

Abstract
Spinal muscular atrophy (SMA) is one of the most common inherited forms of neurological disease leading to infant mortality. Patients exhibit selective loss of lower motor neurons resulting in muscle weakness, paralysis, and often death. Although patient fibroblasts have been used extensively to study SMA, motor neurons have a unique anatomy and physiology which may underlie their vulnerability to the disease process. Here we report the generation of induced pluripotent stem (iPS) cells from skin fibroblast samples taken from a child with SMA. These cells expanded robustly in culture, maintained the disease genotype, and generated motor neurons that showed selective deficits compared to those derived from the child's unaffected mother. This is the first study to show human iPS cells can be used to model the specific pathology seen in a genetically inherited disease. As such, it represents a promising resource to study disease mechanisms, screen novel drug compounds, and develop new therapies.
Spinal muscular atrophy (SMA) is an autosomal recessive genetic disorder caused by mutations in the survival motor neuron 1 gene (SMN1) significantly reducing SMN protein expression 1, 2 and resulting in the selective degeneration of lower α-motor neurons 3. Clinically, patients with SMA 1 typically show symptoms at 6 months of age and die by age 2 4. The SMN2 gene is an almost identical copy of SMN1 except that SMN2 has a single nucleotide difference that results in only 10% of full-length protein being produced and high levels of a truncated, unstable protein lacking exon 7 (SMNΔ7) 5. However, patients with multiple copies of SMN2 produce more full-length protein and have a less severe phenotype 6. While current model systems using worms, flies, or mice have provided invaluable data concerning the genetic cause of SMA, mechanisms of motor neuron death, and potential drug therapies 7, they have important limitations. For example, mice, flies, and worms lack the SMN2 gene thus requiring complicated knockout and over-expression strategies in animal models 8-12. As some therapies aim to target activation of endogenous SMN2 as a potential disease modifier, a human cell-based assay system is required. Although SMA patient fibroblasts are available, fibroblasts do not show the same vulnerability as motor neurons, and the processing and functioning of the SMN protein is likely to have unique features in a neural context.
Induced pluripotent stem (iPS) cells, which show striking similarities to embryonic stem cells, can now be derived from human adult somatic tissues 13-17, and recent studies have been successful in generating patient-specific iPS cells from a variety of diseases including amyotrophic lateral sclerosis, muscular dystrophy, and Huntington's disease 18, 19. However, none of these studies have shown any disease specific changes in cell survival or function. In the current study, we successfully established iPS cells from a type 1 SMA patient and his unaffected mother and showed that these cells retained the capacity to generate differentiated neural tissue and motor neurons while maintaining a lack of SMN1 expression and the disease phenotype of selective motor neuron death. These cells also responded to compounds known to increase SMN protein. Together, these results will allow disease modeling and drug screening for SMA in a far more relevant system 20.
Characterization of iPS cells
We generated iPS cells from primary fibroblasts from a type 1 SMA patient and his unaffected mother following infection with lentiviral constructs encoding OCT 4, SOX 2, NANOG, and LIN 28 16 (Suppl Fig 1). Two SMA clones (3.5 and 3.6) and one WT clone (4.2) propagated robustly when maintained on mouse embryonic fibroblasts. Quantitative RT-PCR, teratoma formation, DNA fingerprinting, and microarray analysis all indicated reprogramming of WT and SMA fibroblasts to a pluripotent state occurred, along with repression of the exogenously introduced genes (Fig 1 f-j, Suppl Tables 1-3, and Suppl Figs 2-4). Only the 3.6 clone (iPS-SMA) and the 4.2 clone (iPS-WT; Fig 1 a-c) were used for further evaluation in this study. Both the iPS-SMA and iPS-WT cells grew at similar rates and maintained a normal karyotype for at least 12 weeks (Fig 1 d,e).
Figure 1.
