

Abstract
Influenza infection remains a significant cause of pulmonary morbidity and mortality worldwide with the highest hospitalization and mortality rates occurring in infants and elder adults. The mechanisms inducing this considerable morbidity and mortality are largely unknown. To address this question, we established a neonatal mouse model of influenza infection to test the hypothesis that the immaturity of the neonatal immune system is responsible for the severe pulmonary disease observed in infants. Seven-day-old mice were infected with influenza A virus (H1N1) and allowed to mature. As adults, these mice showed enhanced airway hyperreactivity, chronic pulmonary inflammation, and diffuse emphysematous-type lesions in the lungs. The adaptive immune responses of the neonates were much weaker than those of adults. This insufficiency appeared to be in both magnitude and functionality and was most apparent in the CD8+ T cell population. To determine the role of neonatal CD8+ T cells in disease outcome, adult, naive CD8+ T cells were adoptively transferred into neonates prior to infection. Neonatal mice receiving the adult CD8+ T cells had significantly lower pulmonary viral titers and greatly improved pulmonary function as adults (airway resistance similar to SHAM). Additional adoptive transfer studies using adult CD8+ T cells from IFN-γ-deficient mice demonstrated the importance of IFN-γ from CD8+ T cells in controlling the infection and in determining disease outcome. Our data indicate that neonates are more vulnerable to severe infections due to immaturity of their immune system and emphasize the importance of vaccination in infants.
Keywords: Rodents; T Cells, Cytotoxic; Viral infection; Inflammation; Lung
Introduction
Each year influenza viruses cause significant morbidity and mortality (1). Influenza virus A, in particular, has been shown to elicit respiratory illnesses such as pneumonia and bronchiolitis and to exacerbate underlying respiratory diseases such as chronic obstructive pulmonary disease (COPD) and asthma (2). In fact, last winter (Oct 2006 to May 2007), 23,753 specimens tested positive for influenza viruses in collaborating laboratories of the World Health Organization and the National Respiratory and Enteric Virus Surveillance System in the United States (http://www.cdc.gov/flu/weekly/weeklyarchives2006-2007/06-07summary.htm). Of the influenza positive specimens, 79% were influenza A viruses and 62% of these were of the H1 subtype (http://www.cdc.gov/flu/weekly/weeklyarchives2006-2007/06-07summary.htm). Interestingly, the highest incidence of illness occurs in pre-school and school age children with 20 to 30% of children in this age group being diagnosed with influenza infections each year (3-6). Infected infants younger than six months of age have higher hospitalization and mortality rates than do older children (7, 8). Complications of influenza infections in infants typically include lower respiratory tract involvement and although these infections occur less frequently than respiratory syncytial virus (RSV) infections in infants, they are also a significant cause of wheeze (9). Epidemiological data demonstrate that infection of infants with influenza can lead to chronic pulmonary distress and furthermore that the effects of influenza in this population are underestimated and may be responsible for previously unexplained chronic pulmonary dysfunction (10).
To understand how influenza induces pulmonary illness, various animal models have been used, including ferrets, rats, chickens, and mice (11). While neutralizing antibodies to influenza are important for protection from viral infection, CD8+ T cells have been shown to play a pivotal role in viral clearance and recovery from the illness in adult mice. CD8+ T cell deficient mice exhibit delayed viral clearance and significantly higher mortality when challenged with a sub-lethal dose of influenza (12). In addition, in B cell deficient mice, adoptive transfer of CD8+ T cells promotes more rapid clearance of the virus and recovery from illness (13, 14).
Despite the global burden of influenza infection in infants and children, very few studies have examined the pathogenesis of infection in an infant model. The infant and neonatal immune systems are immature and this immaturity contributes to the pathogenesis of various lower respiratory tract infections including influenza, RSV, and others. Lower respiratory tract viral infections in infants are often associated with acute and persistent pulmonary dysfunction, which is characterized by increased airway resistance and hyperresponsiveness (15-17). Previously, we and other groups have described a neonatal mouse model (i.e. 7d of age at initial infection) of RSV infection (18-20). In this model, the age of primary infection determined the immunological nature (Th1 or Th2) of the secondary infection. Th2 immune responses dominated upon secondary infection, if the primary infection occurred at 1 week or less of age; while Th1 responses dominated if the mice were primarily infected as adults (8 wk old) (18). Interestingly, a single infection with RSV in neonatal mice induced long-term pulmonary dysfunction (20); which was exacerbated upon secondary infection with RSV (19). The RSV neonatal model demonstrated that the age at which the initial infection occurred is critical in determining the subsequent host immune response to the pathogen; and furthermore, it suggested that if the infection occurs too early, the immature immune response contributes to pathogenesis instead of protection.
A recent study in human infants suggests that a failure to develop a cytotoxic T lymphocyte response is responsible for the high rate of infant morbidity and mortality caused by respiratory viruses (21). The article demonstrated that in both fatal infant influenza and RSV infection, CD4+ and CD8+ T cells were present in the lung at very low frequency. In addition, granzyme-producing cells (either cytotoxic CD8+ T cells or NK cells) were not observed in the lungs of infants with fatal influenza virus infection and correlated with massive viral replication and apoptosis of inflammatory cells.
To understand the relationships among the type of respiratory virus, the pulmonary immune response and the long-term pulmonary pathophysiology after infection in infancy, we established a neonatal (7d of age) mouse model of influenza A virus infection. In this model, neonatal infection induced long-term pulmonary inflammation and airway injury and dysfunction accompanied with a weak CD8+ T cell response (lower number and lower IFN-γ production in CD8+ T cells). To examine if the weak CD8+ T cell response played a role in the pathogenesis of influenza infection in neonates, we adoptively transferred adult CD8+ T cells into neonatal mice prior to infection with influenza. Our data demonstrate that the insufficient production of IFN-γ by neonatal CD8+ T cell may be responsible for the long-term pulmonary inflammation and lung injury in influenza infected neonatal mice.
Materials and Methods
Mice
BALB/c mice were purchased as breeders from Harlan Sprague Dawley, Inc. IFN-γ knockout mice (C.129S7(B6)-Ifngtm1Ts/J) were purchased from the Jackson Laboratory. Mice were maintained under specific pathogen-free conditions within the vivarium at Louisiana State University Health Sciences Center (New Orleans, LA). Sentinel mice within each colony were monitored and were negative for specific known mouse pathogens. Breeders were time-mated and 7 d old pups (neonates) were used for experiments. All animal protocols were prepared in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996) and approved by the Institutional Animal Care and Use Committee at Louisiana State University Health Sciences Center.
Viral infection and viral titer determination
Human Influenza A/PR/8/34 (H1N1) was purchased as a sucrose-gradient purified virus from Advanced Biotechnologies, Inc. The virus preparation was determined to be free of bacteria, yeast, and fungi. Viral titer was measured in whole lung homogenates at different time points (Fig 1) using the TCID50 method of Spearman-Kärber (22, 23). Mardin-Darby canine kidney cells were seeded on a 96-well plate and then inoculated with a series of 10 fold dilutions of lung homogenates. Cells were then incubated at 37°C and 5% CO2 for four days; wells showing cytopathic effects were counted and TCID50 were calculated.
Figure 1.

The study protocol for the experiments is outlined. A) 7 d old mouse pups were infected intranasally with 10 TCID50 H1N1 / g whole body weight (FLU); while a control group of mice were mock-infected with vehicle (SHAM). BALF was isolated at 0.2, 2, 4, 10, and 109 dpi. Lungs were isolated for viral titer assays at 2, 5, and 10 dpi; for histopathology at 10 and 109 dpi; and for evaluation of T cell subpopulations (T pop) at 7 dpi. Pulmonary function (PFT) was tested at 109 dpi. B) For the adoptive transfer study, 6 d old pups were administered CD8 T cells purified from the spleens of naïve neonatal (7 d old, CD8N), adult (4 wk old, CD8A), or adult IFN-γ deficient (4 wk old, CD8AKO) mice. Pups were infected with influenza (FLU) at 7 d of age and allowed to mature until 37 d of age (30 dpi). Four control groups were included: neonatal or adult non-infected mice (SHAM and ASHAM, respectively) and neonatal or adult influenza infected (FLU and AFLU, respectively). Viral titers were measured at 4 and 30 dpi. Pulmonary function test, BALF cellularity and lung histopathology were measured at 30 dpi.
