

Abstract
‘Mitochondrial dysfunction’, which may result from an accumulation of damaged mitochondria in cells due to a slowed-down rate of mitochondrial turnover and inadequate removal of damaged mitochondria during aging, has been implicated as both cause and consequence of the aging process and a number of age-related pathologies. Despite growing interest in mitochondrial function during aging, published data on mitochondrial turnover are scarce, and differ from each other by up to one order of magnitude. Here we demonstrate that re-utilization of the radioactively labelled precursor in pulse-chase assays is the most likely cause of significant overestimation of mitochondrial turnover rates. We performed a classic radioactive label pulse-chase experiment using 14C NaHCO3, whose 14C is incorporated into various amino acids, to measure mitochondrial turnover in mouse liver. In this system, the activity of the urea cycle greatly limited arginine dependent label re-utilization, but not that of other amino acids. We used information from tissues that do not have an active urea cycle (brain and muscle) to estimate the extent of label re-utilization with a dynamic mathematical model. We estimated the actual liver mitochondrial half life as only 1.83 days, and this decreased to 1.16 days following 3 months of dietary restriction, supporting the hypothesis that this intervention might promote mitochondrial turnover as a part of its beneficial effects.
Keywords: dietary restriction, half life, liver, mathematical model, mitochondria, mice, turnover
Recent years have seen a surge of interest in the role of mitochondrial dysfunction, reactive oxygen species production and mitochondrial DNA mutation as driving factors in the aging process (Balaban et al., 2005; Trifunovic et al., 2005; Bender et al., 2006; Passos et al., 2007). Damaged mitochondria might accumulate in cells due to a slowed-down turnover with aging (de Grey, 1997; Kowald & Kirkwood, 2000; Terman & Brunk, 2005). Conversely, it was proposed that the ‘anti-aging’ function of dietary restriction (DR) might be at least partially due to stimulation of molecular and, specifically, mitochondrial turnover (Donati et al., 2001; Bergamini et al., 2003; Del Roso et al., 2003; Cuervo et al., 2005). Surprisingly, essentially all information on mitochondrial turnover relies on data that are not only at least 20 years old but that vary by one order of magnitude between different estimates. These differences are largely methodological. Mitochondrial protein degradation is generally measured by radio-isotope pulse-chase assay to calculate the rate constant of degradation (and so the half life), assuming that the data follow a simple exponential decay. Label re-utilization (i.e. the re-incorporation of labelled precursor arising from broken down products of the pulse-labelled protein) can be a major problem with this approach, leading to significant lengthening of the apparent half life. An ideal precursor would allow fast protein labelling (i.e. the level of the specific activity of the precursor pool should quickly decrease to zero) thereby avoiding label re-utilization. In essence, the best precursor will give the shortest half life estimate.
Labelling of liver mitochondria with 14C NaHCO3 (which is converted to arginine in the liver) (Lipsky & Pedersen, 1981; Lavie et al., 1982; Saikumar & Kurup, 1985) or 14C arginine (Aschenbrenner et al., 1970; Gross, 1971; Glass & Doyle, 1972) has consistently given the shortest half life in contrast to the use of other amino acid precursors, suggesting that label re-utilization of non-arginine amino acid precursors was a major problem. This is because 14C NaHCO3 is converted to 14C arginine (the 14C label being in the guanidine position; 6-14C arginine) in the liver through urea cycle activity, while NaHCO3 turnover is fast in vivo and the specific activity of 14C NaHCO3 decreases rapidly (Millward, 1970). Re-utilization of labelled arginine is minimized by the high activity of arginase found in the liver, which quickly decomposes arginine into urea (which inherits the labelled 14C) that is readily excreted. Unlike 14C arginine, 14C NaHCO3 will not substantially label proteins in nonhepatic tissues and can thus avoid 14C label re-utilization of broken down products deriving from nonhepatic tissues.