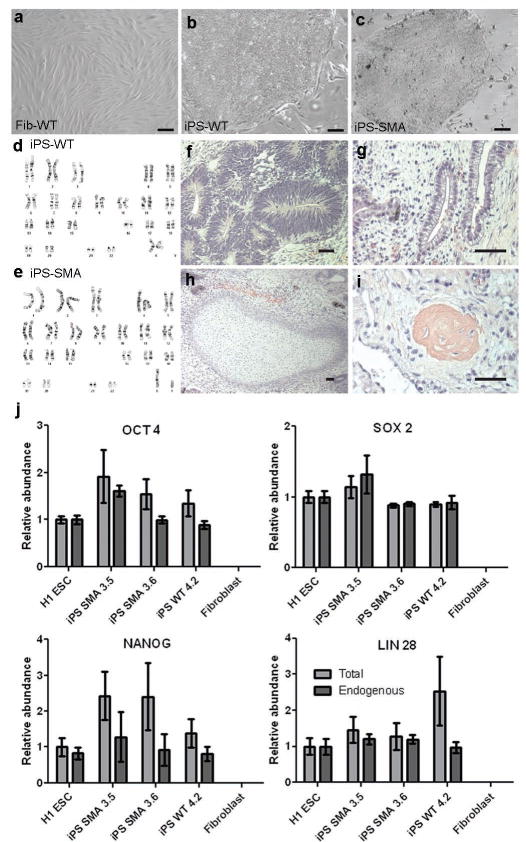
Newly generated iPS cells were fully reprogrammed. a-c, iPS-WT and iPS-SMA cells formed tightly packed colonies in contrast to the spindle morphology of fibroblasts. d,e, No karyotypic abnormalities were observed. Following transplantation, all iPS cells generated teratomas showing f, neural tissue (ectoderm), g, primitive gut (endoderm), h, cartilage (mesoderm), and i, bone (mesoderm). j, Quantitative RT-PCR showed induction of endogenous transcripts of OCT 4, SOX 2, NANOG, and LIN 28. “Endogenous” refers to primers recognizing the 3′ untranslated region, whereas “Total” identifies both the endogenous and exogenously expressed transgene. Data are expressed as mean ± s.e.m. Scale bar = 50μm
Cells from SMA patients have significantly reduced levels of SMN transcripts that contain all 9 exons (full-length (FL) transcripts) due to the loss of the SMN1 gene 1, 2. To test whether the derivation of iPS cells affected SMN production, iPS and fibroblast SMN RNA was analyzed. RT-PCR analysis revealed that iPS-WT had SMN levels comparable to the Fib-WT whereas iPS-SMA had lower levels similar to the Fib-SMA cells (Fig 2 a). As expected due to maintenance of SMN2 function in this disorder 1, 2, transcripts for some FL SMN and the alternatively spliced product lacking exon 7 (Δ7) were identified in all samples (Fig 2 a). Furthermore, specific digestion of FL bands produced the expected SMN2 cleavage products in all samples, confirming that SMN2 produces FL SMN in both the WT and SMA cells (Fig 2 b). Intact SMN1 was detected only in the WT cells, thus verifying the absence of SMN1 in SMA cells and recapitulating SMA and carrier transcript patterns 21 (Fig. 2 b). Quantitative RT-PCR further confirmed the significantly reduced level of FL SMN transcript in SMA cells (32-39% reduced compared to WT, Fig. 2 c). These data are consistent with FL SMN mRNA levels observed in SMA peripheral blood mononuclear cells 22. Taken together, these data demonstrate that the generation of iPS cells does not alter the critical gene expression profiles or alternative splicing events of SMN1 and SMN2 in unaffected or disease-specific contexts.
Figure 2.
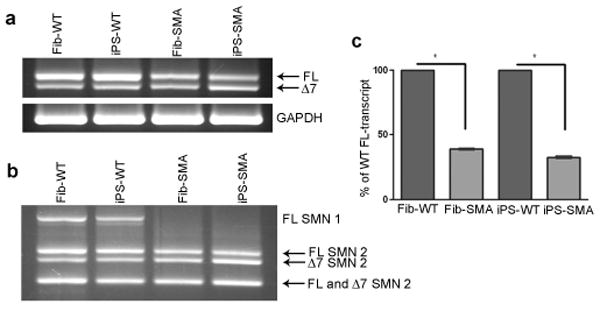
iPS-SMA show decreased SMN transcripts and lack of gems. a, iPS-WT and iPS-SMA cells had levels of full-length (FL) and truncated, exon 7 deleted (Δ7) SMN transcripts similar to their respective fibroblast lines (Fib-WT and Fib-SMA, respectively). b, Additionally, both Fib-SMA and iPS-SMA cells showed a specific lack of SMN1, although SMN2 FL and □7 transcripts were still present in all samples. c, Quantitative RT-PCR showed reduced FL-SMA transcript in both fibroblasts (61.1 % ± 0.6 reduced) and iPS cells (67.4 % ± 0.8 reduced) when compared to WT cultures (Student's t-test *p<0.001). Data are presented as mean ± s.e.m.
Neuronal differentiation of iPS cells
To establish whether the lack of SMN1 may affect neuronal differentiation or survival in this new model, we next generated neurons and astrocytes from both iPS-SMA and iPS-WT cells (Suppl Fig 1). Traditional embryoid body formation was found to be very inefficient for neural differentiation from iPS cultures and so an alternate differentiation protocol was developed. The iPS cultures were first removed from their feeder layers and grown in a defined media as iPS spheres for a minimum of two weeks. These could be continually passaged using a novel chopping method that avoids losing cell/cell contact know to be important for maintaining both neural and embryonic stem cell proliferation 23, 24. These cultures were then dissociated and plated onto laminin coated coverslips. The iPS-SMA and iPS-WT spheres generated nestin positive cells indicative of a neural stem cell phenotype 25 (Fig 3 a,b). Upon further differentiation, Tuj1 positive neurons and GFAP positive astrocytes were also found (Fig 3 c,d). The iPS spheres were simple to expand, remarkably stable over time and maintained the ability to produce neural progeny for over 20 passages.