Experimental design
Seven day old pups were infected intranasally with 10 TCID50/g body weight of influenza (FLU) in 10 μl of DPBS or sham infected with 10 μl DPBS (SHAM). Mouse pups were then allowed to mature and various assays performed at the indicated time points as outlined in Fig 1A. For adoptive transfer studies, CD8+ T cells from naïve wild-type adults (FLU/CD8A), wild-type neonates (FLU/CD8N), or IFN-γ knockout mice (FLU/CD8AKO) were administered intraperitoneally to 6 d old pups; and at 7 d of age, these pups were then infected with influenza (Fig 1B). Four groups of mice were included as controls: vehicle treated pups (SHAM), influenza infected pups (FLU), vehicle treated adult mice (ASHAM), and influenza infected adult mice (AFLU). A range of assays were performed at indicated time points as shown in Fig 1B.
Pulmonary function test
Pulmonary function, specifically the respiratory mechanics, were measured using an invasive method as previously described (20). Briefly, anesthetized mice were intubated and mechanically ventilated by a computer controlled piston ventilator (FlexiVent, SCIREQ). Mice were then challenged by an aerosolized bronchoconstrictor methacholine (Sigma) at increasing doses (MeCh: 0, 25, and 50 mg/ml). At each dose, lung resistance and compliance were calculated using the single compartment model.
Lung histopathology
At different time points, lungs were perfused, inflated by gentle infusion of HistoChoice Tissue Fixative (Amresco, Inc) to tidal volume (6 ml/kg), and isolated (Fig 1). The fixed lungs were then dehydrated, embedded in paraffin, and sectioned at 4 μm. Each lung section was stained with either hematoxylin and eosin (H&E) or periodic acid-Schiff (PAS). Specific histopathological diagnoses were performed by unbiased pathologists (D. T. and D. S.).
Initial observations of the influenza infected lungs suggested emphysematous-type lesions. Morphometric analyses of the lung sections were used to quantify these changes in lung architecture including airspace enlargement (i.e. mean linear intercept; Lm) and destruction of the alveolar walls (i.e. destructive index; DI). Lm was quantified using NIH-Image (http://rsb.info.nih.gov/nih-image). The number of alveolar wall intersections was counted on 12 non-overlapping lung fields and was expressed as μm using a protocol adapted from Thurlbeck (24). To measure DI, a grid with 42 equidistant points (100 μm between each point) was placed at the center of and superimposed on the lung field. Structures lying under these points were classified as normal (N) or destroyed (D) alveolar and/or duct spaces. Points falling over other structures, such as duct walls, alveolar walls, etc. were excluded from the calculations. The DI was calculated from the formula: DI = D / (D + N) × 100. (25)
Bronchoalveolar lavage fluid cellularity and cytokine measurement
Bronchoalveolar lavage fluid (BALF) was isolated in 1 ml of PBS containing 2% heat inactivated FBS at the indicated time points (Fig 1). The cells were then centrifuged onto slides and stained using a Hema-3 staining kit (Fisher). Two unbiased readers counted a total of 300 cells per slide and recorded the differential cell counts based on the morphology and staining of the cells. Cytokine levels were measured from 50 μl of cell-free BALF using a high-throughput multiplex cytokine assay system (X-Plex Mouse Assay; BioRad) according to the manufacturer’s instructions. Each sample was analyzed in triplicate on the Bio-Plex 200 system (BioRad). A broad sensitivity range of standards ranging from 1.21 to 37,312 pg/ml (depending on the analyte) was used to quantitate a dynamic range of cytokine concentrations. The concentrations of analytes in these assays were quantified using a standard curve and a 5-parameter logistic regression was performed to derive an equation that was then used to predict the concentration of the unknown samples. The following cytokines were assayed: IL-2, IL-4, IL-5, IL-6, IL-12(p40),IL-12 (p70), IL-13, IL-17, IFN-γ and TNF-α. The data presented here excluded any number below the range of sensitivity for the particular analyte.
Assessment of pulmonary T cell populations
A single cell suspension of lung cells was prepared using a standardized protocol (26). Briefly, lungs were perfused, excised, cut into small pieces and incubated at 37°C for 1 hour in RPMI-1640 media supplemented by 2% heat inactivated FBS, 1 mg/ml Collagenase I (Invitrogen), and 150 μg/ml DNase I (Sigma). After incubation, single cells were obtained by mashing the lung pieces through a 40 μm cell strainer (BD Biosciences). Red blood cells were lysed using 1X RBC lysis buffer (eBioscience) and the remaining cells were stimulated for 5 hours with 5 ng/ml PMA and 500 ng/ml ionomycin (Sigma) in the presence of a protein transport inhibitor (1μl / 1 × 106 cells; GolgiPlug, BD Biosciences). After stimulation, cells were harvested, fixed and permeabilized (fixation and permeabilization buffer; eBioscience), and stained with the following antibodies purchased from eBioscience: Pacific Blue-CD3 (17A2), PerCP-CD4 (RM4-5), Alexa Fluor 488-CD8a (53-6.7), PE-IFN-γ (XMG1.2) and PE-Cy7-IL-4 (11B11). In order to evaluate influenza specific CD8+ T cell response, lung cells were stained with APC labeled H-2Kd tetramer complexed with the immunodominant epitope TYQQRTRALV (H-2Kd/TYQQRTRALV, Immunomics) from influenza A nucleoprotein (NP) without PMA / ionomycin stimulation. Cell staining was determined with an LSRII (BD Biosciences) flow cytometer after gating on the lymphocyte population as determined by forward and side scatter properties. Flow data were analyzed and plotted using FlowJo software (Version 7.2.2 for windows, Tree Star, Inc).
Adoptive transfer of CD8+ T cells
The protocol is modified from the method developed by Wells et al (27). In brief, single cell suspensions were prepared from the spleens of young adult (4 wks of age), adult IFN-γ knockout mice (4 wks old) or neonatal (7 d old) mice using a standardized protocol as above. CD8+T cells were then isolated using a negative selection strategy according to the manufacturer’s directions (Mouse CD8+ T cell enrichment kit, Stem Cell). Neonatal, adult, or IFN-γ knockout adult CD8+ T cells (4 × 106 in 25 μl of PBS) were then injected intraperitoneally into 6 d old pups (CD8N CD8A, CD8AKO, respectively).
Statistical analysis
All data were plotted as mean ± SEM and analyzed using GraphPad Prism (GraphPad Software Inc., Version 5.0.0). Two-way ANOVA and Bonferroni post-tests were used to test for differences between the groups for the pulmonary function, BALF cellularity, cytokine assays and T cell populations. Student’s T test was used to analyze the differences in lung histopathology parameters (Lm and DI). Differences were considered statistically significant if p < 0.05.
Results
Neonatal influenza infection resulted in acute pulmonary inflammation
BALB/c pups were infected at 7 d of age with 10 TCID50 of influenza A virus/gram body weight (FLU); controls received sterile DPBS (SHAM). Mice infected with this sublethal dose (>90% survival) of influenza developed mild-to-moderate illness, which was characterized by ruffled fur and significant reductions in weight gain compared to SHAM. By 18 dpi, mice infected with influenza weighed 12% less than SHAM animals (p<0.05). Infectious influenza virus was detected in whole lung homogenates in neonatal mice as early as 2 dpi (104.49±0.03 TCID50 /g lung tissue) and peaked at 5 dpi (106.93±0.25 TCID50 /g lung tissue). No infectious viruses were detected in neonatal or adult mice after 7 dpi.