However, in a standard 14C NaHCO3 pulse-chase protocol as described by Lipsky & Pedersen (1981, 1982) (Fig. 1A), we still found evidence for label re-utilization: the fit of a single exponential decay curve to the data was not good. In particular, using this model, the estimated half lives increased systematically if later time points were included in the data set (Fig. 1B). In addition, when comparing DR animals to controls, the estimated half lives appeared faster or slower depending on the length of the chase period (Fig. 1B). This indicated that the decay in specific activity of 14C in liver mitochondria followed a more complex pattern consisting of a fast component, which is the degradation of mitochondrial 6-14C-labelled arginine, and another slower component. The slow component could be non-arginine-derived-14C, which would not escape the re-utilization problem. In fact, a small fraction of exogenous carbon is incorporated into non-arginine amino acids in the liver, and this is similar to the incorporation in skeletal muscles and intestine (Swick et al., 1953; Swick & Handa, 1956), which do not have complete and active urea cycle enzymes to convert 14C NaHCO3 to 6-14C arginine. Accordingly, we found low 14C labels in skeletal muscles and brain mitochondria, which were similar to each other (Fig. 2A). Moreover, both the absolute values and the rates of label decay in muscle and brain mitochondria were very similar to the estimated slow component in liver mitochondria (estimated by a two-component decay model). Therefore, we propose to use data on the slow component obtained from different tissues of the same animals as a surrogate estimate for the slow component in the liver. This significantly improves the statistical power of the obtainable fit in comparison to a two-component decay model which is fit to liver data alone.
Fig. 1.

A single exponential decay model is not adequate to measure mouse liver mitochondrial half life. (A) Changes in specific activity of 14C in liver mitochondria from control mice with time. Results from two sets of independent experiments are shown (full and open circles). Each data point represents an individual animal. (B) Effect of chase period on half lives of mitochondria from dietary restricted (DR) and control mice. Apparent half life time of mitochondria λ depends on the chase period if calculated as single exponential decay by logarithmic transfer. Data are mean ± SEM. • = DR;  = controls. These estimates in control animals are in good agreement with published data with corresponding chase periods (Lipsky & Pedersen, 1981; Lavie et al., 1982; Saikumar & Kurup, 1985).
= controls. These estimates in control animals are in good agreement with published data with corresponding chase periods (Lipsky & Pedersen, 1981; Lavie et al., 1982; Saikumar & Kurup, 1985).
Fig. 2.

Liver mitochondrial half life is short and decreases further by dietary restriction. (A) Observed (circles) and median simulated (solid lines) decay of specific activity of 14C label in liver (left), muscle (centre) and brain (right) mitochondria. Top panels are from 6-month-old control mice and the bottom panels are from mice dietary restricted for 3 months. Dashed lines are 5% and 95% quantiles of samples from simulated label count posterior distributions (20 000 simulations sampled after discarding 10 000 ‘run-in’ simulations). (B) Posterior frequency distributions of calculated half lives (λFast, in days) of liver mitochondria from control and dietary restricted mice. In 99.96% of the 20 000 sampled simulations λDR < λControl, which is a statistically significant result. The median values for λDR and λControl are 1.16 days and 1.83 days, respectively.
Thus, we describe nonspecific 14C label and its decay in liver mitochondria (the slow component) by the average of the brain and muscle 14C counts. Modelling the slow decay in all three tissues as a linear decrease, i.e. the simplest dynamic model possible (an exponential decay model changed the results very little), we obtain:
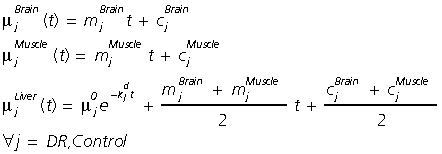 |
where 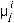 (count · mg−1) are the total 14C counts at time t (day) for tissue i and experimental condition j (control and DR),
(count · mg−1) are the total 14C counts at time t (day) for tissue i and experimental condition j (control and DR), 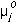 (count · mg−1) represents the value of the exponential ‘fast’ component in the liver at t= 0,
(count · mg−1) represents the value of the exponential ‘fast’ component in the liver at t= 0, 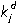 (day−1) are rate parameters describing the exponential decay of the specific label (6-14C arginine) with time in the liver, and
(day−1) are rate parameters describing the exponential decay of the specific label (6-14C arginine) with time in the liver, and 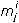 (count mg−1 · t−1) and
(count mg−1 · t−1) and 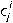 (count · mg−1) are the slope and intercept of the slower linear processes observed in tissue i and experimental condition j, respectively. Note that the amount of label in the liver at time t= 0 (the amount incorporated initially) can be estimated as:
(count · mg−1) are the slope and intercept of the slower linear processes observed in tissue i and experimental condition j, respectively. Note that the amount of label in the liver at time t= 0 (the amount incorporated initially) can be estimated as:
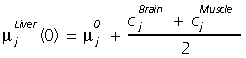 |
We estimated model parameter values for both control and DR mice by Bayesian inference using Gibbs sampling (using Markov chain Monte Carlo methods) as implemented in the OpenBUGS software package (Thomas et al., 2006) with starting conditions as indicated in the Supporting Information (script available: http://www.cisban.ac.uk/downloads/Miwa2008.odc). The main advantage of this method over more traditional ones such as least squares is that it generates parameter and model estimate distributions instead of just a single ‘best’ value. This allows us to test for the significance of differences observed between treatments (i.e. DR and control). Modelling process dynamics in all three tissues simultaneously allows us to use information in the brain and muscle data sets to increase the precision of the parameter estimates in the liver model beyond that which we would have using the liver data alone, and to assess the difference in liver mitochondrial half lives between control and DR mice more rigorously.