Figure 3.
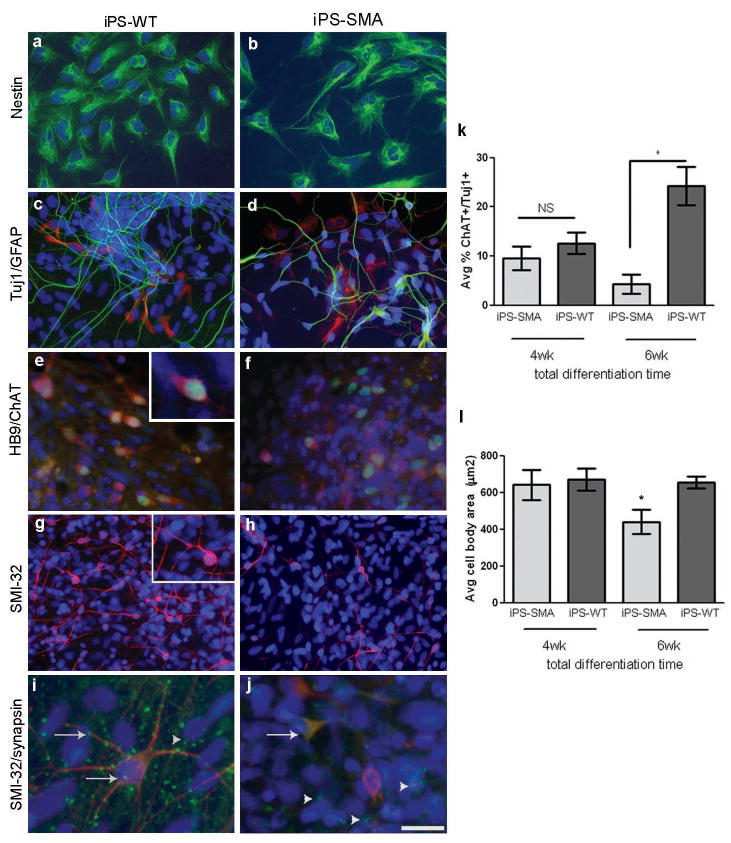
iPS-WT and iPS-SMA cells can generate cells in the neural lineage. a,b, iPS-WT and iPS-SMA cells generated nestin positive neural progenitor cells (green); c,d, Tuj1 positive neurons (green) and GFAP positive astrocytes (red); e,f, HB9 (green)/ChAT (red) double positive motor neurons, and g,h, SMI-32 (red) positive motor neurons at 4-6 weeks of differentiation i, At 8 weeks, punctate synapsin staining (green) on SMI-32 positive motor neurons (red) was identified on iPS-WT cells. j, However, only diffuse synapsin staining (green) was observed on SMI-32 positive motor neurons (red) in iPS-SMA cells. Arrowheads in i and j show punctate synapsin staining on SMI-32 negative cells. Nuclei are labeled with Hoechst nuclear dye (blue). k,l, iPS-SMA-derived motor neurons are significantly reduced in number and size at 6 weeks compared to iPS-WT cells (n=3 ANOVA, * p<0.05). All data are presented as mean ± s.e.m. Scale bar = 50μm (a-h), 25μm (i,j).
As SMA adversely affects motor neurons, we next asked whether the iPS cells could be lineage restricted toward a motor neuron fate (Suppl Fig 1). Basing our differentiation paradigm on a previously published report using human embryonic stem cells 26, iPS spheres were grown in a neural induction medium containing retinoic acid (RA) for 1 week followed by an additional week of RA and sonic hedgehog (SHH). Spheres were then seeded onto laminin coated coverslips for an additional 2-6 weeks (totaling 4-8 weeks of differentiation) and grown in the presence of RA, SHH, cyclic AMP, ascorbic acid (AA), glial cell line-derived neurotrophic factor (GDNF), and brain derived neurotrophic factor (BDNF). One week after plating, long fine processes resembling neuronal axons were observed and neural-like cells were seen migrating out from the sphere. Both iPS-SMA and iPS-WT spheres generated cells that expressed the motor neuron transcription factors HoxB4, Olig 2, Islet 1, and HB9 27, 28 during the differentiation process (Fig 3 e,f and Suppl Fig 5). The presumptive motor neurons were then immunostained with SMI-32 and choline acetyltransferase (ChAT), which are established markers for mature motor neurons 29. Both SMI-32 and ChAT staining identified positive neurons in all cultures after 4 weeks of differentiation (Fig 3 e-h). At this point there was no significant difference between the iPS-SMA and iPS-WT cultures in the number of motor neurons (12.6 % ± 2.2 and 9.5 % ± 2.4, respectively) or their size (641.0 μm2 ± 81.3 and 669.8 μm2 ± 59.1, respectively, Fig 3 k,l) suggesting that iPS-SMA cells are capable of generating motor neurons, which is similar to the human condition and mouse models in which functional motor neurons are generated at early developmental times 30.