To measure pulmonary inflammation, bronchoalveolar lavage fluid (BALF) cellularity, BALF cytokine levels, and lung histopathology were monitored throughout the course of the infection (Figs 2 & 3). All results from SHAM mice at different time points (5 hours, 1, 2, 4, and 10 days post-infection) were similar and showed no difference (data not shown), therefore only data from 10dpi were presented as a representative. Mice infected with influenza recruited significantly more inflammatory cells to the lung as observed in both the lung and the BALF. Monocytes / AMs were shown to be recruited to the bronchoalveolar space as early as 5 hours post-infection (hpi, Fig 2A), and significantly more cells compared to SHAM were observed at 4 and 10 dpi with the peak at 10dpi (44.09 ± 6.83 vs. 13.19 ± 1.92 × 104). Neutrophils in the BALF showed the same trend. They peaked at 2 dpi (3.20 ± 1.11 vs. 0.12 ± 0.02 × 104); and remained elevated at 10 dpi compared to SHAM (2.58 ± 0.75 vs. 0.08 ± 0.03 × 104). A significant increase in lymphocytes was observed at 10 dpi (8.47 ± 3.79 vs. 0.01 ± 0.01 × 104). Total BALF cell numbers were five-fold higher in the FLU group compared to the SHAM group at 10 dpi (61.30 ± 10.70 vs. 13.30 ± 1.92 × 104, p<0.05).
Figure 2.
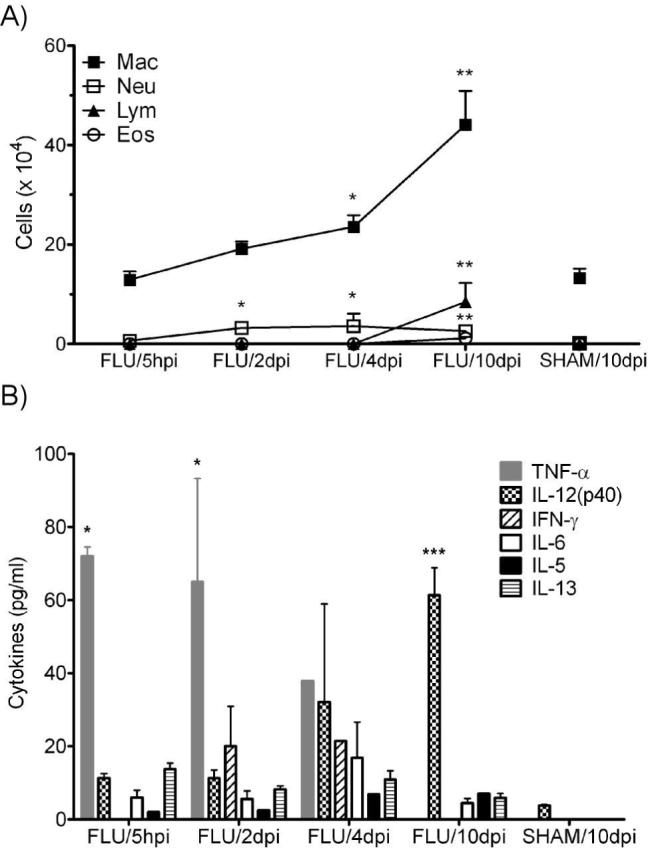
BALF cellularity and cytokine levels in neonatal mice after infection. A) BALF was isolated and the cellularity determined at 5 hpi and at 2, 4, and 10 dpi. Absolute cell numbers were plotted versus time post-infection. n = 4-5 / group. B) Cytokine levels in the BALF supernatant was determined for the following cytokines: IL-2, -4, -5, -6, -12(p40) -12(p70),-13, -17, IFN-γ and TNF-α. Only detected cytokines are displayed on the graph. n = 3-5 / group. Data are expressed as mean ± SEM, *: p<0.05, **: p<0.01, ***: p<0.001 compared to SHAM.
Figure 3.
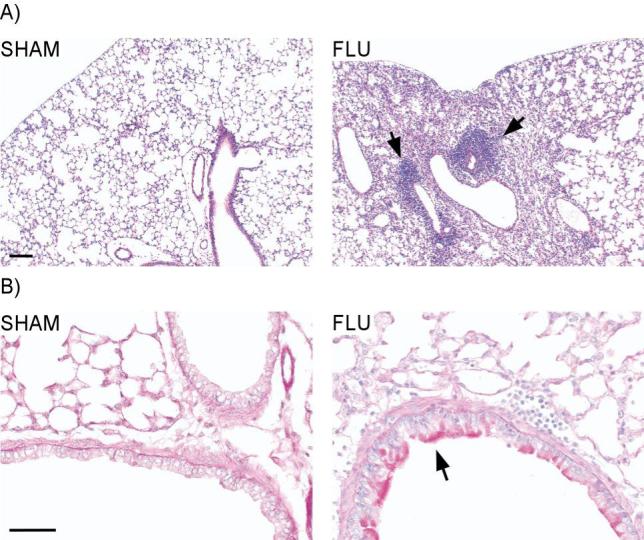
Lung histopathology following neonatal influenza infection. Lungs were isolated, fixed and stained with H&E and PAS at 10 dpi. A) H&E staining showed marked inflammation in the perivascular, peribronchial, and alveolar spaces of the lung (right panel). Diffuse emphysematous changes were also observed in the infected lungs along with slight thickening of the alveolar walls. SHAM mice showed no inflammation or emphysematous changes (left panel). Scale bar = 100 μm. B) PAS staining demonstrated widespread mucus production and proliferation of peribronchial glands within the hilum in the influenza infected mice (right panel). SHAM mice showed no mucus staining. Scale bar = 50μm. Each picture is representative of 3 different animals.
Cytokines in the BALF peaked at various times during the infection (Fig 2B). Although a total of ten cytokines (IL-2, -4, -5, -6, -12(p40), -12(p70),-13, -17, IFN-γ and TNF-α) were measured, only six (IL-5, -6, -12(p40), -13, IFN-γ and TNF-α) were detectable at the time points tested. Of those cytokines detected (with the exception of IFN-γ), all were elevated at 5 hpi. TNF-α was one of the earliest cytokines detected and was present in the highest concentration at 5 hpi (72.0 ± 2.55 pg/ml). It remained significantly elevated at 2 dpi (65.0 ± 28.3 pg/ml) and was undetectable by 10 dpi. IL-12(p40) gradually increased from 5 hpi to 10 dpi; where it became significantly elevated over SHAM (61.4 ± 7.51 pg/ml). IFN-γ was only detectable at 2 (20.1 ± 10.9 pg/ml) and 4 dpi (21.5 ± 0.00 pg/ml). The other three cytokines (IL-5, -6, and -13) were detected at all time points but not significantly different than in SHAM treated mice.
Histopathologic examination of lungs from neonatally infected mice revealed marked pulmonary inflammation that was observed in the perivascular, peribronchial, and alveolar spaces of the lung at 10 dpi (Fig 3A, right panel). The inflammatory infiltrates consisted of lymphocytes and occasional plasma cells. In addition, extensive mucus production was observed in the goblet cells of the infected lungs, along with proliferation of peribronchial glands within the hilum (Fig 3B, right panel). Diffuse emphysematous changes with a slight thickening of the alveolar walls and vascular congestion were also observed. In contrast, SHAM lungs showed normal thickening of the alveolar wall with no inflammation (Fig 3A, left panel) and no mucus staining (Fig 3B, left panel).
Neonatal influenza infection led to long-term pulmonary dysfunction and injury
In order to investigate the long-term effects of neonatal influenza infection, a group of 7 d old pups were infected with the virus and allowed to mature to 116 d of age (109 dpi, Fig 1A). As with the acute studies, BALF cellularity, BALF cytokine levels, and lung histopathology were recorded at 109 dpi.
