

Abstract
We recently reported the identification and characterization of a novel BH3-only pro-death protein, apolipoprotein L1 (ApoL1), that, when overexpressed, induces autophagic cell death (ACD) in a variety of cells, including those originated from normal and cancerous tissues. ApoL1 failed to induce ACD in autophagy-deficient Atg5−/− and Atg7−/− MEF cells, suggesting that ApoL1-induced cell death is indeed autophagy-dependent. In addition, a BH3 domain deletion allele of ApoL1 was unable to induce ACD, demonstrating that ApoL1 is a bona fide BH3-only pro-death protein. To further investigate regulation of ApoL1 expression, we showed that ApoL1 is inducible by interferon-γ and tumor necrosis factor-α in human umbilical vein endothelial cells, suggesting that ApoL1 may play a role in cytokine-induced inflammatory response. Moreover, we observed that ApoL1 is a lipid-binding protein with high affinity for phosphatidic acid and cardiolipin and less affinity for various phosphoinositides. Functional genomics analysis identified 5 nonsynonymous single nucleotide polymorphisms (NSNPs) in the coding exons of the human ApoL1 structural gene– all the 5 NSNPs may cause deleterious alteration of ApoL1 activity. Finally, we discuss the link between ApoL1 and various human diseases.
Keywords: trypanosomal lytic factor, apolipoprotein L1 (ApoL1), autophagy, autophagic cell death, BH3-only protein, cardiolipin, lipid binding, PIPs, phosphatidic acid, schizophrenia, SNPs
Macroautophagy (hereafter referred to as autophagy) is a lysosome-dependent mechanism of intracellular degradation that is used for the turnover and recycling of cytoplasmic constituents. It involves the sequestration of a fraction of cytosol and organelles within a de novo synthesized, double-membraned organelle called the autophagosome, which then fuses with the endosome and/or lysosome to create the amphisome and autolysosome, respectively. Eventually, the inner membrane of the autophagosome, together with the enclosed cargo, is degraded by the lysosomal hydrolases.1 Autophagy in mammals serves multiple purposes: it functions as a survival mechanism during periods of starvation and stress, it is directly involved in eliminating aberrant protein aggregates, removing damaged/aged organelles, and defending against pathogens, and it serves an essential role in early neonatal development.2 Evidently, autophagy also functions as type II programmed cell death (PCD), or autophagic cell death (ACD), in multi-cellular organisms.3,4 It has been reported that autophagy is involved in physiological cell death during development of Drosophila and Dictyostelium discoideum, however, ACD has not yet been observed/demonstrated in mammals in vivo.5,6
In the past two decades, yeast has been used as an experimental model in dissecting the molecular bases of autophagy. Currently 31 ATG (autophagy-related) genes have been identified in yeast.3,6 Recent identification and characterization of human orthologs/homologs of yeast ATG genes and the link between autophagy and human diseases further emphasize the importance of autophagy in health and disease. Deficiency of autophagy correlates with diseases such as cancer, cardiomyopathies, inflammatory bowel diseases and neurodegenerative diseases.5,7 In addition, autophagy involves unique membrane dynamics and biosynthesis, requiring de novo synthesis of double-membrane autophagosomes, in which both protein and lipid components are required. Regarding protein components, aside from Atg proteins, Vps34, a class III phosphatidylinositol-3-phosphate kinase (PI3K), the proteins that are part of its complex, and the Bcl-2 family members play critical roles in regulating autophagosome formation. Interestingly, Beclin 1, the mammalian ortholog of yeast Atg6 that is required for autophagy, is a BH3-only protein. In addition, other Bcl-2 family members, such as Bcl-2, Bax, Bid and BNIP3, regulate autophagy.6,8 By contrast, the lipid composition of autophagosomal membranes remains unknown because it is technically difficult to isolate pure autophagosomes. It is evident however that some lipid species, for example, phosphatitylinositol-3-phosphate or PI3P, are required for autophagosomal membranes.1,9
In an effort to identify and characterize novel BH3-only proteins in ACD, and discover molecular regulators that dictate autophagy for death or survival, we examined a novel BH3-only protein, apolipoprotein L1 (ApoL1). We show that wild-type ApoL1 induces ACD in a variety of cells, whereas a BH3 deletion allele of ApoL1 fails to induce cell death, indicating that ApoL1 is a bona fide BH3-only pro-death protein. We further demonstrate that ApoL1 is a potent and specific inducer of ACD that stimulates time-dependent accumulation and translocation of lipidated LC3-II, formation of autophagosomes and subsequently cell death. We also show that ApoL1 is inducible by p53 during p53-induced cell death. p53, a well-known tumor suppressor, regulates both apoptosis and ACD.4 Interestingly, a recent study shows that p53 plays a dual role in the control of autophagy: on the one hand, nuclear p53 can induce autophagy through transcriptional effects, whereas on the other hand, cytoplasmic p53 may act as a master repressor of autophagy.10 Our results suggest that ApoL1 is a novel BH3-only protein and is a p53 downstream effector functioning as a molecular determinant that determines whether autophagy plays a role in cell death.4 Importantly, ApoL1 has been found only in humans and African green monkeys (NCBI Entrez Gene, ApoL1), suggesting that ApoL1-induced ACD is primate-specific.