To further develop the system, we cultured the cells for another 2 weeks and again analyzed motor neuron number and size. Importantly, at this time the iPS-SMA cultures had significantly fewer motor neurons (4.3 % ± 2.0) with a reduced size (383.1 μm2 ± 38.6) compared to iPS-WT cultures (24.2 % ± 4.0, 654.8 μm2 ± 32.6, Fig 3 k,l). However, there was no difference in the number of total Tuj1 positive neurons in either iPS-WT or iPS-SMA at 6 weeks of differentiation (15.78 % ± 2.9 and 15.55 % ± 2.8, respectively) suggesting a specific effect of the SMA phenotype on motor neurons. Taken together, these data show that iPS-SMA cells can produce similar numbers of neurons and motor neurons initially, but that the disease phenotype is selectively hindering motor neuron production and/or increasing motor neuron degeneration at later time points. Although synapses were not observed after 6 weeks of differentiation, by 8 weeks synapses were identified by punctate synapsin staining on iPS-WT SMI-32 positive motor neurons and non-motor neurons (Fig 3 i and Suppl Fig 6) suggesting pre-synaptic maturation of the neurons was occurring in this system. Interestingly, synapsin staining remained diffuse on the iPS-SMA SMI-32 positive motor neurons (Fig 3 j), although some punctate synapsin staining was observed on SMI-32 negative cells (Fig 3 j), again suggesting a specific motor neuron deficit in the SMA cultures.
Drug induced increase of SMN protein
Finally, we assessed whether SMN-inducing compounds could elevate SMN levels in the iPS-SMA cellular context as this would be an important proof of principle for further development of drug screens. SMN protein is found both in the cytoplasm and in nuclear aggregate structures called gems, and the number of gems present is inversely correlated to disease severity 2. We therefore assessed nuclear gem localization in iPS-SMA and iPS-WT derived neurons and astrocytes in the presence or absence of 1mM valproic acid or 320μM tobramycin, two compounds shown to increase SMN protein levels 31-33. Using an antibody against SMN, untreated Fib-WT and iPS-WT showed an abundance of nuclear gems which characterize the normal distribution of this protein (Fig. 4 a,c). Both untreated Fib-SMA and iPS-SMA cells showed the expected lack of nuclear gems (Fig 4 b,d) providing further support for reliable disease modeling using iPS-SMA cells. Following 2 days of drug treatment, there was no significant increase in gem localization in treated iPS-WT (Fig 4 e,g,i) compared to untreated iPS-WT (Fig 4 c,i). However, valproic acid and tobramycin significantly increased the number of gems in treated compared to untreated iPS-SMA cells (Fig 4 d,f,h,i). We further verified this increase by examining SMN protein by western blot analysis. Two days after drug treatment, total SMN protein was far higher in iPS-WT cells than seen in iPS-SMA cells (Fig 4 j,k) due to the lack of SMN1 expression in the latter (Fig 2 b). iPS-SMA cells treated with either valproic acid or tobramycin showed a significant 2 to 3 fold increase, respectively, in SMN protein levels compared to untreated iPS-SMA cells (Fig 4 j,k). We are currently assessing gem formation in differentiating motor neurons and have shown that they can indeed be detected (Suppl Fig 7). Together these data suggest that iPS-SMA cells respond to drug treatment in a similar fashion as Fib-SMA and could be useful for novel drug screening specifically on motor neurons in future studies.
Figure 4.