Significantly more total cells were present in the BALF of previously infected mice compared to SHAM animals at 109 dpi (6.05 ± 1.89 vs. 2.59 ± 0.70 × 105, p<0.05). As shown in Fig 4, there were significantly more monocytes / AMs (51.02 ± 8.79 × 104) present in the BALF of infected animals compared to SHAM mice (25.08 ± 5.17 × 104). Neutrophils (14.42 ± 6.47 × 104) and lymphocytes (6.20 ± 3.24 × 104) were higher than SHAM (1.08 ± 0.53 and 0.25 ± 0.07 × 104, respectively), although not significantly different (p=0.074). Eosinophils were not observed in the BALF of either FLU or SHAM mice. None of the cytokines assayed in the BALF at 109 dpi were significantly different than that of controls (data not shown).
Figure 4.

BALF cellularity in adult mice infected with influenza as neonates. BALF was isolated at 109 dpi and leukocyte populations determined. Data were expressed as mean ± SEM, n = 4-5/group. *: p<0.05.
The persistent change in BALF cellularity at 109 dpi suggested that there might also be unresolved pulmonary inflammation in the lungs of the adult mice initially infected with influenza as neonates. Indeed, lung histopathologic examination (Fig 5A, right panel) confirmed mild-to-moderate peribronchial chronic inflammation consisting mostly of lymphocytes. Furthermore, moderate mucus production was still present in the goblet cells of infected lungs (Fig 5B, right panel). Diffuse emphysematous changes with focal areas of minimal thickening within the alveolar walls were also observed in the infected lungs. Emphysematous lesions were quantified by evaluating both the airspace enlargement in terms of Lm and the destruction of the alveolar walls by measurement of DI. Influenza infected lungs exhibited a marked increase of alveolar enlargement (Lm 79.0 ± 2.89 μm) compared to SHAM (60.0 ± 5.15 μm) and a significant increase destroyed alveolar walls (DI: 57.1 ± 2.91 μm) versus SHAM (14.5 ± 0.58 μm).
Figure 5.
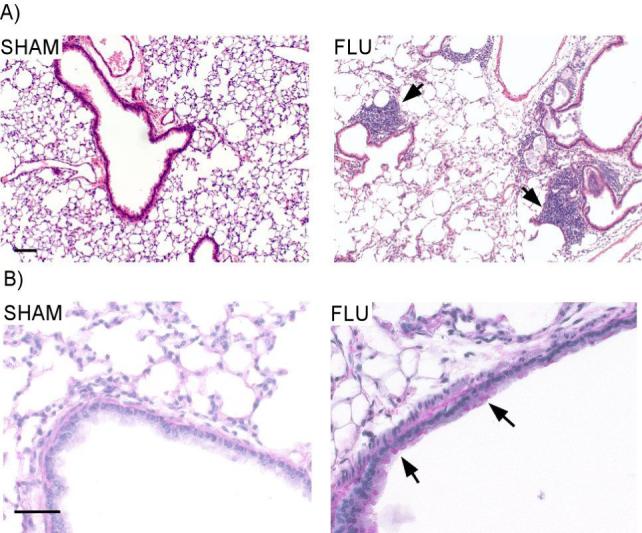
Adult lung histopathology of mice infected with influenza as neonates. Lungs were isolated, fixed and stained by H&E and PAS at 109 dpi. A) Mild-to-moderate peribronchial chronic inflammation consisting mostly of lymphocytes and diffuse emphysematous changes were observed in the lungs of mice infected with influenza as neonates (right panel). No inflammation or emphysematous changes was observed in SHAM mice (left panel). Scale bar = 100 μm. B) Moderate mucus production persisted in airway epithelial cells of influenza infected mice (right panel); while no mucus staining was observed in the lungs of SHAM infected mice. Scale bar = 50 μm. Each picture is a representative of 3 animals.
The change in lung structure associated with the persistence of inflammatory cells in the lung and the BALF suggested alterations in pulmonary function. To determine if neonatal infection with influenza produced long-term effects on lung function, lung mechanics were measured in mechanically ventilated animals at 109 dpi. Mice infected with influenza as neonates had significantly impaired lung function compared to SHAM as evidenced by increased airway hyperreactivity with decreased dynamic compliance in response to MeCh (Fig 6A, B). Baseline resistance was similar in these two groups at this time point (0.49 ± 0.03 vs. 0.47 ± 0.08 cm H2O·s/ml). Although there was no significant shift to the left in the MeCh dose response curve; influenza infection increased pulmonary resistance in response to MeCh (4.12 ± 0.93 vs. 1.25 ± 0.21 cm H2O·s/ml, Fig 6A) and therefore, airway hyperreactivity.
Figure 6.

Persistent airway dysfunction in adult mice infected with influenza as neonates. Pulmonary function was assayed at 109 dpi. A) Influenza infected mice showed a significant increase in lung resistance compared to sham-inoculated mice when challenged by 50 mg / ml methacholine (MeCh). There was no difference in baseline resistance between the FLU and SHAM groups. Data were normalized to baseline (0 mg /ml MeCh) and are expressed as mean ± SEM, n = 5 / group. ***: p<0.001. B) These same mice exhibited a marked decrease in lung compliance if they were infected. For simplicity in interpretation, data are presented as normalized to baseline (0 mg / ml MeCh) and expressed as mean ± SEM, n=5/group. **: p<0.01.
Reduced IFN-γ+ T cells are observed in the neonate in response to influenza infection
To dissect the possible mechanism(s) responsible for the observed long-term pulmonary dysfunction, pulmonary T cell responses were measured using intracellular cytokine staining in influenza infected neonates (FLU) and adults (AFLU; 4wk old) at 7 dpi (Fig 7). Approximately, two-fold more CD8+ T cells (9.7 ± 1.1 vs. 5.2 ± 1.3 × 105) and slightly more CD4+ T cells (1.67 ± 0.18 vs. 1.41 ± 0.48 × 106) were present in the adult lungs compared to the neonatal lungs (Fig 7B, left panel). In addition, neonates infected with influenza appeared to mount a weak IFN-γ response to the virus, compared to that of adults (Fig 7A) (Fig 7B, middle panel). Adult mice infected with influenza (AFLU) were able to recruit almost six-fold more CD8+IFN-γ+ T cells (12.3 ± 1.92 vs. 2.76 ± 1.92 % of CD8+ T cells) and two-fold more CD4+IFN-γ+ T cells (4.04 ± 0.67 vs. 1.92 ± 0.34 % of CD4+ T cells) to the lung as compared to the neonates. CD4+ IFN-γ+ T cells in neonates were statistically different from SHAM levels (0.61 ± 0.07 % of CD4+ T cells). No difference was observed in the number of CD4+IL-4+ T cells recruited to the lungs of mice infected with influenza as neonates or adults (data not shown). To examine the specificity of the CD8+ T cells recruited to the lung, we stained lung lymphocytes with H-2Kd tetramers containing the immunodominant epitope from the influenza nucleoprotein (Fig 7B right panel). Influenza infection of adult mice resulted in the recruitment of about two-fold more CD8+ T cells that bound the influenza tetramer compared to infection of neonatal mice (2.29 ± 0.16% vs 1.24 ± 0.04%).
Figure 7.