Employing a protein-lipid overlay assay, we demonstrate that ApoL1 is an intracellular lipid-binding protein that binds strongly with phosphatidic acid (PA) and cardiolipin (CL), and with less affinity with different species of phosphoinositides (PIPs) in the following order PI(3,5)P2>PI4P>PI5P>PI3P>PI(4,5)P2>PI(3,4,5)P3>PI(3,4)P2.4 PIPs are phosphorylated derivatives of the membrane phospholipids phosphatidylinositols and play critical roles in regulating receptor signaling, cytoskeleton function and membrane trafficking. They are highly concentrated and localized in distinct pools in the plasma membrane, endosomes, nucleus and autophagosomal membranes, where they function as ligands, adaptors or docking sites for PIP-binding proteins. In addition, the homeostatic equilibrium of PIPs determines cellular proliferation, differentiation and PCD.11,12 Vps34 is required for autophagy, implying an essential role of its product PI3P in this process.13 Recently, Obara and colleagues9 showed that PI3P is highly enriched and delivered to autophagosomal membranes but not as a cargo enclosed in autophagosomes, implying direct involvement of PI3P in autophagosome formation. They also report a possible enrichment of PI3P on the inner autophagosomal membranes compared to the outer membrane.9 Regarding binding with PIPs, we show that ApoL1 possesses higher affinity for PI(3,5)P2, PI4P, PI5P, and PI3P, implying that intracellular accumulation of ApoL1 may alter the homeostasis of PIPs leading to ACD. In addition, we conducted a protein domain homology search and found no homology between ApoL1 and any known PIP-binding domains, including FYVE (Fab1, YOTB, Vac1 and EEA1), PX (Phox homology), PH (Pleckstrin homology), ENTH (Epsin N-terminal homology), FERM (Band4.1, Ezrin, Radixin, and Moesin), GRAM (Glucosyltransferase, Rab-like GTPase Activator, and Myotubularins), ANTH (AP180/CALM), and WD-repeats of WIPIs, human orthologs of yeast Atg18 and/or Atg20.7,14,15 In fact, no specific binding domain for PI(3,5)P2 has been identified thus far. Taken together, ApoL1 may represent a novel class of PIP-binding protein. Importantly, in autophagy, at least 2 Atg proteins are involved in PIP synthesis (Atg6 and Atg14) and 5 Atg proteins bind PIP (Atg18, Atg20, Atg21, Atg24 and Atg27).1,7 Evidently, yeast mutant strains deficient in PIP binding will be useful for functional complementation and biochemical analysis of human ApoL1.
Interestingly, ApoL1 also binds PA and CL. PA is a mitogenic messenger that serves as a critical component of mTOR signaling. PA directly interacts with the domain in mTOR that is targeted by rapamycin, and this interaction positively correlates with mTOR's ability to activate downstream effectors. mTOR is a negative regulator of autophagy;16,17 thus, binding of PA by ApoL1 might inhibit mTOR activation and subsequently induce autophagy. In addition, CL, a negatively charged phospholipid abundant in the mitochondrial inner membrane, is important in maintaining protein (for example cytochrome c) and membrane integrity of mitochondria. During apoptosis, CL functions as the docking site for t-Bid and Bad and induces mitochondrial membrane permeability and cytochrome c release.18,19 In addition, CL relocates to the plasma membrane and other subcellular organelles during cell death.20 Sequestering of PA and CL with ApoL1 may alter the homeostasis between survival and death leading to ACD. To our knowledge, this is the first BH3-only protein with lipid binding activity that, when overproduced intracellularly, induces ACD.