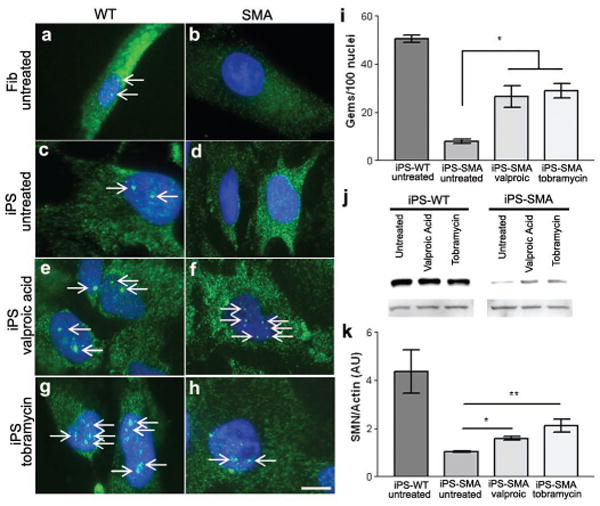
iPS-WT and iPS-SMA cells increase SMN protein in response to drug treatment. a,c, Untreated Fib-WT and iPS-WT cells show nuclear gem localization, whereas b,d, untreated Fib-SMA and iPS-SMA lack nuclear gems. f,h,i, Following valproic acid or tobramycin treatment, iPS-SMA cells show a significant increase the number of gems (ANOVA, p<0.05). Gems are indicated by arrows. j,k, Western blot analysis following valproic acid or tobramycin treatment showed a significant 2-3 fold increase in SMN protein in iPS-SMA cells compared to iPS-SMA cells (n=3 ANOVA, *p<0.05; **p<0.01). Data are presented as mean ± s.e.m. Scale bar = 50μm.
Discussion
Previous efforts to understand the mechanisms of SMA in human tissues have relied on fibroblasts from patients or immortalized non-motor neuron cell lines. However, one of the most fascinating aspects of the disease is that a ubiquitous loss of SMN1 protein from all cells in the body results in the specific degeneration of motor neurons. SMN1 has been shown to form complexes involved in the production of small nuclear RNA proteins that make up the splicesosome 34-36. More recently, SMN1 has also been shown to traffic to neuronal processes of motor neurons and may have other important roles in motor axons yet to be fully elucidated 37, 38. Using human motor neurons carrying the SMA phenotype generated from a virtually limitless source of iPS cells described here should help further clarify this new role for SMN1 in disease initiation and progression.
This is the first report to observe disease-specific effects on human motor neuron survival and drug induced increases in protective proteins - thus validating that the iPS model can recapitulate at least some aspects of this genetically inherited disorder. While the motor neurons generated in the current study show appropriate morphology, specific markers, and synapsin staining, experiments are currently underway to more fully assess their function including electrophysiology and co-cultures with muscle fibers. More clones from this and other patient sources also need to be studied. These are important next steps to ensure that the reprogramming has not subtly affected the ability of the motor neurons to function normally. However, this new model should provide a unique platform for studies aimed at both understanding SMA disease mechanisms that lead to motor neuron dysfunction and death, and the potential discovery of new compounds to treat this devastating disorder. It also points a future of using iPS technology to better understand and develop treatments for a number of other genetically inherited diseases.
Methods Summary
Detailed methods are included in the Methods section. SMA and WT fibroblast cell lines were from Coriell Institute for Medical Research (Camden, New Jersey). Lentiviral infection of the fibroblasts and iPS cell culture was performed as described previously 16. PCR was performed according to standard procedures using specific primers for OCT 4, SOX 2, NANOG, LIN 28, HoxB4, SMN, and GAPDH as published previously 16, 22 and shown in the Supplementary Material. Gene expression profiling, DNA fingerprinting, and microarray analysis were performed following standard protocols. Neural induction was modified from previously published methods 26, and immunological analyses were performed using standard protocols for nestin (Chemicon, 1:10,000), Tuj1 (Sigma, 1:5000), GFAP (Dako, 1:1000), Olig2 (Santa Cruz, 1:1000), HB9 (Hybrodoma bank, 1:100), Islet-1 (Hybrodoma bank, 1:100), ChAT (Chemicon, 1:250), SMI-32 (Covance, 1:500), SMN (4B7 33, 1:10), and synapsin (Calbiochem, 1:250). Fluorescent images were acquired using a Nikon Eclipse E600 microscope and Spot image software. Neuron counts and measurements were analyzed using Metamorph software, and statistical calculations were performed using Prizm software.
Methods
iPS cell culture and lentiviral infection
iPS cells were maintained on irradiated mouse embryonic fibroblasts (MEF) as previously described 16. Fibroblast cells (GM03813 and GM03814, Coriell Inst.) were cultured in Minimum Essential Medium (Eagle) (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (HyClone Laboratories). Lentiviral transduction of fibroblast cells was performed as previously described 16.