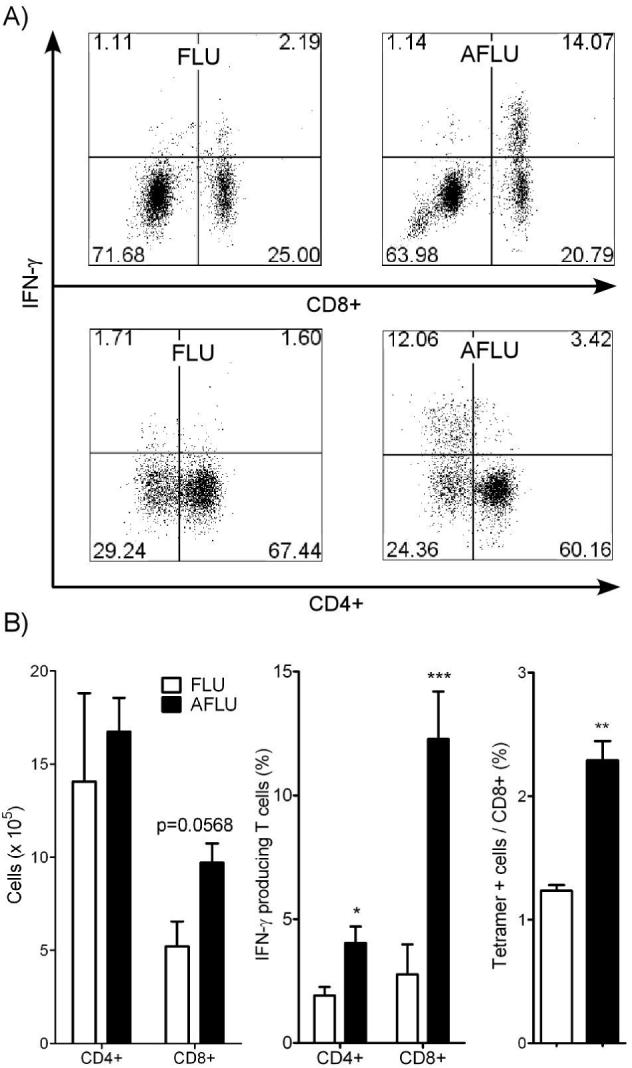
Lymphocyte populations in the lungs of mice infected with influenza as neonates and adults. T cell subpopulations were analyzed in lung homogenates at 7 dpi in mice infected at either 7 d (FLU) or 4 w (AFLU) of age using intracellular cytokine staining and flow cytometry. A) A representative flow cytometry dot plot of IFN-γ+ CD4+ and CD8+ T cells in infected adult or neonatal mice. B) Both CD4+ and CD8+ T cells appeared to be elevated in influenza infected adults (AFLU) compared to mice infected as neonates (FLU, left panel). IFN-γ+ CD4+ and CD8+ T cells were significantly greater in influenza infected adults as compared to infected neonates (middle panel). Finally, CD8+ T cells specific for an immunodominant epitope of influenza in AFLU mice were also significantly greater than in FLU mice (right panel).
Reversal of long-term pulmonary dysfunction after adoptive transfer of adult CD8+T cells but not neonatal CD8+ T cells
To determine whether the persistent pulmonary inflammation, altered lung structure, and long-term lung dysfunction were due to the functional immaturity of host lymphocytes or other factors (e.g. IFN-γ), CD8+ T cells were purified from the spleen of naïve neonatal, adult, or adult IFN-γ deficient mice and adoptively transferred to 6 d old pups (Fig 1B). One day later, these mice were then infected with 10 TCID50/g body weight of influenza (FLU/CD8N, FLU/CD8A, or FLU/CD8AKO, respectively). In addition, neonatal and adult mice were simply infected with influenza (FLU and AFLU, respectively). Pulmonary function testing, BALF cellularity and lung histopathology were performed at 30 dpi.
Both FLU/CD8N and FLU/CD8A groups showed an improvement in pulmonary function, as evidenced by lower airway hyperreactivity compared to control FLU mice (2.89 ± 0.94 and 1.45 ± 0.46 vs. 5.17 ± 1.35 H2O·s/ml at 50 mg/ml MeCh, Fig 8A). Since non-infected control mice, either adult or neonate at time of sham infection, exhibited almost identical responses (data not shown), therefore only the non-infected control mice that were sham infected as neonates (SHAM) are presented. Compared to the FLU/CD8N group, the FLU/CD8A group showed significantly lower pulmonary resistance at 50 mg/ml MeCh (1.00 ± 0.17 cm H2O·s/ml). Moreover, the resistance of FLU/CD8A mice was comparable to that of SHAM mice (1.25 ± 0.21 cm H2O·s/ml). In contrast, FLU/CD8N mice had higher resistance compared to SHAM mice, although pulmonary resistance was lower than that of the FLU mice (2.05 ± 0.39 vs. 4.12 ± 0.93 cm H2O·s/ml). Interestingly, AFLU mice showed medium airway hyperreactivity - lower than mice infected as neonates (FLU) and higher than FLU/CD8A mice. Finally, adoptive transfer of adult CD8+ T cells deficient in IFN-γ prior infection (FLU/CD8AKO) was unable to reverse the effects on pulmonary dysfunction observed in mice infected with influenza as neonates (resistance at 50 mg/ml MeCh of 5.80 ± 1.83 vs. 5.17 ± 1.35 H2O·s/ml).
Figure 8.
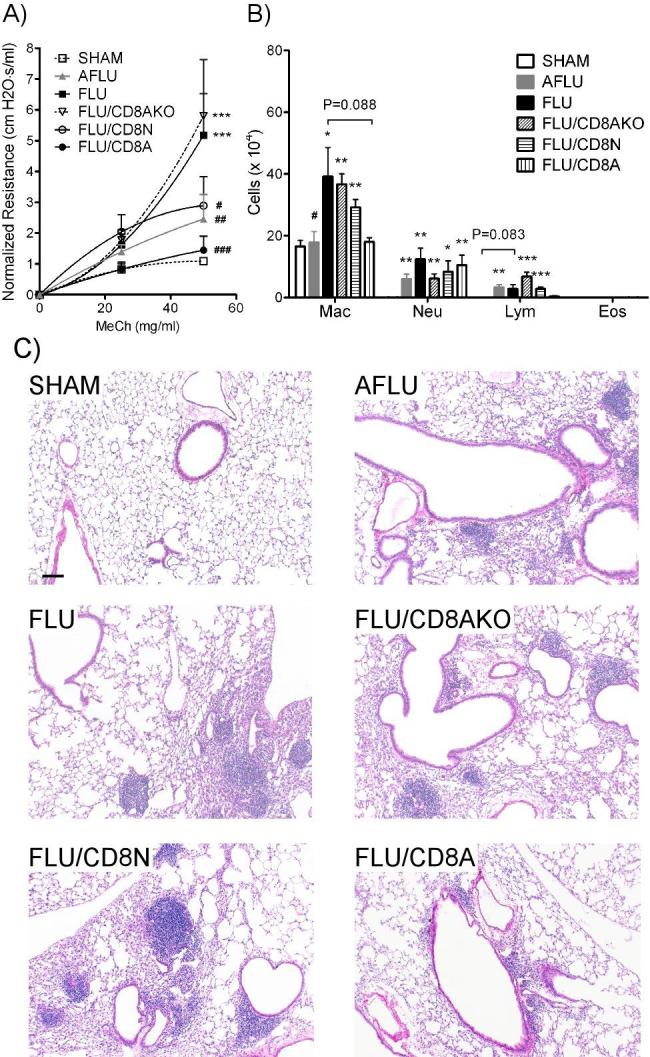
Pulmonary function, BALF cellularity and lung histopathology following adoptive transfer of CD8+ T cells. Neonatal (FLU/CD8N), adult (FLU/CD8A), or adult IFN-γ deficient CD8+ T cells (FLU/CD8AKO) were administered one day prior to influenza infection of neonatal mice. A) Pulmonary function was assayed at 30 dpi. Mice receiving either adult or neonatal CD8+ T cells exhibited improved pulmonary function as demonstrated by a reduction in airway hyperreactivity compared to control mice infected with influenza (FLU), while mice receiving adult IFN-γ deficient CD8+ T cells showed no improvement in lung function. Adoptive transfer of adult CD8+ T cells completely reversed airway hyperreactivity to SHAM levels, while FLU/CD8N mice still showed increased airway hyperreactivity compared to SHAM. Data were normalized to baseline (0 mg / ml MeCh) and are expressed as mean ± SEM, n=4-7/group. ***: p<0.001, compared to SHAM; #: p<0.05, ##: p<0.01, ###: p<0.001, compared to FLU. B) BALF cellularity. There were less monocytes / AMs and lymphocytes in FLU/CD8A mice compared to FLU mice. BALF cellularity for FLU/CD8N and FLU/CD8AKO mice was similar to FLU mice. Data were expressed as mean ± SEM, n=4-7/group. *: p<0.05, **:p<0.01, ***:p<0.001, compared to SHAM; #: p<0.05 compared to FLU mice. C) Lung pathology was assessed at 30 dpi. Substantial reductions in pulmonary infiltrates are observable in the lungs of mice receiving adult CD8+ T cells (FLU/CD8A) prior to infection as compared to all other groups. Other groups included: mice infected as neonates (FLU); mice receiving CD8+ T cells from naïve wild-type adults (FLU/CD8A), wild-type neonates (FLU/CD8N), or IFN-γ knockout mice (FLU/CD8AKO) prior to infection; and adult infected mice (AFLU). Scale bar = 100 μm.