Roles of ApoL1 in human diseases
African Sleeping sickness
The parasites Trypanosoma brucei rhodesiense and Trypanosoma brucei gambiense cause human sleeping sickness in Africa. It has been known for over a century that human serum contains trypanosomal lytic factor (TLF) that can lyse most of the invading trypanosome species. TLF, residing in a minor subclass of human high-density lipoprotein particles, contains apolipoprotein A-I (ApoA-I) and two primate-specific proteins, ApoL1 and hepatoglobin-related protein (Hrp), both of which are toxic to trypanosomes.21,22 The molecular basis of this disease is that pathological trypanosomes produce the serum resistance-associated protein (SRA), an inhibitory protein that binds specifically to ApoL1 to prevent trypanosomal lysis. Unsequestered and engulfed ApoL1 promotes the formation of anion (Cl−) channels on lysosomal membranes of trypanosomes and induces osmotic swelling of lysosomes due to massive influx of chloride ions from the cytoplasmic compartment, and subsequently kills the parasite.21 In addition to ApoL1 and Hrp, Wildener and colleagues23 recently provided in vitro evidence that hemoglobin is a cofactor of human TLF that increases the affinity of TLF for its receptor, and therefore the internalization efficiency of TLF to the parasite. More importantly, ApoL1 alone is sufficient to confer trypanolytic activity in transgenic mice but both ApoL1 and Hrp are required to increase the specific activity of TLF.24 It is worth noting that a recent case report documents that an Indian patient whose serum lacked trypanolytic activity due to the absence of ApoL1 was infected by a serum-sensitive T. evansi, a strain that would normally not infect humans. Mutation analysis shows that the patient possesses two frameshift mutations in both ApoL1 alleles, further emphasizing the critical role of ApoL1 in innate immunity against trypanosomal infection.22 Nevertheless, it is evident that ApoL1-induced cell death in mammalian cells is through autophagy, not through Cl− influx and osmotic swelling of the lysosome that happens in trypanosomes and in E. coli.22,25 Our recent result showed that ApoL1 is localized in the cytosol, not in lysosomes, in mammalian cells (Wan et al., unpublished observation). In addition, ApoL1-induced trypanosomal lysis can be delayed by the anion blocker DIDS (4,4-diisothiocyanatostilbene-2,2-disulfonic acid). It is thought that ApoL1-mediated influx of Cl− from the cytoplasm to the lysosome causes a change in the cytosolic Cl− concentration that in turn causes a compensatory movement of extracellular Cl− across the plasma membrane through DIDS-sensitive channels in trypanosomes.21 However, our recent observation indicated that DIDS fails to delay or block ApoL1-induced ACD in mammalian cells (Wan, unpublished observation). As indicated earlier, ApoL1 has been found only in humans and African green monkeys, suggesting that ApoL1-induced ACD is primate specific.
Schizophrenia
The human gene encoding ApoL1 is localized to chromosome 22q13.1, a region with loci that might be linked to susceptibility of schizophrenia. Importantly, expression of ApoL1 is significantly upregulated (>1.41 fold) in the brains of schizophrenics.26 Takahashi and colleagues27 recently performed a family-based association study using 377 families with schizophrenia and 130 SNPs (single nucleotide polymorphisms) associated with the ApoL gene family. They show that seven SNPs have p-values <0.05 in the ApoL1, ApoL2 and ApoL4 regions in 226 families (approximately 60%), suggesting ApoL genes should be more extensively studied in schizophrenia. It will be interesting to see whether overexpression of ApoL1 induces ACD in CNS cells, and if so, what role ApoL1 plays in the etiology of schizophrenia.