Neural differentiation
iPS spheres were generated by lifting intact iPS colonies from the feeder layers following collagenase treatment (1mg/ml, Gibco) and placing them directly into a human neural progenitor growth medium (Stemline, Sigma) supplemented with 2% B27 (Gibco), 100ng/ml basic fibroblast growth factor (bFGF, Chemicon), 100ng/ml epidermal growth factor (EGF, Chemicon), and 5μg/ml heparin (Sigma) in polyhema coated flasks to prevent attachment and were passaged weekly using a chopping technique 24. To induce neuron and astrocyte differentiation, spheres were dissociated with accutase (Chemicon) and plated onto poly-ornithine/laminin (Sigma) coated coverslips in Stemline/2 % B27 without bFGF, EGF, and heparin for 1 week. To induce motor neuron differentiation, spheres were placed in neural induction medium (1:1 DMEM/F12 and 1% N2 supplement (Gibco) in the presence of retinoic acid (RA, 0.1 μM) for 1 week followed by the addition of sonic hedgehog (SHH, 100ng/ml, R&D) for another week. Spheres were then plated onto polyornithine/laminin coated coverslips in RA/SHH medium supplemented with cAMP (1μM), ascorbic acid (200ng/ml), BDNF, and GDNF (both 10 ng/ml, PeproTech Inc.) for an additional 2-6 weeks.
RNA isolation and PCR analysis
Total RNA was isolated using the RNeasy Mini Kit (Qiagen) with on-column DNase I digestion or Tri-Reagent (Sigma). cDNA was generated from 1-4μg total RNA using SuperScript III (Invitrogen). RT-PCR and/or qRT-PCR were performed using specific primer sequences (Suppl Table 3). FL and Δ7 SMN products were gel purified and digested with Dde1 specifically cleaving SMN2 giving expected band sizes of: 713nt (undigested FL), 436nt and 277nt (SMN2 FL), and 382nt and 277nt (SMN2 Δ7).
Karyotyping and DNA fingerprinting
Standard G-banding chromosome analysis was performed in the Cytogenetics Lab at WiCell Research Institute (Madison, WI). To confirm the fibroblast origins of the iPS-SMA and iPS-WT cells, short tandem repeat (STR) analysis was performed by Cell Line Genetics (Madison, WI).
Teratoma formation
Two 10-cm dishes of iPS-WT clone 4.2, iPS-SMA clone 3.5, and clone 3.6 (∼ 50% confluent) grown on irradiated MEF were injected into the hind limb muscle of two mice. All iPS clones gave rise to teratomas. Control mice injected with ∼8.5 × 106 Fib-WT and 11 × 106 Fib-SMA failed to form teratomas. Hematoxylin and eosin staining of teratoma sections was performed after 7-10 weeks.
Microarray analysis
Human genome U133 Plus 2.0 GeneChip arrays carrying 54,675 probe sets (Affymetrix) were used for microarray hybridizations to examine the global gene expression. Approximately 3 μg of RNA from each sample was labeled using the MessageAmp™ Biotin II-Enhanced IVT kit (Ambion) following manufacturer's instructions. All arrays were hybridized at 45°C for 16 hours and scanned using an AFX GC3000 G7 scanner.
The gene expression raw data were extracted using the AFX Expression Console software. Quality control was done based on Affymetrix quality control metrics. The qualified data sets were then analyzed in the R statistical environment, freely available under the GNU General Public Licence (http://www.r-project.org) using bioconductor libraries (http://www.bioconductor.org). All chips were normalized by the quantile method 39 and background corrected using robust multi-array analysis (RMA) 40 followed by summarization using median polish 40 to get the probe set level measurement. 54,675 probe sets were collapsed to 51,337 transcripts by taking the average log intensity values for probe sets representing common accession numbers. Hierarchical cluster analyses were carried out with 1-PCC (Pearson Correlation Coefficient) as the distance measurement. The maximum distance between cluster members was used as the basis to merge lower-level clusters (complete linkage) into higher-level clusters. Multiscale bootstrap resampling (10,000 bootstraps) was applied to the hierarchical clustering, p-values of hypotheses were calculated, and bootstrap probabilities were determined for each cluster 41.
HeatMap Generation and Data Visualization
For each gene, average expression level was calculated across five normal human ES cell lines and the two SMA/WT fibroblast cell lines. The fold changes were calculated for all the genes in the SMA/WT fibroblast cell lines over the corresponding average in human ES cells. The top 25 genes most specifically expressed in SMA/WT fibroblasts and 30 genes that are known to be enriched in human ES cells were selected for the heatmap generation. The log intensities of these 55 genes were standardized, so that their expression values across all samples have mean 0 and standard deviation 1. The standardized values were reordered and displayed in a heat map, with the spectrum ranging from green (low level) to red (high level).