Lung histopathology was also greatly improved in the FLU/CD8A mice as compared to FLU, FLU/CD8N, and FLU/CD8AKO mice. These improvements included reduced pulmonary inflammation (Fig 8C) and mucus production (data not shown) in the infected lungs at 30 dpi. Adoptive transfer of adult CD8+ T cells was not able to completely reverse pulmonary inflammation at 30 dpi, since small foci of inflammatory cells were still observed in the peribronchial and perivascular areas of the lung (Fig 8C). Although the lungs from AFLU mice exhibited similar levels of inflammation as observed in the FLU, FLU/CD8N, and FLU/CD8AKO mice (Fig 8C), there were no emphysematous-type lesions and little-to-no mucus (data not shown).
BALF cellularity showed the same trend (Fig 8B). FLU/CD8A had less monocytes/AMs (18.07 ± 1.32 vs. 39.24 ± 9.35 × 104) and lymphocytes (0.37 ± 0.24 vs. 2.82 ± 1.41) in BALF compared to FLU controls, although a significant amount of neutrophils were still present (10.54 ± 3.23 vs. 12.40 ± 3.61 × 104). Conversely, transfer of neonatal CD8 + T cells (FLU/CD8N) or adult IFN-γ deficient CD8+ T cells (FLU/CD8AKO) prior infection did not reduce cellular infiltrates in BALF, as there were no differences observed between these two groups and FLU controls. Interestingly, adult mice infected with influenza (AFLU) had significant elevated numbers of neutrophils and lymphocytes, but not monocytes / AMs, in their BALF compared to SHAM mice.
The ability of neonatal mice to resolve infection following adoptive transfer of the adult or neonatal CD8+ T cells was then analyzed. Viral titers were determined at 4 and 30 dpi from lung homogenates. The FLU/CD8A group showed a significantly lower viral load compared to FLU mice (102.80± 0.040 vs. 106.42 ± 0.53 TCID50 /g lung tissue). AFLU mice also had lower viral titer than FLU mice (103.57± 0.18 vs. 106.42 ± 0.53 TCID50 /g lung tissue). In addition, FLU/CD8A mice had a significantly lower viral load than AFLU mice at 4dpi (102.80± 0.040 vs. 103.57± 0.18 TCID50 /g lung tissue). No significant differences were observed between FLU/CD8N and FLU or FLU/CD8AKO and FLU mice, although titers in both groups were slightly lower than FLU group (data not shown). At 30 dpi, virus was no longer detectable in any group.
Discussion
In the present study, we described a neonatal mouse model of influenza A infection. In this model, BALB/c mice were infected with influenza virus at 7 d of age and allowed to mature. The mice developed acute, severe pulmonary inflammation, which remained unresolved four months later and long after influenza virus was no longer detectable. This correlated with significant increases in pulmonary resistance in response to increasing doses of methacholine (i.e. airway hyperreactivity) and decreases in compliance at four months. Histopathologic analysis of the lungs from adult mice infected as neonates revealed emphysematous-type lesions characterized by airspace enlargement and destruction of alveolar walls. Adoptive transfer of adult CD8+ T cell into the pups before infection with influenza reversed the effects of neonatal influenza infection as evidenced by enhanced pulmonary function (returning airway resistance to SHAM levels) and reduced pulmonary inflammation and reduced viral load compared to pups receiving neonatal CD8+ T cells. Adoptive transfer of adult CD8+ T cells deficient in IFN-γ indicated that IFN-γ was critical in determining disease outcome.
The inflammatory cells in the BALF during the acute phase (5 hpi to 10 dpi) and chronic phase (109 dpi) included monocytes/AMs, lymphocytes, and neutrophils. Interestingly, although there were more cells in the BALF at 10 dpi than at 109 dpi (∼50% more), the composition of the BALF was not significantly altered (i.e. both were comprised of ∼80% of monocytes/AMs and 20% of lymphocytes and neutrophils) suggesting the continued presence or secretion of chemokines responsible for neutrophil and lymphocyte recruitment even in the absence of detectable infectious virus or viral antigen. At 109 dpi, we were unable to detect differences in cytokines between mice infected as neonates and SHAM controls; however, the role of other cytokines or inflammatory mediators (e.g. leukotrienes, etc.) cannot be excluded.
In addition to the chronic inflammation and airway hyperreactivity to MeCh, histopathologic analysis of the lungs from adult mice originally infected as neonates revealed emphysematous-type lesions within the lung architecture. Intriguingly, there were no differences in baseline resistance between this group and SHAM controls at 109 dpi despite significant differences in lung architecture and inflammatory state. The reason(s) for this are unclear, but may represent masking due to the persistent inflammation or a limitation of the model chosen (i.e. single-compartment) to measure respiratory mechanics.
In general, the immune system of a neonate is quite different from that of an adult, in that the innate and adaptive immune responses are immature (28, 29). Naïve, neonatal T cells are also functionally distinct from adult T cells (30); and effector, neonatal T cells are less able to lyse antigen bearing cells or produce cytokines (31). Although neonatal T cells are able to mount comparable proliferative responses to mitogens (32), they have intrinsically lower levels of CD3/TCR, adhesion molecules, and costimulatory molecules (33). Data from our studies indicate that recruitment of T cells is different in the infected neonates versus the adult and although both T cell populations are induced in the neonate, CD8+ T cell numbers are doubled in the adults. This suggested that cell number alone may have been responsible for the pathophysiological impact of neonatal influenza infection. Although adoptive transfer of neonatal CD8+ T cells prior to infection did help to control the infection (i.e. reduced viral load and improved pulmonary function compared to neonates infected with influenza); it was not as effective as adoptive transfer of adult CD8+ T cells (i.e. further reduction in viral load and pulmonary function equivalent to that of SHAM). These data suggest that neonatal CD8+ T cells are functional impaired compared to adult CD8+ T cells.
In addition to the lower magnitude of the T cell response in the neonates, the number of IFN-γ+ CD8+ T cells was significantly lower than that of their adult counterparts after influenza infection. Previous studies have showed that IFN-γ plays an important role in recovery from influenza infection by helping to clear the virus (34, 35) and that adoptive transfer of Tc1 cells (IFN-γhi cytotoxic T cells) promotes clearance of the influenza virus, while transfer of Tc2 cells (IFN-γlo cytotoxic T cells) does not affect viral clearance (36). Our data confirmed that IFN-γ produced by CD8+ T cells was important to effectively clear influenza from the neonatal lung, since mice receiving IFN-γ deficient adult CD8+ T cells showed higher viral loads than mice receiving wild-type adult CD8+ T cells. Moreover, adoptive transfer of IFN-γ deficient CD8+ T cells totally abolished the benefits observed upon administration of adult CD8+ T cells, as demonstrated by increased airway hyperreactivity, BALF cellularity, and lung histopathology (similar to neonatal infection controls). In total, our data further demonstrate the importance of IFN-γ in the resolution of infection and inflammation initiated upon infection of neonatal mice.
Viral load and immune function are inescapably linked. Also, it is readily apparent that CD8+ T cells do not directly affect disease outcome and that they alter the course of pathogenesis by acting against virus-infected cells (i.e. decreasing viral load) through production of IFN-γ. This contention is strengthened by our observation that introduction of poorly functional neonatal CD8 T-cells does not ameliorate disease, or act against virus-infected cells, thereby permitting a higher viral load in the host. Mice receiving wild-type adult CD8+ T cells exhibited lower viral loads, improved pulmonary function, a reduction in total BALF cellularity, and a reduction in pulmonary inflammation compared to mice infected as adults.