The simulations fitted the observed data in brain, skeletal muscle and liver mitochondria from both control and DR mice very well (Fig. 2a). Posterior half life distributions for the exponential-decay-component (the fast component, representing the decay of 6-14C arginine-dependent labels) in liver are shown in Fig. 2b. Thus, the estimated median half life of liver mitochondria is 1.83 days for controls and 1.16 days following 3 months DR, a statistically highly significant difference. Table S1 (Supporting Information) summarizes the parameter estimates and their distribution statistics.
Macro-autophagy is the major pathway of mitochondrial degradation (Kim et al., 2007), which is known to be accelerated by starvation. It has been proposed that DR might act similarly and, by promoting mitochondrial turnover, maintain a healthy population of mitochondria (Bergamini et al., 2003; Kim et al., 2007). We demonstrate here for the first time that DR animals indeed have significantly faster rate of liver mitochondrial turnover compared with controls. It should be noted, however, that starvation-induced autophagy is tissue specific. For example, fasting promoted autophagy in liver and skeletal muscles but not in brain (Mizushima et al., 2004). Thus, it is possible that increased turnover of mitochondria may be one of the beneficial mechanisms of DR in liver, but may not necessarily be so in all cell types.
It should also be noted that the concept of mitochondrial half life in itself is problematic. First, mitochondria form dynamic syncytia in cells. Second, macro-autophagy might be selective (for instance, for damaged mitochondria with low membrane potential). Third, at least some mitochondrial protein turnover is mediated by mitochondrial matrix (Lon) proteases (Bulteau et al., 2006; Ngo & Davies, 2007). Our estimates constitute averages over all the heterogeneity resulting from these various processes.
Acknowledgments
We thank Dr Martin Brand and Professor Darren Wilkinson for advice, the comparative biology unit at the Royal Free and University College London for excellent care and experimental support for the animals, and Professor Tim Cowen and Dr Vernon Skinner for use of the facility. This work was supported by a Biotechnology and Biological Sciences Research Council (BBSRC) Systems Biology grant (Centre for Integrative Systems Biology of Ageing and Nutrition (CISBAN)).
Supporting information
Additional supporting information may be found in the online version of this article:
Appendix S1 Supplementary experimental procedures
Table S1 Parameter estimates and their distribution statistics.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the Aging Cell Central Office.