Immunocytochemistry
Cells were fixed in 4% paraformaldehyde or 1:1 acetone/methanol for 20 minutes at room temperature and rinsed with PBS. Nonspecific labeling was blocked and the cells permeabilized with 5% normal goat serum and/or 5% normal donkey serum and 0.2% Triton X-100 in PBS for 30 minutes at room temperature. Cells were rinsed with PBS and then incubated with primary antibodies for one hour at room temperature or overnight at 4°C. Cells were then labeled with the appropriate fluorescently-tagged secondary antibodies. Hoechst nuclear dye was used to label nuclei. Gems were counted in 100 nuclei, and positively stained neurons were counted and measured on 5 areas on each of 3 coverslips using Metamorph software. All data were analyzed using Prizm statistical software.
Protein isolation and western blot analysis
Cells were isolated, suspended in 1% Triton X-100 lysis buffer supplemented with 1% protease inhibitor cocktail (Sigma), triturated, and centrifuged at 13,000 rpm for 10 min at 4°C. Ten to twenty μg of protein was separated on 12% SDS-polyacrylamide gel and transferred to nitrocellulose membrane and probed with primary antibody toward SMN (mouse monoclonal), followed by a horseradish peroxidase conjugated secondary antibody (Promega), and then visualized using ECL chemiluminescence (Amersham). As a control, the membrane was stripped and re-probed for β-actin. For semi-quantitative analysis, SMN signal intensity was analyzed and corrected with respect to β-actin.
Supplementary Material
Acknowledgments
The authors thank Jason Meyer for helpful discussions, and Brandon Shelley, Brittany Heins, and Erin McMillan for technical assistance. We also thank WiCell Research Institute, Madison, WI, for karyotype analysis; Cell Line Genetics, Madison, WI, for DNA fingerprinting; Ron Stewart, Shulan Tian, and Victor Ruotti at the Morgridge Institute for Research, Madison, WI, for microarray analysis; and Promega Corp., Madison, WI, for qRT-PCR analysis. The MNR2/HB9 (81.5C10) and the Islet-1 (40.2D6) monoclonal antibodies (both developed by Dr. Thomas Jessell) were obtained from the Developmental Studies Hybridoma Bank. Funding support was provided by the ALS Association and the NIH/NINDS (PO1NS057778 to CNS).
Footnotes
Contributions: ADE participated in all aspects and prepared the manuscript; JY generated and aided in characterization of iPS-SMA and iPS-WT clones; FR, VBM, and CLL performed SMN analysis and manuscript preparation; JAT participated in the generation of the iPS clones; CNS conceived the project and participated in planning, data analysis, and manuscript preparation. The authors declare no competing financial interest.
Supplementary Information accompanies the paper on www.nature.com/nature. A schematic outlining the main results is included as Supplementary Figure 1.
References
- 1.Lefebvre S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 2.Coovert DD, et al. The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet. 1997;6:1205–1214. doi: 10.1093/hmg/6.8.1205. [DOI] [PubMed] [Google Scholar]
- 3.Crawford TO, Pardo CA. The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis. 1996;3:97–110. doi: 10.1006/nbdi.1996.0010. [DOI] [PubMed] [Google Scholar]
- 4.Munsat TL, Davies KE. Neuromuscul Disord; International SMA consortium meeting; 26-28 June 1992; Bonn, Germany. 1992. pp. 423–428. [DOI] [PubMed] [Google Scholar]
- 5.Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lefebvre S, et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet. 1997;16:265–269. doi: 10.1038/ng0797-265. [DOI] [PubMed] [Google Scholar]
- 7.Schmid A, DiDonato CJ. Animal models of spinal muscular atrophy. J Child Neurol. 2007;22:1004–1012. doi: 10.1177/0883073807305667. [DOI] [PubMed] [Google Scholar]
- 8.Schrank B, et al. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci U S A. 1997;94:9920–9925. doi: 10.1073/pnas.94.18.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DiDonato CJ, et al. Cloning, characterization, and copy number of the murine survival motor neuron gene: homolog of the spinal muscular atrophy-determining gene. Genome Res. 1997;7:339–352. doi: 10.1101/gr.7.4.339. [DOI] [PubMed] [Google Scholar]