Inflammation in atherosclerosis
Atherosclerosis is a chronic inflammatory disease of the vasculature with lesions developing in the arterial wall, and it is governed by a complex network of inter- and intra-cellular signaling pathways. The pleiotropic, pro-inflammatory cytokine interferon-γ (INF-γ) and tumor necrosis factor-α (TNF-α) are key mediators that are highly expressed in atherosclerotic lesions.28 The endothelium, the monolayer covering the inner surface of blood vessels, plays a pivotal role in the regulation of vascular tone and structure, as well as vascular inflammation. Dysfunction of endothelial cells is an early sign of atherosclerosis. Interestingly, previous studies show ApoL1 is strongly induced by IFN-γ and TNF-α in endothelial cells at the mRNA level.29 Using human umbilical vein endothelial cells (HUVEC), a model for endothelial cell physiology and pathophysiology, we recently showed that expression of ApoL1 at the protein level was significantly increased 6 h and 12 h after INF-γ and TNF-α treatment, respectively (Figure 1), suggesting that ApoL1 plays a role in inflammatory and cell death response in endothelial cells. It would be of interest to investigate whether expression of ApoL1 is upregulated in human atherosclerotic tissues, and if so, how and why.
Figure 1.
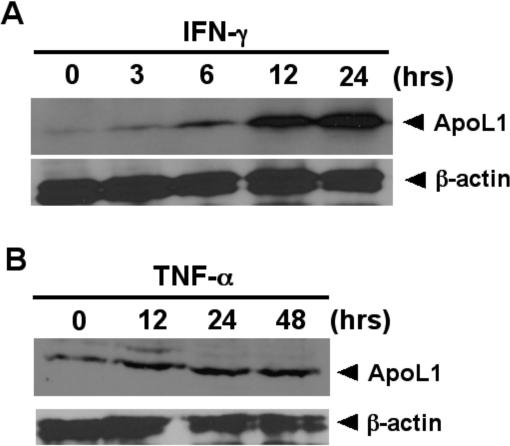
Expression of ApoL1 is inducible by interferon-γ (INF-γ) and tumor necrosis factor-α (TNF-α) in HUVEC cells. Immunoblot analysis of the time-dependent expression of ApoL1 under the influence of INF-γ and TNF-α was assayed. Hours (hrs) after treatment are as indicated. Induction of ApoL1 was detected 6 hrs and 12 hrs after INF-γ and TNF-α treatment, respectively. β-actin was used as a loading control.
Regarding other possible roles of ApoL1 in human disease, in a microarray study Kang and colleagues30 show that ApoL1 is one of the senescence-associated genes that are induced greater than 14-fold in normal human oral keratinocytes undergoing senescence. However, the function of ApoL1 in aging/senescence has not been further explored. To explore possible genetic alterations in ApoL1 that may correlate with human disease, we conducted a SNP analysis by searching/mining the NCBI SNP database. We found NSNPs, G96R, E150K, M228I, R255K, and D337N, in the coding sequence of human ApoL1 that alter amino acid sequences (Figure 2). All 5 of these changes are considered to be drastic. It is not unreasonable to postulate that some of these NSNPs may be associated with human disease state. Future investigation of ApoL1 will focus on its disease association and lipid binding. For example, whether the BH3 domain of ApoL1 is necessary for lipid binding, and whether binding of different lipid species by ApoL1 is essential for the induction of ACD. Finally, ApoL1-induced ACD provides an excellent model to dissect the molecular basis of autophagy and other molecular determinants, both protein and lipid species, that dictate the control of autophagy for cell survival or death.
Figure 2.

Human ApoL1 and its nonsynonymous SNPs. The polypeptide sequence of ApoL1 is shown. Five newly identified NSNPs, G96R, E150K, M228I, R255K, and D337N, are indicated. The BH3 (amino acids 158−166) and leucine zipper (amino acids 365−392) domains are in bold and underlined.
Acknowledgements
This work was supported in part by NCI RO1 (5RO1CA106644-01A1) and to C.-A. A. Hu, and by DOD PCRP predoctoral fellowship (W81XWH-08-1-0183) to R. Kaini.