Influenza and another common respiratory virus, RSV, infect the same human population (infants) but elicit different pulmonary diseases. It has been reported that Th2 (IL-4+ T helper cells) responses dominate in neonatal immunity; while Th1 (IFN-γ+ T helper cells) responses dominate in adults (37-41). However, studies from our lab and other groups clearly demonstrate that the immune response initiated by neonates is more complex (20). A previous study showed that RSV infected neonatal mice mount a Th2 biased response when rechallenged as adults with RSV (18). Although a mixture of Th1 and Th2 cells is elicited in lungs during reinfection, there were significantly more (4 fold) Th2 cells in lungs compared to mice primarily infected as adults (18). Data from our lab showed that even at primary infection, neonatal RSV infection mounts a Th2-skewed response (20) compared to influenza infection (data presented here). In fact, both infections mount a mixed Th1/Th2 response. Following influenza infection about 5 fold more Th1 cells than Th2 cells were recruited to the lungs, while RSV infection recruited similar numbers of Th1 and Th2 cells. These data suggest that the immune response initiated in neonates is not predestined toward a Th2 response, as previously implied, and appears to depend on the antigen encountered. Besides the differences in responses of helper T cells to RSV and influenza, both viruses induce a weak CD8+ T cell response similar in magnitude and function. Finally during neonatal RSV infections, although airway remodeling is present (i.e. increased basement membrane thickness, smooth muscle hypertrophy, subepithelial fibrosis), there is relatively no tissue destruction (20). In contrast, neonates infected with influenza exhibited a tremendous amount of tissue destruction, which may be the principal determinant of the severity of airway symptoms. Taken together, there is similarity and disparity in the immune responses induced by RSV and influenza, and the immune and cytopathic differences may explain the specific pulmonary diseases elicited by these two viruses.
In summary, our data demonstrate that infection of newborn mice with influenza has long-term consequences for the host inducing diffuse emphysematous changes in the lung and marked pulmonary inflammation. These alterations were persistent and associated with increased airway resistance and reduced compliance. The adaptive T cell response was markedly reduced in the neonates, with the most striking difference being observed among the CD8+ T cell population. Our adoptive transfer data suggest that the immaturity of this cell population is an important factor in determining disease outcome in the context of the pulmonary microenvironment. These data, along with recent data suggesting that one lung infection has the potential to modify immunity for extended periods of time(42), emphasize the importance of delaying the time of initial influenza infection, and therefore the importance of vaccination in infants and young children. Future studies to elucidate the molecular mechanisms responsible for the persistent inflammation and structural alterations observed with neonatal influenza infection should identify important therapeutic targets capable of controlling long-term complications due to viral bronchiolitis in infancy. Our observations (i.e. that CD8+ T cell responses in neonate are functional different than that of adults) have significant implications for human infants beyond just influenza infection including infant responses to nosocomial infections and even responses to vaccination.
Acknowledgement
We wish to thank Drs. K. Varner, A. Aiyar, P. Zhang, and J. Shellito for review of the manuscript and insightful comments. We also thank Louisiana State University Health Sciences Center Morphology and Imaging Core, Flow Cytometry Core, Division of Animal Care and the office staff of the Department of Pharmacology and Experimental Therapeutics.
Footnotes
This work was supported in part by NIH grant numbers P20 RR020159 from the LSU/Tulane COBRE-CEIDR Program of the National Center for Research Resources and RO1 ES015050 from the National Institutes of Environmental Health to SAC. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
- COPD
- chronic obstructive pulmonary disease
- RSV
- respiratory syncytial virus
- DI
- destructive index
- Lm
- mean linear intercept
- N
- normal
- D
- destroyed
- BALF
- bronchial alveolar lavage fluid
- Eos
- eosinophils
- Mac
- macrophages
- AM
- alveolar macrophages
- Lym
- lymphocytes
- Neu
- neutrophils
Disclosures
The authors declare that they have no competing interests.
References
- 1.Epidemiology and virology of influenza illness. Based on a presentation by Arnold, S. Monto, MD. Am J Manag Care. 2000;6:S255–264. [PubMed] [Google Scholar]
- 2.Upshur RE, Moineddin R, Crighton EJ, Mamdani M. Interactions of viral pathogens on hospital admissions for pneumonia, croup and chronic obstructive pulmonary diseases: results of a multivariate time-series analysis. Epidemiol Infect. 2006;134:1174–1178. doi: 10.1017/S0950268806006236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glezen WP. Emerging infections: pandemic influenza. Epidemiol Rev. 1996;18:64–76. doi: 10.1093/oxfordjournals.epirev.a017917. [DOI] [PubMed] [Google Scholar]
- 4.Glezen WP, Taber LH, Frank AL, Gruber WC, Piedra PA. Influenza virus infections in infants. Pediatr Infect Dis J. 1997;16:1065–1068. doi: 10.1097/00006454-199711000-00012. [DOI] [PubMed] [Google Scholar]
- 5.Collins SD, Lehmann J. Trends and epidemics of influenza and pneumonia: 1918-1951. Public Health Rep. 1951;66:1487–1516. [PMC free article] [PubMed] [Google Scholar]
- 6.Chin TD, Foley JF, Doto IL, Gravelle CR, Weston J. Morbidity and mortality characteristics of Asian strain influenza. Public Health Rep. 1960;75:148–158. [PubMed] [Google Scholar]
- 7.Bhat N, Wright JG, Broder KR, Murray EL, Greenberg ME, Glover MJ, Likos AM, Posey DL, Klimov A, Lindstrom SE, Balish A, Medina MJ, Wallis TR, Guarner J, Paddock CD, Shieh WJ, Zaki SR, Sejvar JJ, Shay DK, Harper SA, Cox NJ, Fukuda K, Uyeki TM. Influenza-associated deaths among children in the United States, 2003-2004. N Engl J Med. 2005;353:2559–2567. doi: 10.1056/NEJMoa051721. [DOI] [PubMed] [Google Scholar]
- 8.Ajayi-Obe EK, Coen PG, Handa R, Hawrami K, Aitken C, McIntosh ED, Booy R. Influenza A and respiratory syncytial virus hospital burden in young children in East London. Epidemiol Infect. 2007:1–13. doi: 10.1017/S0950268807009557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neuzil KM, Zhu YW, Griffin MR, Edwards KM, Thompson JM, Tollefson SJ, Wright PF. Burden of interpandemic influenza in children younger than 5 years: A 25-year prospective study. Journal of Infectious Diseases. 2002;185:147–152. doi: 10.1086/338363. [DOI] [PubMed] [Google Scholar]
- 10.Laraya-Cuasay LR, DeForest A, Huff D, Lischner H, Huang NN. Chronic pulmonary complications of early influenza virus infection in children. The American review of respiratory disease. 1977;116:617–625. doi: 10.1164/arrd.1977.116.4.617. [DOI] [PubMed] [Google Scholar]
- 11.Eichelberger MC. The cotton rat as a model to study influenza pathogenesis and immunity. Viral Immunol. 2007;20:243–249. doi: 10.1089/vim.2007.0017. [DOI] [PubMed] [Google Scholar]