- 10.Hsieh-Li HM, et al. A mouse model for spinal muscular atrophy. Nat Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- 11.Monani UR, et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(-/-) mice and results in a mouse with spinal muscular atrophy. Hum Molec Genet. 2000;9:333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- 12.Le TT, et al. SMN Delta 7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Molec Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- 13.Park IH, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- 14.Jaenisch R, Young R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell. 2008;132:567–582. doi: 10.1016/j.cell.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 16.Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 17.Lowry WE, et al. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc Natl Acad Sci U S A. 2008;105:2883–2888. doi: 10.1073/pnas.0711983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dimos JT, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321:1218–1221. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- 19.Park IH, et al. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jakel RJ, Schneider BL, Svendsen CN. Using human neural stem cells to model neurological disease. Nat Rev Genet. 2004;5:136–44. doi: 10.1038/nrg1268. [DOI] [PubMed] [Google Scholar]
- 21.Gavrilov DK, Shi XY, Das K, Gilliam TC, Wang CH. Differential SMN2 expression associated with SMA severity. Nat Genet. 1998;20:230–231. doi: 10.1038/3030. [DOI] [PubMed] [Google Scholar]
- 22.Sumner CJ, et al. SMN mRNA and protein levels in peripheral blood: biomarkers for SMA clinical trials. Neurology. 2006;66:1067–1073. doi: 10.1212/01.wnl.0000201929.56928.13. [DOI] [PubMed] [Google Scholar]
- 23.Fox V, et al. Cell-cell signaling through NOTCH regulates human embryonic stem cell proliferation. Stem Cells. 2008;26:715–723. doi: 10.1634/stemcells.2007-0368. [DOI] [PubMed] [Google Scholar]
- 24.Svendsen CN, et al. A new method for the rapid and long term growth of human neural precursor cells. J Neurosci Methods. 1998;85:141–52. doi: 10.1016/s0165-0270(98)00126-5. [DOI] [PubMed] [Google Scholar]
- 25.Lendahl U, Zimmerman LB, McKay RDG. Cns Stem-Cells Express A New Class of Intermediate Filament Protein. Cell. 1990;60:585–595. doi: 10.1016/0092-8674(90)90662-x. [DOI] [PubMed] [Google Scholar]
- 26.Li XJ, et al. Specification of motoneurons from human embryonic stem cells. Nat Biotechnol. 2005;23:215–221. doi: 10.1038/nbt1063. [DOI] [PubMed] [Google Scholar]
- 27.Jessell TM. Neuronal specification in the spinal cord: Inductive signals and transcriptional codes. Nat Rev Genet. 2000;1:20–29. doi: 10.1038/35049541. [DOI] [PubMed] [Google Scholar]
- 28.Wichterle H, Lieberam I, Porter JA, Jessell TM. Directed differentiation of embryonic stem cells into motor neurons. Cell. 2002;110:385–397. doi: 10.1016/s0092-8674(02)00835-8. [DOI] [PubMed] [Google Scholar]
- 29.Carriedo SG, Yin HZ, Weiss JH. Motor neurons are selectively vulnerable to AMPA/kainate receptor-mediated injury in vitro. J Neurosci. 1996;16:4069–4079. doi: 10.1523/JNEUROSCI.16-13-04069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Monani UR. Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron. 2005;48:885–896. doi: 10.1016/j.neuron.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 31.Brichta L, et al. Valproic acid increases the SMN2 protein level: a well-known drug as a potential therapy for spinal muscular atrophy. Hum Molec Genet. 2003;12:2481–2489. doi: 10.1093/hmg/ddg256. [DOI] [PubMed] [Google Scholar]
- 32.Sumner CJ, et al. Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann Neurol. 2003;54:647–654. doi: 10.1002/ana.10743. [DOI] [PubMed] [Google Scholar]
- 33.Wolstencroft EC, Mattis V, Bajer AA, Young PJ, Lorson CL. A non-sequence-specific requirement for SMN protein activity: the role of aminoglycosides in inducing elevated SMN protein levels. Hum Molec Genet. 2005;14:1199–1210. doi: 10.1093/hmg/ddi131. [DOI] [PubMed] [Google Scholar]
- 34.Pellizzoni L, Yong J, Dreyfuss G. Essential role for the SMN complex in the specificity of snRNP assembly. Science. 2002;298:1775–1779. doi: 10.1126/science.1074962. [DOI] [PubMed] [Google Scholar]
- 35.Fischer U, Liu Q, Dreyfuss G. The SMN-SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell. 1997;90:1023–1029. doi: 10.1016/s0092-8674(00)80368-2. [DOI] [PubMed] [Google Scholar]
- 36.Liu Q, Fischer U, Wang F, Dreyfuss G. The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell. 1997;90:1013–1021. doi: 10.1016/s0092-8674(00)80367-0. [DOI] [PubMed] [Google Scholar]
- 37.Carrel TL, et al. Survival motor neuron function in motor axons is independent of functions required for small nuclear ribonucleoprotein biogenesis. J Neurosci. 2006;26:11014–11022. doi: 10.1523/JNEUROSCI.1637-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang HL, et al. Multiprotein complexes of the survival of motor neuron protein SMN with gemins traffic to neuronal processes and growth cones of motor neurons. J Neurosci. 2006;26:8622–8632. doi: 10.1523/JNEUROSCI.3967-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 40.Irizarry RA, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 41.Suzuki R, Shimodaira H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics. 2006;22:1540–1542. doi: 10.1093/bioinformatics/btl117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.