Abbreviations
- ANTH
AP180/CALM
- ACD
autophagic cell death
- AV
autophagic vacuoles
- BH
Bcl-2 homology domain
- CL
cardiolipin
- ENTH
epsin N-terminal homology
- FERM
band4.1, ezrin, radixin, and moesin
- FYVE
Fab1p, YOTB, Vac1p and EEA1
- GRAM
Glucosyltransferase, Rab-like GTPase activator, and myotubularins
- LC3
microtubule associated proteins light chain 3
- 3-MA
3-methyladenine
- PA
phosphatidic acid
- PH
pleckstrin homology
- PI
phosphatidylinositol
- PI3K
phosphatidylinositol-3-phosphate kinase
- PIPs
phosphoinositides
- PX
Phox homology
- PCD
programmed cell death
- SNP
single nucleotide polymorphism
Footnotes
Addendum to: Wan G, Zhaorigetu S, Liu Z, Kaini R, Jiang Z, Hu CA. Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. J Biol Chem. 2008; 283:21540−49.
References
- 1.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–09. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- 2.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–77. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 3.Scarlatti F, Granata R, Meijer AJ, Codogno P. Does autophagy have a license to kill mammalian cells? Cell Death Differ. 2008 doi: 10.1038/cdd.2008.101. in press. [DOI] [PubMed] [Google Scholar]
- 4.Wan G, Zhaorigetu S, Liu Z, Kaini R, Jiang Z, Hu CA. Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. J Biol Chem. 2008;283:21540–49. doi: 10.1074/jbc.M800214200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang J, Klionsky DJ. Autophagy and human disease. Cell Cycle. 2007;6:1837–49. doi: 10.4161/cc.6.15.4511. [DOI] [PubMed] [Google Scholar]
- 6.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kundu M, Thompson CB. Autophagy: basic principles and relevance to disease. Annu Rev Pathol. 2008;3:427–55. doi: 10.1146/annurev.pathmechdis.2.010506.091842. [DOI] [PubMed] [Google Scholar]
- 8.Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy. 2008;4:600–6. doi: 10.4161/auto.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Obara K, Noda T, Niimi K, Ohsumi Y. Transport of phosphatidylinositol 3-phosphate into the vacuole via autophagic membranes in Saccharomyces cerevisiae. Genes Cells. 2008;13:537–47. doi: 10.1111/j.1365-2443.2008.01188.x. [DOI] [PubMed] [Google Scholar]
- 10.Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D'Amelio M, Criollo A, Morselli E, Zhu C, Harper F, Nannmark U, Samara C, Pinton P, Vicencio JM, Carnuccio R, Moll UM, Madeo F, Paterlini-Brechot P, Rizzuto R, Szabadkai G, Pierron G, Blomgren K, Tavernarakis N, Codogno P, Cecconi F, Kroemer G. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676–87. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonsalvez F, Gottlieb E. Cardiolipin: setting the beat of apoptosis. Apoptosis. 2007;12:877–85. doi: 10.1007/s10495-007-0718-8. [DOI] [PubMed] [Google Scholar]
- 12.Sasaki T, Sasaki J, Sakai T, Takasuga S, Suzuki A. The physiology of phosphoinositides. Biol Pharm Bull. 2007;30:1599–1604. doi: 10.1248/bpb.30.1599. [DOI] [PubMed] [Google Scholar]
- 13.Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3'-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275:992–98. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- 14.Lemmon MA. Phosphoinositide recognition domains. Traffic. 2003;4:201–13. doi: 10.1034/j.1600-0854.2004.00071.x. [DOI] [PubMed] [Google Scholar]
- 15.Proikas-Cezanne T, Waddell S, Gaugel A, Frickey T, Lupas A, Nordheim A. WIPI-1α (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene. 2004;23:9314–25. doi: 10.1038/sj.onc.1208331. [DOI] [PubMed] [Google Scholar]
