICI)1097-0177(200006)218:2<213::AID-DVDY1>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 97.Miner JH. Renal basement membrane components. Kidney Int. 1999;56:2016–2024. doi: 10.1046/j.1523-1755.1999.00785.x. [DOI] [PubMed] [Google Scholar]
- 98.Miner JH. Developmental biology of glomerular basement membrane components. Curr Opin Nephrol Hypertens. 1998;7:13–19. doi: 10.1097/00041552-199801000-00003. [DOI] [PubMed] [Google Scholar]
- 99.St John PL, Abrahamson DR. Glomerular endothelial cells and podocytes jointly synthesize laminin-1 and -11 chains. Kidney Int. 2001;60:1037–1046. doi: 10.1046/j.1523-1755.2001.0600031037.x. [DOI] [PubMed] [Google Scholar]
- 100.Miner JH, Li C. Defective glomerulogenesis in the absence of laminin a5 demonstrates a developmental role for the kidney glomerular basement membrane. Dev Biol. 2000;217:278–289. doi: 10.1006/dbio.1999.9546. [DOI] [PubMed] [Google Scholar]
- 101.Abrahamson DR, St John PL, Isom K, Robert B, Miner JH. Partial rescue of glomerular laminin a5 mutation by wildtype endothelial cells produce hybrid glomeruli. J Am Soc Nephrol. 2007;18:2285–2297. doi: 10.1681/ASN.2007020207. [DOI] [PubMed] [Google Scholar]
- 102.Miner JH, Go G, Cunningham J, Patton BL, Jarad G. Transgenic isolation of skeletal muscle and kidney defects in laminin beta2 mutant mice: implications for Pierson syndrome. Development. 2006;133:967–975. doi: 10.1242/dev.02270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hudson BG. The molecular basis of Goodpasture and Alport syndromes: beacons for the discovery of the collagen IV family. J Am Soc Nephrol. 2004;15:2514–2527. doi: 10.1097/01.ASN.0000141462.00630.76. [DOI] [PubMed] [Google Scholar]
- 104.Hudson BG, Tryggvason K, Sundaramoorthy M, Neilson EG. Alport's syndrome, Goodpasture's syndrome, and type IV collagen. N Engl J Med. 2003;348:2543–2556. doi: 10.1056/NEJMra022296. [DOI] [PubMed] [Google Scholar]
- 105.Desjardins M, Bendayan M. Ontogenesis of glomerular basement membrane: Structural and functional properties. J Cell Biol. 1991;113:689–700. doi: 10.1083/jcb.113.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ninomiya Y, Kagawa M, Iyama K, et al. Differential expression of two basement membrane collagen genes, COL4A6 and COL4A5, demonstrated by immunofluorescence staining using peptide-specific monoclonal antibodies. J Cell Biol. 1995;130:1219–1229. doi: 10.1083/jcb.130.5.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Butkowski RJ, Langeveld JPM, Wieslander J, Hamilton J, Hudson BG. Localization of the Goodpasture epitope to a novel chain of basement membrane collagen. J Biol Chem. 1987;262:7874–7877. [PubMed] [Google Scholar]
- 108.Tryggvason K, Zhou J, Hostikka SL, Shows TB. Molecular genetics of Alport syndrome. Kidney Int. 1993;43:38–44. doi: 10.1038/ki.1993.8. [DOI] [PubMed] [Google Scholar]
- 109.Kashtan CE. Alport syndrome and thin glomerular basement membrane disease. J Am Soc Nephrol. 1998;9:1736–1750. doi: 10.1681/ASN.V991736. [DOI] [PubMed] [Google Scholar]
- 110.Kashtan CE. Alport syndrome. An inherited disorder of renal, ocular and cochlear basement membranes. Medicine. 1999;78:338–360. doi: 10.1097/00005792-199909000-00005. [DOI] [PubMed] [Google Scholar]
- 111.Kalluri R, Shield CF, Todd P, Hudson BG, Neilson EG. Isoform switching of type IV collagen is developmentally arrested in X-linked Alport syndrome leading to increased susceptibility of renal basement membranes to endoproteolysis. J Clin Invest. 1997;99:2470–2478. doi: 10.1172/JCI119431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gunwar S, Ballester F, Noelken ME, Sado Y, Ninomiya Y, Hudson BG. Glomerular basement membrane. Identification of a novel disulfide-cross-linked network of alpha3, alpha4 and alpha5 chains of type IV collagen and its implications for the pathogenesis of Alport syndrome. J Biol Chem. 1998;273:8767–8775. doi: 10.1074/jbc.273.15.8767. [DOI] [PubMed] [Google Scholar]
- 113.Cosgrove D, Meehan DT, Grunkemeyer JA, Kornak JM, Sayers R, Hunter WJ, Samuelson GC. Collagen COL4A3 knockout: A mouse model for autosomal Alport syndrome. Genes Dev. 1996;10:2981–2992. doi: 10.1101/gad.10.23.2981. [DOI] [PubMed] [Google Scholar]
- 114.Miner JH, Sanes JR. Molecular and functional defects in kidneys of mice lacking collagen alpha3(IV): Implications for Alport syndrome. J Cell Biol. 1996;135:1403–1413. doi: 10.1083/jcb.135.5.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lu W, Phillips CL, Killen PD, Hlaing T, Harrison WR, Elder FF, et al. Insertional mutation of the collagen genes Col4a3 and Col4a4 in a mouse model of Alport syndrome. Genomics. 1999;61:113–124. doi: 10.1006/geno.1999.5943. [DOI] [PubMed] [Google Scholar]
- 116.Abrahamson DR, Prettyman AC, Robert B, St John PL. Laminin-1 reexpression in Alport mouse glomerular basement membranes. Kidney Int. 2003;63:826–834. doi: 10.1046/j.1523-1755.2003.00800.x. [DOI] [PubMed] [Google Scholar]
- 117.Cosgrove D, Rodgers K, Meehan D, Miller C, Bovard K, Gilroy A, et al. Integrin alpha1beta1 and transforming growth factor-beta1 play distinct roles in Alport glomerular pathogenesis and serve as dual targets for metabolic therapy. Am J Pathol. 2000;157:1649–1659. doi: 10.1016/s0002-9440(10)64802-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Abrahamson DR, Hudson BG, Stroganova L, St Borza DB, John PL. Cellular origins of glomerular basement membrane type IV collagen networks. J Am Soc Nephrol. 2009 in press. [Google Scholar]
- 119.Morello R, Zhou G, Dreyer SD, Harvey SJ, Ninomiya Y, Thorner PS, et al. Regulation of glomerular basement membrane collagen expression by LMX1B contributes to renal disease in nail patella syndrome. Nat Genet. 2001;27:205–208. doi: 10.1038/84853. [DOI] [PubMed] [Google Scholar]