- 12.Bender BS, Croghan T, Zhang L, Small PA., Jr. Transgenic mice lacking class I major histocompatibility complex-restricted T cells have delayed viral clearance and increased mortality after influenza virus challenge. J Exp Med. 1992;175:1143–1145. doi: 10.1084/jem.175.4.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Graham MB, Braciale TJ. Resistance to and recovery from lethal influenza virus infection in B lymphocyte-deficient mice. J Exp Med. 1997;186:2063–2068. doi: 10.1084/jem.186.12.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Epstein SL, Lo CY, Misplon JA, Bennink JR. Mechanism of protective immunity against influenza virus infection in mice without antibodies. J Immunol. 1998;160:322–327. [PubMed] [Google Scholar]
- 15.Pullan CR, Hey EN. Wheezing, asthma, and pulmonary dysfunction 10 years after infection with respiratory syncytial virus in infancy. Br Med J (Clin Res Ed) 1982;284:1665–1669. doi: 10.1136/bmj.284.6330.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sigurs N, Gustafsson PM, Bjarnason R, Lundberg F, Schmidt S, Sigurbergsson F, Kjellman B. Severe respiratory syncytial virus bronchiolitis in infancy and asthma and allergy at age 13. Am J Respir Crit Care Med. 2005;171:137–141. doi: 10.1164/rccm.200406-730OC. [DOI] [PubMed] [Google Scholar]
- 17.Sigurs N, Bjarnason R, Sigurbergsson F, Kjellman B. Respiratory syncytial virus bronchiolitis in infancy is an important risk factor for asthma and allergy at age 7. Am J Respir Crit Care Med. 2000;161:1501–1507. doi: 10.1164/ajrccm.161.5.9906076. [DOI] [PubMed] [Google Scholar]
- 18.Culley FJ, Pollott J, Openshaw PJ. Age at first viral infection determines the pattern of T cell-mediated disease during reinfection in adulthood. J Exp Med. 2002;196:1381–1386. doi: 10.1084/jem.20020943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dakhama A, Park JW, Taube C, Joetham A, Balhorn A, Miyahara N, Takeda K, Gelfand EW. The enhancement or prevention of airway hyperresponsiveness during reinfection with respiratory syncytial virus is critically dependent on the age at first infection and IL-13 production. J Immunol. 2005;175:1876–1883. doi: 10.4049/jimmunol.175.3.1876. [DOI] [PubMed] [Google Scholar]
- 20.You D, Becnel D, Wang K, Ripple M, Daly M, Cormier SA. Exposure of neonates to respiratory syncytial virus is critical in determining subsequent airway response in adults. Respir Res. 2006;7:107. doi: 10.1186/1465-9921-7-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Welliver TP, Garofalo RP, Hosakote Y, Hintz KH, Avendano L, Sanchez K, Velozo L, Jafri H, Chavez-Bueno S, Ogra PL, McKinney L, Reed JL, Welliver RC., Sr. Severe human lower respiratory tract illness caused by respiratory syncytial virus and influenza virus is characterized by the absence of pulmonary cytotoxic lymphocyte responses. J Infect Dis. 2007;195:1126–1136. doi: 10.1086/512615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karber G. Beitrag zur kollektiven behandlung pharmakologischer reihenversuche. Arch exp Path Pharmak. 1931;162:480–483. [Google Scholar]
- 23.Spearman C. The Method of Right and Wrong Cases (constatn stimuli) Without Gauss’s Formulae. Brit J Psychol. 1908;2:227–242. [Google Scholar]
- 24.Thurlbeck WM. Measurement of pulmonary emphysema. The American review of respiratory disease. 1967;95:752–764. doi: 10.1164/arrd.1967.95.5.752. [DOI] [PubMed] [Google Scholar]
- 25.Saetta M, Shiner RJ, Angus GE, Kim WD, Wang NS, King M, Ghezzo H, Cosio MG. Destructive index: a measurement of lung parenchymal destruction in smokers. The American review of respiratory disease. 1985;131:764–769. doi: 10.1164/arrd.1985.131.5.764. [DOI] [PubMed] [Google Scholar]
- 26.Ormerod MG. Preparing suspensions of single cells. Flow Cytometry: A Practical Approach. 2000:39–40. [Google Scholar]
- 27.Wells MA, Ennis FA, Albrecht P. Recovery from a viral respiratory infection. II. Passive transfer of immune spleen cells to mice with influenza pneumonia. J Immunol. 1981;126:1042–1046. [PubMed] [Google Scholar]
- 28.Billingham RE, Brent L, Medawar PB. Actively acquired tolerance of foreign cells. Nature. 1953;172:603–606. doi: 10.1038/172603a0. [DOI] [PubMed] [Google Scholar]
- 29.Bona C. Phenotypic Characteristics of Neonatal T CellsNeonatal Immunity. 2005 [Google Scholar]
- 30.Adkins B. T-cell function in newborn mice and humans. Immunol Today. 1999;20:330–335. doi: 10.1016/s0167-5699(99)01473-5. [DOI] [PubMed] [Google Scholar]
- 31.Granberg C, Hirvonen T, Toivanen P. Cell-mediated lympholysis by human maternal and neonatal lymphocytes: mother’s reactivity against neonatal cells and vice versa. J Immunol. 1979;123:2563–2567. [PubMed] [Google Scholar]
- 32.Yarchoan R, Nelson DL. A study of the functional capabilities of human neonatal lymphocytes for in vitro specific antibody production. J Immunol. 1983;131:1222–1228. [PubMed] [Google Scholar]
- 33.Velilla PA, Rugeles MT, Chougnet CA. Defective antigen-presenting cell function in human neonates. Clin Immunol. 2006;121:251–259. doi: 10.1016/j.clim.2006.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bruder D, Srikiatkhachorn A, Enelow RI. Cellular immunity and lung injury in respiratory virus infection. Viral Immunol. 2006;19:147–155. doi: 10.1089/vim.2006.19.147. [DOI] [PubMed] [Google Scholar]
- 35.Moskophidis D, Kioussis D. Contribution of virus-specific CD8+ cytotoxic T cells to virus clearance or pathologic manifestations of influenza virus infection in a T cell receptor transgenic mouse model. J Exp Med. 1998;188:223–232. doi: 10.1084/jem.188.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiley JA, Cerwenka A, Harkema JR, Dutton RW, Harmsen AG. Production of interferon-gamma by influenza hemagglutinin-specific CD8 effector T cells influences the development of pulmonary immunopathology. Am J Pathol. 2001;158:119–130. doi: 10.1016/s0002-9440(10)63950-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adkins B, Bu Y, Cepero E, Perez R. Exclusive Th2 primary effector function in spleens but mixed Th1/Th2 function in lymph nodes of murine neonates. J Immunol. 2000;164:2347–2353. doi: 10.4049/jimmunol.164.5.2347. [DOI] [PubMed] [Google Scholar]
- 38.Adkins B, Chun K, Hamilton K, Nassiri M. Naive murine neonatal T cells undergo apoptosis in response to primary stimulation. J Immunol. 1996;157:1343–1349. [PubMed] [Google Scholar]
- 39.Adkins B, Ghanei A, Hamilton K. Developmental regulation of IL-4, IL-2, and IFN-gamma production by murine peripheral T lymphocytes. J Immunol. 1993;151:6617–6626. [PubMed] [Google Scholar]
- 40.Min B, Legge KL, Pack C, Zaghouani H. Neonatal exposure to a self-peptide-immunoglobulin chimera circumvents the use of adjuvant and confers resistance to autoimmune disease by a novel mechanism involving interleukin 4 lymph node deviation and interferon gamma-mediated splenic anergy. J Exp Med. 1998;188:2007–2017. doi: 10.1084/jem.188.11.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pack CD, Cestra AE, Min B, Legge KL, Li L, Caprio-Young JC, Bell JJ, Gregg RK, Zaghouani H. Neonatal exposure to antigen primes the immune system to develop responses in various lymphoid organs and promotes bystander regulation of diverse T cell specificities. J Immunol. 2001;167:4187–4195. doi: 10.4049/jimmunol.167.8.4187. [DOI] [PubMed] [Google Scholar]
- 42.Didierlaurent A, Goulding J, Patel S, Snelgrove R, Low L, Bebien M, Lawrence T, van Rijt LS, Lambrecht BN, Sirard JC, Hussell T. Sustained desensitization to bacterial Toll-like receptor ligands after resolution of respiratory influenza infection. J Exp Med. 2008;205:323–329. doi: 10.1084/jem.20070891. [DOI] [PMC free article] [PubMed] [Google Scholar]