- 16.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. 2001;294:1942–1945. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 17.Hornberger TA, Chu WK, Mak YW, Hsiung JW, Huang SA, Chien S. The role of phospholipase D and phosphatidic acid in the mechanical activation of mTOR signaling in skeletal muscle. Proc Natl Acad Sci U S A. 2006;103:4741–46. doi: 10.1073/pnas.0600678103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lutter M, Fang M, Luo X, Nishijima M, Xie X, Wang X. Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat Cell Biol. 2000;2:754–61. doi: 10.1038/35036395. [DOI] [PubMed] [Google Scholar]
- 19.Hekman M, Albert S, Galmiche A, Rennefahrt UE, Fueller J, Fischer A, Puehringer D, Wiese S, Rapp UR. Reversible membrane interaction of BAD requires two C-terminal lipid binding domains in conjunction with 14−3−3 protein binding. J Biol Chem. 2006;281:17321–36. doi: 10.1074/jbc.M600292200. [DOI] [PubMed] [Google Scholar]
- 20.Sorice M, Circella A, Cristea IM, Garofalo T, Di Renzo L, Alessandri C, Valesini G, Esposti MD. Cardiolipin and its metabolites move from mitochondria to other cellular membranes during death receptor-mediated apoptosis. Cell Death Differ. 2004;11:1133–45. doi: 10.1038/sj.cdd.4401457. [DOI] [PubMed] [Google Scholar]
- 21.Perez-Morga D, Vanhollebeke B, Paturiaux-Hanocq F, Nolan DP, Lins L, Homble F, Vanhamme L, Tebabi P, Pays A, Poelvoorde P, Jacquet A, Brasseur R, Pays E. Apolipoprotein L-I promotes trypanosome lysis by forming pores in lysosomal membranes. Science. 2005;309:469–72. doi: 10.1126/science.1114566. [DOI] [PubMed] [Google Scholar]
- 22.Vanhollebeke B, Truc P, Poelvoorde P, Pays A, Joshi PP, Katti R, Jannin JG, Pays E. Human Trypanosoma evansi infection linked to a lack of apolipoprotein L-I. N Engl J Med. 2006;355:2752–6. doi: 10.1056/NEJMoa063265. [DOI] [PubMed] [Google Scholar]
- 23.Widener J, Nielsen MJ, Shiflett A, Moestrup SK, Hajduk S. Hemoglobin is a co-factor of human trypanosome lytic factor. PLoS Pathog. 2007;3:1250–61. doi: 10.1371/journal.ppat.0030129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Molina-Portela MP, Samanovic M, Raper J. Distinct roles of apolipoprotein components within the trypanosome lytic factor complex revealed in a novel transgenic mouse model. J Exp Med. 2008;205:1721–28. doi: 10.1084/jem.20071463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vanhollebeke B, Pays E. The function of apolipoproteins L. Cell Mol Life Sci. 2006;63:1937–44. doi: 10.1007/s00018-006-6091-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mimmack ML, Ryan M, Baba H, Navarro-Ruiz J, Iritani S, Faull RL, McKenna PJ, Jones PB, Arai H, Starkey M, Emson PC, Bahn S. Gene expression analysis in schizophrenia: reproducible up-regulation of several members of the apolipoprotein L family located in a high-susceptibility locus for schizophrenia on chromosome 22. Proc Natl Acad Sci U S A. 2002;99:4680–4685. doi: 10.1073/pnas.032069099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takahashi S, Cui YH, Han YH, Fagerness JA, Galloway B, Shen YC, Kojima T, Uchiyama M, Faraone SV, Tsuang MT. Association of SNPs and haplotypes in APOL1, 2 and 4 with schizophrenia. Schizophr Res. 2008;104:153–64. doi: 10.1016/j.schres.2008.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sana TR, Janatpour MJ, Sathe M, McEvoy LM, McClanahan TK. Microarray analysis of primary endothelial cells challenged with different inflammatory and immune cytokines. Cytokine. 2005;29:256–69. doi: 10.1016/j.cyto.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 29.Horrevoets AJ, Fontijn RD, van Zonneveld AJ, de Vries CJ, ten Cate JW, Pannekoek H. Vascular endothelial genes that are responsive to tumor necrosis factor-alpha in vitro are expressed in atherosclerotic lesions, including inhibitor of apoptosis protein-1, stannin, and two novel genes. Blood. 1999;93:3418–31. [PubMed] [Google Scholar]
- 30.Kang MK, Kameta A, Shin KH, Baluda MA, Kim HR, Park NH. Senescence-associated genes in normal human oral keratinocytes. Exp Cell Res. 2003;287:272–81. doi: 10.1016/s0014-4827(03)00061-2. [DOI] [PubMed] [Google Scholar]