References
- Aschenbrenner B, Druyan R, Albin R, Rabinowitz M. Haem a, cytochrome c and total protein turnover in mitochondria from rat heart and liver. Biochem. J. 1970;119:157–160. doi: 10.1042/bj1190157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- Bergamini E, Cavallini G, Donati A, Gori Z. The anti-ageing effects of caloric restriction may involve stimulation of macroautophagy and lysosomal degradation, and can be intensified pharmacologically. Biomed. Pharmacother. 2003;57:203–208. doi: 10.1016/s0753-3322(03)00048-9. [DOI] [PubMed] [Google Scholar]
- Bulteau AL, Szweda LI, Friguet B. Mitochondrial protein oxidation and degradation in response to oxidative stress and aging. Exp. Gerontol. 2006;41:653–657. doi: 10.1016/j.exger.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Bergamini E, Brunk UT, Droge W, Ffrench M, Terman A. Autophagy and aging: the importance of maintaining ‘clean’ cells. Autophagy. 2005;1:131–140. doi: 10.4161/auto.1.3.2017. [DOI] [PubMed] [Google Scholar]
- Del Roso A, Vittorini S, Cavallini G, Donati A, Gori Z, Masini M, Pollera M, Bergamini E. Ageing-related changes in the in vivo function of rat liver macroautophagy and proteolysis. Exp. Gerontol. 2003;38:519–527. doi: 10.1016/s0531-5565(03)00002-0. [DOI] [PubMed] [Google Scholar]
- Donati A, Cavallini G, Paradiso C, Vittorini S, Pollera M, Gori Z, Bergamini E. Age-related changes in the autophagic proteolysis of rat isolated liver cells: effects of antiaging dietary restrictions. J. Gerontol. A Biol. Sci. Med. Sci. 2001;56:B375–B383. doi: 10.1093/gerona/56.9.b375. [DOI] [PubMed] [Google Scholar]
- Glass RD, Doyle D. On the measurement of protein turnover in animal cells. J. Biol. Chem. 1972;247:5234–5242. [PubMed] [Google Scholar]
- de Grey AD. A proposed refinement of the mitochondrial free radical theory of aging. Bioessays. 1997;19:161–166. doi: 10.1002/bies.950190211. [DOI] [PubMed] [Google Scholar]
- Gross NJ. Control of mitochondrial turnover under the influence of thyroid hormone. J. Cell Biol. 1971;48:29–40. doi: 10.1083/jcb.48.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007;462:245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowald A, Kirkwood TB. Accumulation of defective mitochondria through delayed degradation of damaged organelles and its possible role in the ageing of post-mitotic and dividing cells. J. Theor. Biol. 2000;202:145–160. doi: 10.1006/jtbi.1999.1046. [DOI] [PubMed] [Google Scholar]
- Lavie L, Reznick AZ, Gershon D. Decreased protein and puromycinyl-peptide degradation in livers of senescent mice. Biochem. J. 1982;202:47–51. doi: 10.1042/bj2020047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipsky NG, Pedersen PL. Mitochondrial turnover in animal cells. Half-lives of mitochondria and mitochondrial subfractions of rat liver based on 14C bicarbonate incorporation. J. Biol. Chem. 1981;256:8652–8657. [PubMed] [Google Scholar]
- Lipsky NG, Pedersen PL. Perturbation by clofibrate of mitochondrial levels in animal cells. Implications for a model of mitochondrial genesis. J. Biol. Chem. 1982;257:1473–1481. [PubMed] [Google Scholar]
- Millward DJ. Protein turnover in skeletal muscle. I. The measurement of rates of synthesis and catabolism of skeletal muscle protein using 14C Na2CO3 to label protein. Clin. Sci. 1970;39:577–590. doi: 10.1042/cs0390577. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo JK, Davies KJ. Importance of the lon protease in mitochondrial maintenance and the significance of declining lon in aging. Ann. N.Y. Acad. Sci. 2007;1119:78–87. doi: 10.1196/annals.1404.015. [DOI] [PubMed] [Google Scholar]
- Passos JF, Saretzki G, von Zglinicki T. DNA damage in telomeres and mitochondria during cellular senescence: is there a connection? Nucleic Acids Res. 2007;35:7505–7513. doi: 10.1093/nar/gkm893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saikumar P, Kurup CK. Effect of administration of 2-methyl-4-dimethylaminoazobenzene on the half-lives of rat liver mitochondria and cytochrome oxidase. Biochim. Biophys. Acta. 1985;840:127–133. doi: 10.1016/0304-4165(85)90169-2. [DOI] [PubMed] [Google Scholar]
- Swick RW, Handa DT. The distribution of fixed carbon in amino acids. J. Biol. Chem. 1956;218:577–585. [PubMed] [Google Scholar]
- Swick RW, Buchanan DL, Nakao A. The normal content of fixed carbon in amino acids. J. Biol. Chem. 1953;203:55–61. [PubMed] [Google Scholar]
- Terman A, Brunk UT. The aging myocardium: roles of mitochondrial damage and lysosomal degradation. Heart Lung Circ. 2005;14:107–114. doi: 10.1016/j.hlc.2004.12.023. [DOI] [PubMed] [Google Scholar]
- Thomas A, O’Hara B, Ligges U, Sturtz S. Making BUGS open. R. News. 2006;6:12–17. [Google Scholar]
- Trifunovic A, Hansson A, Wredenberg A, Rovio AT, Dufour E, Khvorostov I, Spelbrink JN, Wibom R, Jacobs HT, Larsson NG. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl Acad. Sci. USA. 2005;102:17993–17998. doi: 10.1073/pnas.0508886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.